

Abstract
The mechanism by which modified single-stranded oligonucleotides (MSSOs) direct base changes in genes is not completely understood, but there is evidence that DNA damage, repair and cell cycle checkpoint proteins are involved in the targeted nucleotide exchange (TNE) process. We are interested in the role of the mismatch repair protein, Msh2 in the repair of a frameshift mutation in both yeast and mammalian cells. We show that this protein exerts different and opposing influences on the TNE reaction in MSH2 deficient yeast compared to MSH2-/- mammalian cells and in wild-type cells that have RNAi silenced Msh2. Data from yeast show a 10 fold decrease in the targeting frequency whereas mammalian cells have an elevated correction frequency. These results show that in yeast this protein is required for efficient targeting and may play a role in mismatch recognition and repair. In mammalian cells, Msh2 plays a suppressive role in TNE reaction by either precluding the oligonucleotide annealing to the target gene or by maintenance of a cell cycle checkpoint induced by the MSSO itself. These results reveal that the mechanism of TNE between yeast and mammalian cells is not conserved, and demonstrate that the suppression of the TNE reaction can be bypassed using RNAi against MSH2 designed to knockdown its expression.
Introduction
Targeted Nucleotide Exchange (TNE) is a process in which modified single-stranded DNA oligonucleotides (MSSOs) are used to direct base changes in genes. The mechanism of the TNE reaction has not been fully elucidated, but there is evidence that the reaction is separated into at least two phases: the pairing phase, which consists of the MSSO pairing to the target site within the gene, and the repair phase, in which the MSSO directs the repair of the targeted base. The repair phase itself may consist of two separate events: the first, the correction of the targeted strand is followed by a second reaction in which the newly modified strand serves as a template to alter the non-targeted strand. Mechanistic studies of TNE have revealed that a number of repair proteins play a role in the correction process by acting through various pathways that include mismatch repair (MMR), and nucleotide excision repair (NER) (Cole-Strauss et al., 1999; Dekker et al., 2003; Rice et al., 2001).
When oligonucleotides are transferred into the cell, mammalian cells respond by activating several DNA damage pathways, including homologous recombinational repair. This response also involves proteins that modulate cell cycle, DNA repair and DNA replication through the action of ATM, and Chk1 and Chk2 (Ferrara and Kmiec, 2004). In the process of studying the mechanism of TNE, we have become interested in the role of the mismatch repair protein, Msh2. Msh2 has several well-known cellular roles including mismatch recognition in a complex by itself, with Msh3 or Msh6, acting as an anti-recombinase during HR, and interacting with proteins involved in the regulation of the cell cycle (Alani et al., 1994; Alani et al., 1995; Brown et al., 2003; Chen and Jinks-Robertson, 1999; Datta et al., 1996; de Wind et al., 1995; Marsischky et al., 1996; Selva et al., 1995; Zhang et al., 2000). The potential involvement of Msh2 in the latter phases of the TNE reaction based on the endogenous functions of Msh2 suggests that this protein maybe active at several steps in the targeting process.
Initial data from experiments in yeast extracts indicated that Msh2 was not required to promote the TNE reaction in vitro (Rice et al., 2001). Furthermore, results from studies in mammalian cells revealed that ES cells deficient in MSH2 actually exhibited higher frequencies of TNE than the rate observed in wild-type cells (Dekker et al., 2003). These two observations created a paradox in the development of mechanistic models of TNE. The results in mammalian cells can be explained by the anti-recombinase activity of Msh2 where this protein has been shown to inhibit recombination between sequences containing more than 1% sequence divergence (Datta et al., 1996). The same protein is known to interact with recombination proteins in a “repairosome” structure called BASC (Wang et al., 2000), although a full characterization of this complex and its relation to TNE has yet to be completed.
We report here that MSH2 is involved in the TNE or gene repair reaction in both yeast and mammalian cells, but has different effects and perhaps directs different reactions in each system. In yeast, the correction efficiency is decreased in the absence of MSH2 in vivo, but in a mammalian MEF cell line, lacking the functional protein, the targeting efficiency is significantly increased. Therefore, we speculate that the role of MSH2 in yeast is more direct and possibly active at the level of the DNA, whereas in mammalian cells, MSH2 regulates the frequency of repair by suppressing the reaction through its anti-recombinase function.
Materials and Methods
Yeast strains, oligonucleotide vectors, and plasmids
The yeast strain LSY678 (Mat a leu2-3,112 trp1-1 ura3-1 his3-11,15 ade2-1can1-100) (a gift from Dr. Lorraine Symington, Colombia University) contains the plasmid pAUR101 HygeGFP (del) integrated at the AUR1 locus of chromosome IX. LSY814 (LSY678 msh2::TnLUK7-7) is an isogenic derivative of LSY678 containing the target gene at this locus. Both strains were maintained on selection (0.5 μg/ml) of aureobasidin. Oligonucleotide vectors, Hyg3S/74NT-A and Hyg3S/74T-T (HPLC purified), are 74 bases long and have three phosphorothioate linkages at both the 3′ and 5′ termini (Sigma Aldrich, St. Louis MO). The oligonucleotide vector Hyg3S/47NT-A is 47 bases in length and also contains three terminal phosophorothioate linkages to protect against nuclease degradation. The plasmid pEGFPN3, wild type for the enhanced green fluorescent protein (EGFP), has been mutated by site-directed mutagenesis to delete a cytosine at nucleotide position 875 (pEGFP N3 del1), which will result in a stop codon (TAG) at this position and the production of a truncated and nonfluorescent protein. Oligonucleotides, eGFP3S/47NT and eGFP3S/74NT, were designed to align to this mutated region within the EGFP gene and direct the insertion of a C, which will restore its reading frame and allow for translation of the full length protein product.
Targeted nucleotide exchange in S. cerevisiae
Plasmids and oligonucleotides were transformed into yeast by electroporation. Briefly, cells were grown in YPD at 30°C to a density of approximately 2×107 cells/ml; they were then harvested and treated with 25 μM DTT for 20 minutes at 30°C with shaking. Cells were washed twice with distilled H20 and once with 1 M sorbitol. They were resuspended in 120 μl of 1 M sorbitol and aliquots (40 μl; 2×108 cells) were electroporated by using a Bio-Rad gene pulser apparatus at the following settings:1.5 kV,25 μF,200 Ω, one pulse, and 5-s pulse length. The cells were then allowed to recover in 3 ml of YPD supplemented with 1 M sorbitol and 0.8 μg/ml of aureobasidin for 16 hours, then spread on YPD-hygromycin (300 μg/ml) plates and YPD-aureobasidin plates (0.5μg/ml).
Mammalian cell lines
Mouse embryonic fibroblasts (MEFs), wild-type or mutant (MSH2-/-), were kindly provided by Dr. Niels de Wind (Leiden University Medical Center). The MSH2-/- cell line was generated using a retroviral vector containing the adenovirus E1A oncogene and a neomycin gene. These cells were grown in Dulbecco’s Modified Eagle Medium supplemented with 4.5 g/L glucose,4 mM L-glutamine, pyridoxine hydrochloride, 100 U/ml penicillin,100 μg/ml streptomycin, and 10% FBS and maintained at 37°C and 5% CO2.
Targeted nucleotide exchange in MEF cells
The MEF cells were seeded at density of 2.5×106 cells in a 100 mm polystyrene dish and allowed to grow for 24 hours. The cells were then harvested after 24 hours and collected by centrifugation at 1500 rpm for 6 minutes. They were resuspended in complete DMEM with 10% FBS containing plasmid pEGFP N3 del1, and oligonucleotides, eGFP3S/47NT, to a total volume of 100 μl. This was placed in a 4 mm gap cuvette (Fisher Scientific, Pittsburgh, PA) and electroporated using a BTX Electro Square Porator ECM830 (BTX Genetronics Inc., San Diego, CA) under the following conditions: low voltage (LV), 250 volts, 13 msec, 2 pulses, 1 sec/pulse length. Cells were then transferred to a 60 mm polystyrene dish containing 3 ml of fresh complete DMEM medium with 10% FBS and incubated for 24 hours at 37°C before harvesting for FACS analysis.
Flow cytometric analysis
Green fluorescence (wild-type EGFP and corrected mutant EGFP) was measured using a FACScaliber flow cytometer (Becton Dickinson, San Jose, CA), using a procedure described previously (Brachman and Kmiec, 2004; Ferrara et al., 2004). Briefly, cells were harvested 24 hours after electroporation and resuspended in FACS buffer (0.5% bovine serum albumin [BSA]. 2 mM EDTA, 3 μg/ml propidium iodide [PI] in phosphate-buffered saline [PBS]). In each sample, 50,000 cells were counted and those cells that scored positive for EGFP are marked as corrected. For each sample, a control containing a wild-type EGFP plasmid was performed under the same targeting conditions to establish the transfection efficiency.
Knockdown of MSH2 by RNAi
RNAi, designed to knockdown Msh2, was purchased from Ambion (Ambion, Austin, Texas). The siRNA (ID# 155431) is designed to anneal to the mouse MSH2 RNA at exon 3 NM_008628. During the targeting experiments, the RNAi was electroporated with the oligonucleotide eGFP3S/47NT and the plasmid DNA pEGFP N3 del1 under the conditions described above.
RT PCR and Western Blot analyses
MEF WT cells were seeded at a density of 2.5×106 cells/100 mm dish and electroporated with plasmid, oligonucleotide and 1 nM MSH2 RNAi in a total volume of 100 μl. After electroporation, 25 μl was placed into each of four wells of a 6 well dish (Falcon, Becton Dickenson, Franklin Lakes, NJ) and allowed to recover for various times. All of the cells were harvested and RNA was extracted using the Rneasy Mini Kit from Qiagen (Qiagen Sciences, Maryland, 20874). Total RNA (200 ng) was used to generate cDNA using Superscript first strand synthesis system for RT PCR (Invitrogen, Carlsbad, CA) according to the manufacturers instructions. Four microliters of this reaction was used for PCR amplification using 0.2 μM mouse MSH2 primers: musMSH2/257F 5′agaccctgcagagtgttgtgctta and musMSH2/1010R 5′tgagagccagtggtgtcttcaaca, or mouse β-actin primers: mus β-actin 20F 5′ccgcgagcacagcttctttgc and mus β-actin 425R 5′gatctgggtcatcttttcacgg, under the following conditions: 2 minutes of 94°C for one cycle, than 30 seconds of 94°C, 30 seconds of 55°C, and 1 minute of 72°C for 25 cycles, with a final extension of 1 minute at 72°C.
To determine the level of MSH2 protein after RNAi treatment, cells were electroporated with plasmid, oligonucleotide, and 1 nM MSH2 RNAi and the samples were seeded into a 60 mm dish. After 8 and 12 hours, the cells were collected and total protein was extracted in 2X sample buffer at 95°C and incubated at 95°C with intermittent vortexing at full speed for a total of 12 minutes. The samples were then centrifuged for 10 minutes at 13,200 rpm at 4°C and the supernatant was collected. The total protein concentration was measured with the BioRad microBCA assay using BSA as a standard. Protein concentrations were determined, separated on a 10% SDS-PAGE gel according to standard procedures, and proteins were transferred to a PVDF membrane (Amersham Biosciences, UK). The blot was blocked for 2 hours at RT in 5% nonfat milk with 0.5% Tween-20 and then incubated with a primary antibody against MSH2 (rabbit polyclonal, Santa Cruz Biotechnology) or a goat polyclonal antibody against β-actin (Sigma). The appropriate species specific HRP-conjugate IgG was used as a secondary antibody to visualize proteins.
Results
Modified single-stranded oligonucleotides direct the repair of a frameshift mutation in the yeast chromosome
Modified single-stranded oligonucleotides (MSSOs) were used to direct the alteration of a deletion mutation in a Saccharomyces cerevisiae model system. This assay has previously enabled an investigation of optimal targeting vectors and proteins essential for the pairing phase of the TNE reaction (Liu et al., 2001; Liu et al., 2002a; Liu et al., 2002b; Liu et al., 2003; Liu et al., 2004; Parekh-Olmedo et al., 2002). Studies have shown that an MSSO is capable of directing the repair of a chromosomal insertion mutation, but the repair of a chromosomal deletion mutation was not investigated. Therefore, studies were undertaken to determine whether an MSSO could direct the repair of a deleted base. The yeast strain LSY678 contains plasmid pAUR101 Hyg eGFP (del) integrated at the AUR1 locus of chromosome IX (Figure 1A). The hygromycin gene has been mutated by a 1bp deletion at nucleotide 137 changing a tyrosine codon, TAT, to a stop codon, TAG (Figure 1B). Oligonucleotides, Hyg3S/74NT-A and Hyg3S/74T-T, were designed to correct this codon to the wild-type sequence TAT or ATA on the nontranscribed or transcribed strands, respectively (Figure 1C).
Figure 1.
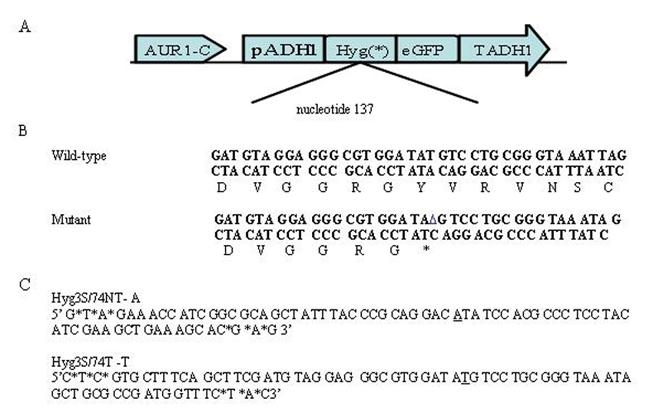
Yeast model system for TNE: A) The plasmid pAUR101 carrying a mutant hygromycin gene was integrated at the AUR1C locus in yeast strains and these were maintained under aureobasidin selection throughout the targeting assay. B) The hygromycin gene has been mutated at nucleotide position 137 from the wild type tyrosine TAT to an in-frame stop codon TAG. C) A 74 base oligonucleotide was chosen as a targeting vector because it showed the highest level of correction activity in this system. This vector is modified on either end by the addition of three terminal phosphorothioated bases and is designed to bind to either the nontranscribed (Hyg3S/74NT-A) or transcribed (Hyg3S/74T-T) strand of the hygromycin gene. These oligonucleotides will direct the base change to encode the wild type sequence and allow the yeast to confer hygromycin resistance.
Previously, a dose response was observed when a chromosomal point mutation was targeted in LSY678 with an MSSO (Liu et al., 2002a). Therefore, using a similar strategy, we tested different amounts Hyg3S/74NT-A and Hyg3S/74T-T to establish a dose response; Hyg3S/74NT-A targets the nontranscribed strand while Hyg3S/74T-T targets the transcribed strand. The correction efficiency was evaluated by dividing the number of hygromycin resistant (Hygr) colonies by the number of aureobasidin resistant (Aureor x 10-5) colonies.
Hyg3S/74NT-A was chosen, because a 74-mer was shown previously to be an effective vector length in the correction of an episomal frameshift mutation; the nontranscribed strand (NT) was shown to be a better target than the transcribed strand (T) (Liu et al., 2001; Liu et al., 2002b). Lengths shorter than 60-70 nucleotides are generally not productive in directing yeast gene repair. Our results, depicted in Figure2a, show that a deletion mutation can be corrected using a modified single-stranded oligonucleotides with a length of 74 nucleotides. The targeting frequency is increased as the oligonucleotide concentration is elevated in a dose dependent fashion. For comparison, the transcribed strand (T) was also targeted to determine whether there is a strand bias in frameshift correction at the level of the chromosome. We observed that the transcribed strand is less amenable to TNE directed by Hyg3S/74T-T. Thus, a strand bias that favors the NT strand for the repair of a deletion exists with limited dose response using Hyg3S/74T-T as opposed to Hyg3S/74NT-A. We then targeted the mutation with Hyg3S/47NT-A to determine if a shorter oligonucleotide might function in a system containing a frameshift mutation. As shown in Figure 2a, the 47 base oligonucleotide targets at a very low, nearly undetectable efficiency and demonstrates no dose response. These results are consistent with earlier reports (Liu et al. 2002a, Liu et al. 2002b).
Figure 2.
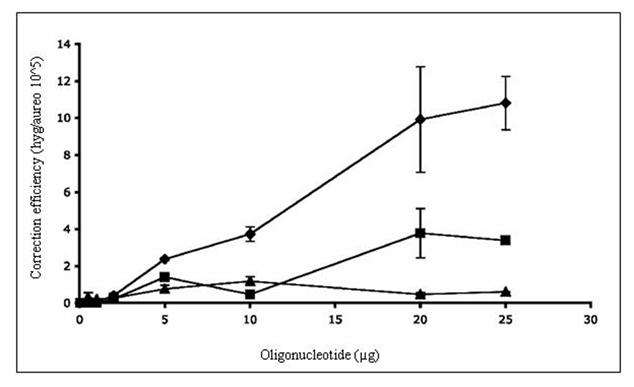
TNE of a chromosomal frameshift mutation in wild-type and mutant yeast. A) The yeast strain LSY678 (MATa leu2-3,112 trp1-1 ura3-1 his3-11,15 ade2-1 can1-100) was used as a host to demonstrate the repair of a chromosomal frameshift mutation. The oligonucleotides Hyg3S/74NT-A (◆), Hyg/3S74T-T (■), or Hyg/3S47NT-A (▲) were introduced over a range of concentrations into the strain by electroporation and after 16 hours of recovery the cells were plated on selective media to assess the level of correction. The correction efficiency (CE) is determined by the number of hygromycin resistant colonies (corrected cells) divided by the number of aureobasidin colonies (total number of viable cells capable of being corrected). B) The targeting oligonucleotides Hyg3S/74NT-A, Hyg3S/47NT-A, and Hyg3S/74T-T were delivered at a range of concentrations to LSY814 (◆,▲, ■, respectively) by electroporation as well as Hyg3S/74NT-A into LSY678 (◆) and the correction efficiency was calculated as described above. The mean and standard deviation for each sample were determined from at least three independent experiments performed in duplicate and are represented in the table below the graph.
Frameshift repair in the absence of Msh2 in Saccharomyces cerevisiae
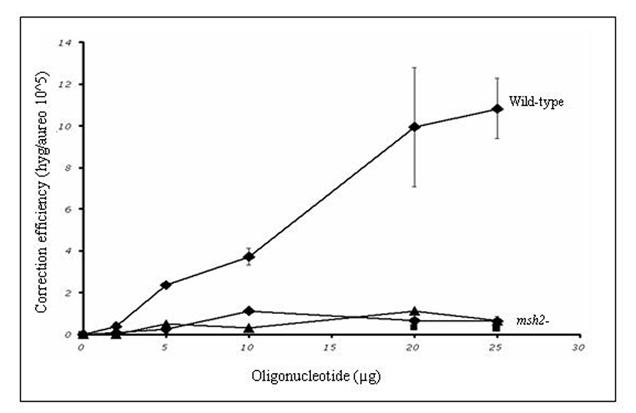
The yeast strain, LSY814, is isogenic to LSY678 except that the MSH2 gene is not functional. We integrated the hygromycin gene into the yeast genome of LSY814 and maintained the strain on aureobasidin selection (see Methods and Materials). A new stock was streaked out after three passages under selection, as this strain has a distinct mutator phenotype and must be transferred routinely (Marsischky et al., 1996). Hyg3S/74NT-A was introduced by electroporation and the correction efficiency was determined by selection on hygromycin and aureobasidin plates as described above. Neither the wild-type nor the msh2 deficient strain show backgroun correction as indicated in Table 1 at the zero oligonucleotide concentration. Compared to the wild type level of correction, the msh2 deficient strain targets less efficiently with Hyg3S/74NT-A (Table 1 and Figure 2B). To determine whether the decrease in correction was due, in part, to the length of the oligonucleotide, we carried out a dose response curve with the 47-mer, Hyg3S/47NT-A. As depicted in Figure 2B, the lower correction efficiency in the msh2 strain is not dependent on the length of the oligonucleotide. We next wanted to determine whether a strand bias of gene repair exists in this strain and targeted the mutation with Hyg3S/74T-T at 20 and 25 μg concentrations. As Figure 2B shows, there is no difference when targeting the T strand versus the NT strand and this is due to the inability of the msh2- strain to target. The 10-fold difference in correction efficiency is maintained at the higher concentrations between the wild-type and msh2 deficient yeast, when targeting with the Hyg3S/74T-T, but there is no difference when targeting with Hyg3S/47NT-A.
Table 1.
Levels of gene repair in msh2 deficient yeast. Wild-type and msh2 deficient yeast strains were targeted with increasing amounts of various oligonucleotides. The targeting oligonucleotide is depicted in the left hand column and CE is provided for both strains.
| ODN | Amount (μg) | Average Correction Efficiency (CE) per 105 | |
|---|---|---|---|
| LSY678 | LSY814 | ||
| Hyg3S/74NT-A | -- | 0.0 | 0.0 |
| 5 | 2.40+/-0.07 | 0.20+/-0.13 | |
| 10 | 3.70+/-0.40 | 1.10+/-0.00 | |
| 20 | 9.90+/-2.80 | 0.70+/-0.04 | |
| 25 | 10.80+/-1.45 | 0.60+/-0.22 | |
| Hyg3S/74T-T | -- | 0.0 | 0.0 |
| 5 | 1.40+/-0.13 | nd | |
| 10 | 0.47+/-0.07 | nd | |
| 20 | 3.80+/-1.33 | 0.30+/-0.09 | |
| 25 | 3.40+/-0.18 | 0.30+/-0.15 | |
| Hyg3S/47NT-A | -- | 0.0 | 0.0 |
| 5 | 0.77+/-0.21 | 0.50+/-0.13 | |
| 10 | 1.18+/-0.24 | 0.30+/-0.09 | |
| 20 | 0.47+/-0.10 | 1.1+/-0.08 | |
| 25 | 0.61+/-0.01 | 0.7+/-0.04 | |
Repair of a frameshift mutation in the absence of functional MSH2
The data obtained from yeast suggest that the MMR proteins are required for generating gene repair activity. These observations run counter to data obtained in mammalian cells where the role of Msh2, in the oligonucleotide mediated gene repair reaction, was characterized (Dekker et al., 2003; Drury et al., 2005). In those studies, Msh2 was found to suppress the reaction. Hence, an investigation was undertaken to determine whether the MSH2 was necessary in mammalian cells for the repair of a frameshift mutation. The plasmid pEGFP N3 del1, which contains a single base deletion at nucleotide 875, was delivered to wild-type and MSH2-deficient MEF cells with the correcting MSSO’s, eGFP3S/47NT or eGFP3S/74NT, by square-wave electroporation (Figure 3A). The cells were allowed to recover for 24 hours in complete media with 10% FBS before preparation for FACS analysis. As shown in Figure 3B, the MEF MSH2-/- cells targeted at a significant (p<0.05) 2 fold higher frequency than those containing a functional MSH2 and there is no dependence on oligonucleotide length in this system. The data, therefore, are in agreement with earlier findings, which indicate that cells lacking MSH2 exhibit higher levels of TNE in the gene repair reaction (Dekker et al., 2003; Drury et al., 2005).
Figure 3.
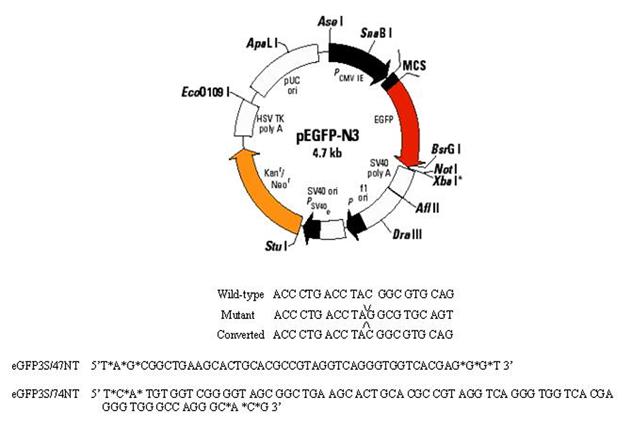
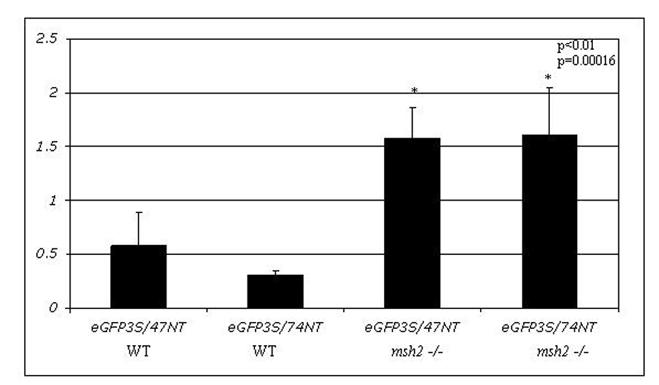
Elevated correction frequency of an episomal frameshift mutation in MSH2 deficient MEF cells: A) Plasmid pEGFPN3 has been mutated to contain a -1 frameshift mutation (pEGFP N3 del1) in the eGFP coding region, which results in a stop codon and causes the production of a truncated and non-functional eGFP protein. An oligonucleotide (eGFP3S/47NT) was designed to align to this sequence and direct the insertion of C, which will restore the reading frame and allow for eGFP protein production. B) The mutant pEGFP N3 del1 plasmid (2.5 μg) and correcting oligonucleotide eGFP3S/47NT (5 μM) were introduced in to mouse embryonic fibroblast (MEF) cells both wild type and deficient for MSH2. The efficiency of correction (CE) was measured as a percentage of EGFP expressing cells in a population of 50,000 by FACS and normalized by the percentage of cells that took up a wild-type eGFP (pEGFPN3) plasmid within the same experiment. The means and standard deviations for each sample were calculated from six individual experiments and a student T-test was performed to evaluate the differences between TNE in these two cell lines.
Reverse complementation of MSH2 in wild-type MEF cells
In order to determine whether this increase in gene repair is due to the absence of MSH2 or, perhaps, due to the inherent mutator phenotype of this type of cell line (de Wind et al., 1999; Reitmair et al., 1997); RNAi technology was used to target MSH2 transcripts and knockdown its expression in the wild type MEF cell line. This cell line has a functional MSH2 gene, which is expressed normally throughout the cell cycle (Reitmair et al., 1997). The cells were seeded at a density of 2.5 x106 cells and allowed to grow for 16 hours. They were then harvested and electroporated with the plasmid pEGFP N3 del1 and eGFP3S/47NT as previously described, except that 1 nM MSH2 RNAi was included in the reaction mixture. Again, the correction frequency was determined by FACS analysis after 24 hours. In the presence of the RNAi, the targeting frequency increased significantly (p<0.05) over the basal targeting level in the wild type line (Figure 4). To determine whether the RNAi itself was having an effect on the correction frequency, we added it to a targeting reaction carried out in MSH2-/- cells. The results indicate (Figure 4) that this addition did not significantly impact the correction frequency in mutant cells (p>0.04).
Figure 4.
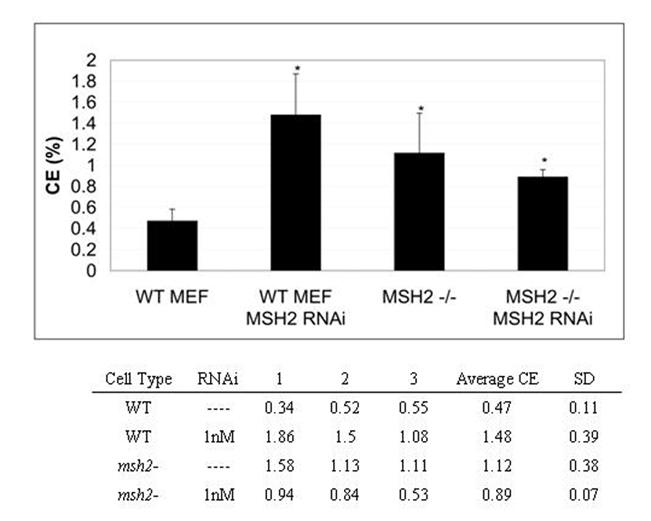
MSH2 knockdown by RNAi in WT MEF cells shows MSH2-/-phenotype. RNAi was introduced to the episomal targeting reaction to knockdown the expression of MSH2 in the WT MEF cell line. WT MEF cells were seeded at a density of 2.5×106 and after 24 hours cells were collected. Plasmid pEGFP N3 del1 (2.5 μg), oligonucleotide (5 μM) and MSH2 RNAi (where indicated) were introduced by electroporation. The cells were allowed to recover for 24 hours in complete media before preparation for FACS analysis and the correction efficiencies were normalized by the percentage of cells that took up the wild type eGFP plasmid under the same conditions. The means and standard deviations for each sample were calculated from three individual experiments and a student T-test was performed to evaluate the significance in differences from the WT MEF line with no MSH2 RNAi.
RT PCR was performed to ensure that the RNAi was reducing Msh2 RNA levels. Total RNA was extracted at 0, 3, 6, and 10 hours after electroporation of the plasmid, oligonucleotide, and Msh2 RNAi. Complementary DNA was synthesized using an oligo dT primer and MSH2 transcripts were amplified in subsequent PCR reactions. Figures 5A and 5B show that the expression level of MSH2 is decreased by three hours after transfection with the RNAi. A Western blot was then carried out to determine whether Msh2 protein levels were lower after introduction of the RNAi. Cells were targeted with or without RNAi in the presence of the mutant plasmid and correcting oligonucleotide. Total protein was extracted at 8 and 12 hours post electroporation, respectively. As shown in Figure 6, Msh2 levels are reduced in samples that received the RNAi for both 8 and 12 hours as compared to the control samples. Thus, the increase in gene repair activity in the wild-type MEF cells is coincident with the knockdown in MSH2 expression and a decrease in the cellular level of Msh2.
Figure 5.
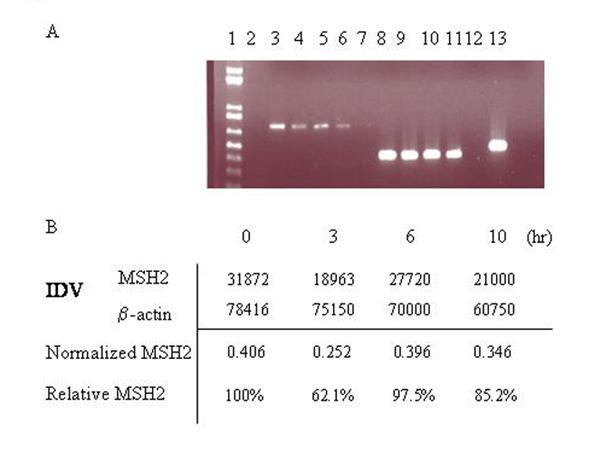
MSH2 mRNA levels are decreased in MEF WT cells treated with RNAi. A)RT PCR was performed on total RNA extracts from cells co-transfected with pEGFP N3 del1, eGFP3S/47NT, and MSH2 RNAi at different time points after electroporation. Lane 1: 1Kb plus ladder, 2: empty, 3: 0h MSH2, 4: 3h MSH2, 5: 6h MSH2, 6: 10h MSH2, 7: neg control dH20. 8: 0h mus β-actin, 9: 3h mus β-actin, 10: 6h mus β-actin, 11: 10h mus β-actin, 12: neg control dH20, 13: HeLa positive control. B) Spot densitometry was performed to determine the relative percentage of MSH2 present over time after treatment with MSH2 RNAi. Integrated Density Values (IDV) (average density per area) was obtained for both MSH2 and β-actin for each time point and the MSH2 value was normalized to the level of β-actin. The percentage of MSH2 present over time was then determined relative to the zero hour time point.
Figure 6.
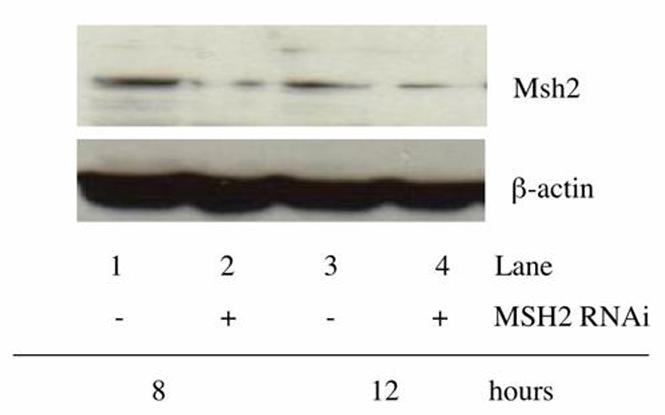
Msh2 protein levels are knocked down in WT MEF cells after treatment with RNAi. The protein level of Msh2 was measured following treatment with RNAi in the WT MEF cells. Cells were co-transfected with the mutant eGFP plasmid (2.5 μg) and correcting oligonucleotide (5 μM), with and without MSH2 RNAi (1 nM) and total cellular protein was extracted after 8 and 12 hours of recovery. Western blot analysis was performed to determine the level of Msh2 protein after this treatment and β-actin was measured as a loading control. Lane 1: 8 hours no MSH2 RNAi.; Lane 2: 8 hours with MSH2 RNAi.; Lane 3: 12 hours no MSH2 RNAi.; Lane 4: 12 hours with MSH2 RNAi.
Discussion
The mechanism of gene repair is not clearly defined with regard to how the mispaired base is actually corrected. It is also not known whether specific repair proteins recognize the mismatch and alter a base using the oligonucleotide as the template. It is plausible that this type of repair is unnecessary and the oligonucleotide simply incorporates into the DNA at progressing replication forks (Parekh-Olmedo et al., 2005). There is substantial evidence however that proteins involved in homologous recombinational repair are active in the TNE or gene repair reaction (Ferrara and Kmiec, 2005; Ferrara and Kmiec, 2004).
It is widely known that the Msh2/Msh6 heterodimer (MutSα) or Msh2/Msh3 (MutSβ) recognizes aberrant structures and signals for the repair of single base mismatches or loops in all types of cells (Marsischky et al., 1996). MMR proteins have also been shown to inhibit recombination between divergent DNA sequences and are responsible for maintenance of the G2 checkpoint by CHK2 following DNA damage (Chen and Jinks-Robertson, 1999; Datta et al., 1996; Franchitto et al., 2003; Selva et al., 1995). MSH2 interacts directly with HR proteins (Brown et al., 2003; Wang et al., 2000), some of which have now been shown to play a direct role in the TNE process (Ferrara and Kmiec, 2004).
In this study, we wanted to understand the role of Msh2 in the TNE reaction in yeast and mammalian cells and we find that a deficiency in this protein affects the targeting frequency in these cells differentially. Repair in yeast requires this protein for efficient activity whereas, in mammalian cells, Msh2 appears to inhibit TNE. When the TNE reaction is carried out in yeast strains lacking MSH2, there is a reproducible 10-fold decrease in correction efficiency. Thus, when comparing the data obtained when targeting with Hyg3S/74NT-A, the yeast MMR proteins could be acting to correct the mispair directly. A negative regulation of TNE is observed in mammalian cells wherein cells defective in MSH2 exhibit 2-fold higher correction efficiency. Consistent with these data is our observation that an increase in targeting occurs when the MSH2 transcript is knocked down with RNAi, suggesting a suppressive role for Msh2 in wild-type mammalian cells. Although, initially, the reaction mechanism was dissected in yeast and some of these data have been important in guiding the field toward understanding the mechanism in mammalian cells, it is now apparent that the role of Msh2 in higher organisms is complex.
The DNA mismatch repair (MMR) pathway recognizes and repairs mismatches that have accumulated during replication and recombination between diverged DNA sequences (see Kunkel and Erie, 2005 for review). In an ATP-driven reaction, Msh2 along with mismatch specific proteins Msh6 or Msh3, recognize and bind single base mismatches or loops and larger insertion-deletion loops, respectively (Kolodner and Marsischky, 1999). Binding at the mismatched base pair signals other proteins to assemble at the site and initiate its repair (Bowers et al., 2000). Msh2 colocalizes with p53 and Rad51, and has been immunoprecipitated in a multiprotein complex termed BASC (BRCA1 associated surveillance complex) (Wang et al., 2000; Yang et al., 2004). Interestingly, we now know that these proteins regulate the TNE reaction (Brachman and Kmiec, 2005; Ferrara and Kmiec, 2005; Ferrara and Kmiec, 2004; Liu et al., 2004). The Msh2/6 heterodimer has been shown to suppress the action of these proteins in the homologous recombinational repair (HRR) reaction (de Wind et al., 1995; de Wind et al., 1999; Modrich and Lahue, 1996). When the 47-mer oligonucleotide creates a single mismatch upon hybridization, a 2% sequence divergence is created at the target site. Additionally, the 74-mer oligonucleotide creates a divergence that is greater than 1%.
Recently, mismatch repair proteins have been implicated in checkpoint activation during S-phase of the cell cycle. Furthermore, Msh2 was shown to bind CHK2 specifically (Brown et al., 2003; Ferrara and Kmiec, 2005), a protein we have reported is activated in the presence of the correcting oligonucleotide. Cells in which an oligonucleotide has been introduced tend not to divide normally (Ferrara and Kmiec, 2005; Olsen et al., 2005) and are apparently slowed through the S and G2 phases of the cell cycle. This delay in division is presumably due to checkpoint activation by DNA damage response proteins including ATM, Chk1 and most interestingly Chk2 (Ferrara and Kmiec, 2005). Thus, a possible role for Msh2 in suppressing TNE lies in its interaction with Chk2. Cells devoid of Msh2 may lack the G2 checkpoint activation enabled by Msh2-Chk2 and are able to stabilize the correction efficiency by bypassing G2 arrest. Alternatively, Msh2 could impede the binding of the Rad51-oligo complex to the target site, specifically if the overall sequence divergence is greater than 1% as discussed above.
It is clear that in mammalian cells, Msh2 is not responsible for directing the repair of the mismatch at the DNA level. The incorporation of the oligonucleotide at a replication fork and subsequent rounds of replication alleviates the need for such traditional repair activity (see Parekh-Olmedo et al., 2005). But, this model of gene repair may be applicable to mammalian cells since there is a clear requirement for Msh2 in the yeast TNE reaction. Since the goal of gene repair is to correct mutations with high efficiency in mammalian cells, MSH2 RNAi could emerge as a useful tool in achieving this goal. Studies coupling the activity of RNAi and MSSO-directed gene repair are well underway.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alani E, Chi NW, Kolodner R. The Saccharomyces cerevisiae Msh2 protein specifically binds to duplex oligonucleotides containing mismatched DNA base pairs and insertions. Genes Dev. 1995;9:234–247. doi: 10.1101/gad.9.2.234. [DOI] [PubMed] [Google Scholar]
- Alani E, Reenan RA, Kolodner RD. Interaction between mismatch repair and genetic recombination in Saccharomyces cerevisiae. Genetics. 1994;137:19–39. doi: 10.1093/genetics/137.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers J, Tran PT, Liskay RM, Alani E. Analysis of yeast MSH2-MSH6 suggests that the initiation of mismatch repair can be separated into discrete steps. J.Mol.Biol. 2000;302:327–338. doi: 10.1006/jmbi.2000.4081. [DOI] [PubMed] [Google Scholar]
- Brachman EE, Kmiec EB. DNA replication and transcription direct a DNA strand bias in the process of targeted gene repair in mammalian cells. J.Cell Sci. 2004;117:3867–3874. doi: 10.1242/jcs.01250. [DOI] [PubMed] [Google Scholar]
- Brachman EE, Kmiec EB. Gene repair in mammalian cells is stimulated by the elongation of S phase and transient stalling of replication forks. DNA Repair (Amst) 2005;4:445–457. doi: 10.1016/j.dnarep.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R. The mismatch repair system is required for S-phase checkpoint activation. Nat.Genet. 2003;33:80–84. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- Chen W, Jinks-Robertson S. The role of the mismatch repair machinery in regulating mitotic and meiotic recombination between diverged sequences in yeast. Genetics. 1999;151:1299–1313. doi: 10.1093/genetics/151.4.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole-Strauss A, Gamper H, Holloman WK, Munoz M, Cheng N, Kmiec EB. Targeted gene repair directed by the chimeric RNA/DNA oligonucleotide in a mammalian cell-free extract. Nucleic Acids Res. 1999;27:1323–1330. doi: 10.1093/nar/27.5.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Adjiri A, New L, Crouse GF, Jinks RS. Mitotic crossovers between diverged sequences are regulated by mismatch repair proteins in Saccaromyces cerevisiae. Mol.Cell Biol. 1996;16:1085–1093. doi: 10.1128/mcb.16.3.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Berns A, Radman M, Te RH. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- de Wind N, Dekker M, Claij N, Jansen L, van Klink Y, Radman M, Riggins G, van d., V, van’t Wout K, Te RH. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat.Genet. 1999;23:359–362. doi: 10.1038/15544. [DOI] [PubMed] [Google Scholar]
- Dekker M, Brouwers C, Te RH. Targeted gene modification in mismatch-repair-deficient embryonic stem cells by single-stranded DNA oligonucleotides. Nucleic Acids Res. 2003;31:E27. doi: 10.1093/nar/gng027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury MD, Skogen MJ, Kmiec EB. A tolerance of DNA heterology in the mammalian targeted gene repair reaction. Oligonucleotides. 2005;15:155–171. doi: 10.1089/oli.2005.15.155. [DOI] [PubMed] [Google Scholar]
- Ferrara L, Kmiec E. Gene Repair activates ATM and stall DNA replication in corrected mammalian cells (submitted) 2005. [DOI] [PubMed] [Google Scholar]
- Ferrara L, Kmiec EB. Camptothecin enhances the frequency of oligonucleotide-directed gene repair in mammalian cells by inducing DNA damage and activating homologous recombination. Nucleic Acids Res. 2004;32:5239–5248. doi: 10.1093/nar/gkh822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara L, Parekh-Olmedo H, Kmiec E. Enhanced oligonucleotide-directed gene targeting in mammalian cells follwoing treatment with DNA damaging agents. Exp.Cell Res. 2004;300:170–179. doi: 10.1016/j.yexcr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Franchitto A, Pichierri P, Piergentili R, Crescenzi M, Bignami M, Palitti F. The mammalian mismatch repair protein MSH2 is required for correct MRE11 and RAD51 relocalization and for efficient cell cycle arrest induced by ionizing radiation in G2 phase. Oncogene. 2003;22:2110–2120. doi: 10.1038/sj.onc.1206254. [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr.Opin.Genet.Dev. 1999;9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu.Rev.Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Liu L, Cheng S, van Brabant AJ, Kmiec EB. Rad51p and Rad54p, but not Rad52p, elevate gene repair in Saccharomyces cerevisiae directed by modified single-stranded oligonucleotide vectors. Nucleic Acids Res. 2002a;30:2742–2750. doi: 10.1093/nar/gkf397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Maguire KK, Kmiec EB. Genetic re-engineering of Saccharomyces cerevisiae RAD51 leads to a significant increase in the frequency of gene repair in vivo. Nucleic Acids Res. 2004;32:2093–2101. doi: 10.1093/nar/gkh506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Parekh-Olmedo H, Kmiec EB. The development and regulation of gene repair. Nat.Rev.Genet. 2003;4:679–689. doi: 10.1038/nrg1156. [DOI] [PubMed] [Google Scholar]
- Liu L, Rice MC, Drury M, Cheng S, Gamper H, Kmiec EB. Strand bias in targeted gene repair is influenced by transcriptional activity. Mol.Cell Biol. 2002b;22:3852–3863. doi: 10.1128/MCB.22.11.3852-3863.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Rice MC, Kmiec EB. In vivo gene repair of point and frameshift mutations directed by chimeric RNA/DNA oligonucleotides and modified single-stranded oligonucleotides. Nucleic Acids Res. 2001;29:4238–4250. doi: 10.1093/nar/29.20.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–420. doi: 10.1101/gad.10.4.407. [DOI] [PubMed] [Google Scholar]
- Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu.Rev.Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- Olsen PA, Randol M, Krauss S. Implications of cell cycle progression on functional sequence correction by short single-stranded DNA oligonucleotides. Gene Ther. 2005;12:546–551. doi: 10.1038/sj.gt.3302454. [DOI] [PubMed] [Google Scholar]
- Parekh-Olmedo H, Drury M, Kmiec EB. Targeted nucleotide exchange in Saccharomyces cerevisiae directed by short oligonucleotides containing locked nucleic acids. Chem.Biol. 2002;9:1073–1084. doi: 10.1016/s1074-5521(02)00236-3. [DOI] [PubMed] [Google Scholar]
- Parekh-Olmedo H, Ferrara L, Brachman E, Kmiec EB. Gene therapy progress and prospects: targeted gene repair. Gene Ther. 2005;12:639–646. doi: 10.1038/sj.gt.3302511. [DOI] [PubMed] [Google Scholar]
- Reitmair AH, Risley R, Bristow RG, Wilson T, Ganesh A, Jang A, Peacock J, Benchimol S, Hill RP, Mak TW, Fishel R, Meuth M. Mutator phenotype in Msh2-deficient murine embryonic fibroblasts. Cancer Res. 1997;57:3765–3771. [PubMed] [Google Scholar]
- Rice MC, Bruner M, Czymmek K, Kmiec EB. In vitro and in vivo nucleotide exchange directed by chimeric RNA/DNA oligonucleotides in Saccharomyces cerevisae. Mol.Microbiol. 2001;40:857–868. doi: 10.1046/j.1365-2958.2001.02407.x. [DOI] [PubMed] [Google Scholar]
- Selva EM, New L, Crouse GF, Lahue RS. Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. Genetics. 1995;139:1175–1188. doi: 10.1093/genetics/139.3.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Zhang R, Wang XW, Linke SP, Sengupta S, Hickson ID, Pedrazzi G, Perrera C, Stagljar I, Littman SJ, Modrich P, Harris CC. The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene. 2004;23:3749–3756. doi: 10.1038/sj.onc.1207462. [DOI] [PubMed] [Google Scholar]
- Zhang H, Marra G, Jiricny J, Maher VM, McCormick JJ. Mismatch repair is required for O(6)-methylguanine-induced homologous recombination in human fibroblasts. Carcinogenesis. 2000;21:1639–1646. doi: 10.1093/carcin/21.9.1639. [DOI] [PubMed] [Google Scholar]