

Abstract
Actin polymerization and depolymerization plays a central role in controlling a wide spectrum of cellular processes. There are many actin-binding proteins in eukaryotic cells. Their roles in the remodeling of the actin architecture and whether they work cooperatively await further study. Caldesmon (CaD) is an actin-binding protein present in nearly all mammalian cells. Cortactin is another actin-binding protein found mainly in the cell cortex. There have been no reports suggesting that CaD and cortactin interact with each other or work as partners. Here we present evidence that CaD binds cortactin directly by overlay, pull-down assays, ELISA and by column chromatography. The interaction involves the N-terminal region of cortactin and the C-terminal region of CaD, and appears to be enhanced by divalent metal ions. Cortactin competes with both full-length CaD and its C-terminal fragment for actin binding. Binding of cortactin partially alleviates the inhibitory effect of CaD on the actomyosin ATPase activity. Not only can binding be demonstrated in vitro, the two proteins also co-localize in activated cells at the cortex. Whether such interactions bear any functional significance awaits further investigation.
Actin polymerization and depolymerization plays a central role in controlling a wide spectrum of cellular phenomena and intracellular processes. This is especially true in cells like smooth muscle cells (SMCs), which undergo cell shape changes during contraction and relaxation. The ability to recover cell shape after either transient or sustained deformation, depends on the dynamic and constant rearrangement of actin cytoskeleton (Bursac et al., 2005; Seow, 2005). Cytoskeletal disassembly is known to contribute to the hyporesponsiveness of SMC and mesangial cells to vasopressors both in vitro and in vivo (Zhou et al., 1995; Cortes et al., 2000). It was also found that under pathological conditions that mimic cytochalasin D treatment, mesangial cells respond with an up-regulation of a group of actin cytoskeleton regulatory proteins in an attempt to restore the cytoskeletal integrity (Clarkson et al., 2002).
The canonical mechanism of actin regulation involves cofilin (an actin disassembly factor), profilin (a G-actin sequester), Arp2/3 (a branching factor), formin (a barbed end capping protein that promotes actin assembly) and capping proteins (that controls the disassembly at the pointed end). However, there are many more other actin-binding proteins in the eukaryotic cells. These include gelsolin (a severing protein) and tropomyosin (a filament stabilizer). Individual actin filaments are further organized into higher order structures by α -actinin (a bundling protein), filamin and spectrin (crosslinking proteins), as well as myosins (motors). The actin filaments are anchored to the cell membrane to provide a framework that allows force-bearing; this is achieved by a group of membrane-associated proteins, such as dystrophin, utrophin, talin, vinculin, paxillin and integrin. While the players are identified, the interactions between them are largely unknown. Like non-muscle cells, SMCs also contain a large number of actin-binding proteins. The roles of these cytoskeleton proteins in the remodeling of the actin architecture of SMC and whether they work in a cooperative fashion are intriguing questions that await further study.
Caldesmon (CaD) is an actin-binding protein present in both smooth muscle and non-muscle cells. In addition to actin, it also binds myosin and calmodulin, making it one of a few unique actin-binding proteins. Binding of actin is weakened by phosphorylation and by calmodulin in the presence of Ca2+ (for recent reviews, see Huber, 1997; Wang, 2001). These biochemical properties equipped CaD to be an ideal candidate for a regulatory element of the actin cytoskeleton. Recent studies have shown that CaD stabilizes actin stress fibers and may control the cell shape change during proliferation and migration (Kordowska et al., 2006).
Cortactin is also an actin-binding protein, mainly found in the cell cortex (Weed and Parsons, 2001). The molecular structure suggests that cortactin functions to promote actin assembly, especially at the branching site, because it also binds and activates Arp2/3 (Daly, 2004). CaD, on the other hand, inhibits Arp2/3-mediated actin nucleation (Yamakita et al., 2003). There have been no reports that CaD and cortactin would interact with each other, despite both bind actin and are involved with Arp2/3. Structurally CaD and cortactin share some common features too: both are elongated molecules with a central region containing repeating sequences. Here we present data to show that CaD is capable of interacting with cortactin directly. Not only binding can be demonstrated in the in vitro experiments, the two proteins are also co-localized at cell cortex. Whether such interactions bear any functional significance remains to be further investigated. It may prove to be another example that cytoskeletal proteins work together to constitute a more versatile regulatory system that allows the cell to weather a wide range of conditions and environmental impacts.
Materials and Methods
Protein Preparations
Cortactin was cloned from mouse smooth muscle RNA (stomach) mixture by RT-PCR using primers derived from the amino acid sequence at both N- and C-terminal segments. After DNA sequencing it was found that the repeating motif was shorter (the 6th kelch repeat is missing), and therefore the isoform was identified as cortactin-B. The cDNA of the His-tagged protein was subcloned into an expression vector (pET28a) and transformed into E. coli cells for expression. Recombinant cortactin-B was purified by Ni2+-column, and further purified by using a CaD-Sepharose affinity column that was prepared by coupling 1.5 mg C-terminal CaD fragment (H32K) to 1 g CNBr-activated Sepharose 4B according to manufacturer’s instructions. Crude preparation of cortactin was loaded onto the H32K-Sepharose column in the presence of 1 mM CaCl2 or MgCl2. After washing with the same buffer, the column was eluted with a buffer containing 5 mM EDTA, and further eluted with a buffer containing 0.5 M NaCl or 50 mM glycine (pH 2,3) and 250 mM NaCl. All fractions were analyzed by SDS-PAGE. Recombinant full-length smooth muscle CaD was expressed in insect cells and purified without heating (Zhuang et al., 1996). Recombinant fragment of the N-terminal region (N240) and the C-terminal region (H32K) of CaD were prepared according to the chicken sequence as previously described (Li et al., 2000; Huang et al., 2003). Purified cortactin-B was also coupled to CNBr-activated Sepharose 4B beads for pull-down experiments.
Antibodies
Anti-CaD (H-C) and anti-phospho-CaD (anti-pS789CaD) polyclonal antibodies, both being raised in this laboratory, have been described previously (Mabuchi et al., 1996; D’Angelo et al., 1999). Monoclonal anti-cortactin (#05-180) was obtained from Upstate.
Western Analysis
The eluted fractions collected from H32K-Sepharose affinity column were separated on SDS-PAGE and immunoblotted with monoclonal anti-cortactin followed by affinity purified anti-mouse secondary antibodies, conjugated with IRDyeTM 800. For detection we used Odyssey Infrared Imaging System by LI-COR, Biosciences (Lincoln, NE) allowing quantitative data presentation by measuring direct infrared fluorescence.
Overlay Assays
Recombinant cortactin-B (20 μg) was first run on 10% SDS-PAGE, and electrophoretically transblotted to PVDF membrane. After washing with PBS buffer, the membrane was blocked with LI-COR Odyssey blocking buffer, cut into strips, and incubated separately with 0.5 μM CaD, H32K or N240 in a solution containing 20 mM Tris-HCl, pH7.5, 50 mM NaCl, 5 μM leupeptin, 1 mM dithiothreitol (DTT) and 1 mM PMSF in the presence of 1mM CaCl2 or EDTA after being washed with the same buffer. After further washing, the membrane strips were rinsed with PBS-Tween, and allowed to react with anti-CaD polyclonal antiserum, followed by fluorescent probe labeled anti-rabbit secondary antibodies and scanning with the LI-COR Odyssey Infrared Image system.
Pull-down assay
Interaction between proteins was assayed by incubating H32K- (or cortactin-) coupled Sepharose beads with cortactin (or full-length CaD, H32K or N240) in a buffer containing 20 mM Tris-Hcl, pH 7.5, 50 mM NaCl and 1 mM CaCl2 for 1 hour, washing with the same buffer four times, each time the beads were spun down at 3000 rpm for 5 min, followed by addition of a buffer containing 2 mM EDTA to dissociate the complex and centrifugation again after 10 min incubation.
ELISA assay
Cortactin was coated on a 96-well microtiter plate at 4 ° C overnight, followed by blocking with 10% milk. After five times of 5 min washing with PBS-Tween, different concentrations of CaD or H32K were added to the plate in the presence of 1mM CaCl2 and incubated at 37 ° C for an hour. After another five times PBS-Tween washing, 1:1000 diluted anti-CaD (H-C) was added into the wells, and incubated at 37 ° C for 1 hour. The antibody was washed thoroughly with PBS-Tween, and the plate was incubated with anti-rabbit secondary antibody at 37 ° C for 1 hour. H2SO4 was added to stop the tetramethylbenzidine staining reaction. The plate was then placed in the Labsystems Multiskan Plus to read the OD at 490 nm.
Mass spectrometric analysis
V8 protease (1:20, w/w; from Sigma) digestion of H32K or cortactin was performed at room temperature for 10 minutes in a buffer containing 20 mM Tris-Cl (pH 7.5), 50 mM NaCl, 1 mM CaCl2, 1 mM DTT, 5 μM leupeptin, and 1 mM PMSF and stopped with diisopropyl fluorophosphate. The digested fragments were mixed with cortactin- or H32K-sepharose resin in the same buffer at room temperature for 1 hour. After several times of washing, the resin was boiled for 10 min and the supernatant was collected by centrifugation. The solution was mixed with α -cyano-4-hydroxycinnamic acid in an acetonitrile/trifluoroacetic acid mixture and spotted onto a 100-well stainless steel plate. Mass spectrometric analysis was then performed on a MALDI-TOF mass spectrometer (PerSeptive Biosystems Model Voyager-Elite with Delayed Extraction Technology) in linear mode.
Actomyosin ATPase activity assay
The actin-activated Mg2+-ATPase activity of myosin was assayed as described previously (Huang et al., 2003), using rabbit skeletal myosin S1 (0.3 μM), rabbit skeletal actin (3 μM), full-length recombinant human smooth muscle CaD or fragment H32K (0.5 μM) in the presence and absence of recombinant cortactin-B (0.5 μM).
Fluorescence microscopy
Rat aorta fibroblast cells grown on glass coverslips were washed in PBS, fixed with 3.7% paraformaldehyde and permeabilized with 0.3% Triton X-100 (in paraformaldehyde and PBS). After antibody treatments (anti-pS789CaD and anti-cortactin) the coverslips were rinsed, mounted on glass slides in Mowiol, and examined with a BioRad confocal microscope.
Results
1. Intracellular co-localization of CaD and cortactin
We have examined the intracellular distribution of CaD in reference to that of cortactin in cultured rat aorta fibroblast (RAF) cells. In quiescent cells CaD typically exhibits a filamentous distribution as it binds to and presumably stabilizes actin stress fibers, whereas cortactin is primarily present in the cytosol. In RAF cells shortly after mitosis, however, CaD and cortactin were found to co-localize at lamellipodia and ruffles (Fig. 1). We have previously reported that under these conditions when the two daughter cells are migrating away from each other, nascent focal contacts are being formed at the leading edges of the cells, while CaD is phosphorylated at ERK sites and becomes transiently associated with these structures (Kordowska et al., 2006). In these active cells cortactin also undergoes translocation and assumes a cortical distribution, although the punctate structures in the cytoplasm are still visible. Notably the distribution of cortactin is wider than that of CaD, but the two coincide very well in the lamella and lamellipodia. The apparent co-localization of CaD and cortactin suggests that these two cytoskeleton proteins may interact with each other. We therefore proceeded to test this possibility using purified proteins.
Fig. 1.

Co-localization of CaD (red) and cortactin (green) in the lamellipodium and ruffles of newly divided and spreading rat aorta fibroblasts. The right panels show the merged image. In these cells CaD is also phosphorylated at ERK sites (Kordowska et al., 2006). For CaD staining, anti-CaD and anti-pS789CaD antibodies were used in the upper and lower panels, respectively. Scale bars: 50 μm.
2. Overlay assays between CaD and cortactin
When full-length chicken gizzard CaD or its C-terminal fragment, H32K, was incubated with cortactin-B transblotted onto PVDF membrane, both CaD and H32K were retained to the membrane at the cortactin band (Fig. 2). Another fragment, N240, which corresponds to the N-terminal region of CaD, however, only poorly bound to the membrane. These observations indicate that interactions between CaD and cortactin exist, and that the major cortactin-binding sites reside in the C-terminal region of CaD. Interestingly, the binding appeared to require Ca2+ ions, since much less CaD or H32K was detected by anti-CaD antibodies on the membrane when the incubation took place in the absence of Ca2+. This is rather surprising, as neither CaD nor cortactin is known to bind Ca2+. It is unlikely that the apparent Ca2+-dependence is mediated by a Ca2+-binding protein, because such contamination, if any, should have been removed upon SDS-PAGE separation.
Fig. 2.

Overlay assays of recombinant cortactin-B blotted with full-length chicken gizzard CaD (Lanes 1 and 1’), CaD fragment H32K (Lanes 2 and 2’) or N240 (Lanes 3 and 3’) in the presence (Lanes 1–3) or absence (Lanes 1’–3’) of Ca2+. Bound CaD peptides were detected by anti-CaD antibody and visualized by Odyssey infrared imaging. M and M’: molecular weight markers.
3. CaD and H32K, but not N240, bind to cortactin-Sepharose
To verify the interaction between CaD and cortactin, we have coupled purified recombinant cortactin-B to CNBr-activated Sepharose 4B beads, and used the resulting material to pull down CaD and its fragments. Indeed, we found both CaD and H32K, but not N240, in the pellet fractions (Fig. 3), in agreement with the results of the overlay assays. The binding occurred in the presence of 100 mM NaCl, suggesting the interaction persists under physiological ionic strength. Again, Ca2+ was added to effect the binding.
Fig. 3.

Pull-down assay results of cortactin-Sepharose beads reacted with the N240 (Lanes 1 and 5), H32K (Lanes 2 and 4) and chicken gizzard CaD (Lanes 3 and 6) in the presence (Lanes 1–3) and absence (Lanes 4–6) of Ca2+. M: molecular weight markers.
The reciprocal experiment was also carried out using immobilized H32K. In this case we found cortactin was retained to the H32K-Sepharose affinity column in the presence of Ca2+, and was eluted with an EDTA-containing buffer (Fig. 4A). The eluted protein band was sufficiently homogeneous that we subsequently used this procedure to purify cortactin (see Materials and Methods). Significantly, almost identical results were obtained when CaCl2 was replaced with MgCl2, indicating that it is divalent metal ion, not Ca2+ alone that is needed for the CaD-cortactin interaction. There apparently were also some non-specific interactions between cortactin and CaD, because additional cortactin, along with other contaminating species, was washed from the column with more drastic conditions (50 mM glycine, pH 2.3 and 250 mM NaCl; or 0.5 M NaCl). As a control, cortactin did not show appreciable binding to calmodulin-Sepharose under the same conditions (data not shown). The eluted cortactin band showed positive reaction toward anti-cortactin antibody (Fig. 4B).
Fig. 4.


Cortactin is retained to the H32K-affinity column. (A) H32K was first coupled with CNBr-activated sepharose 4B beads. Crude cell extract containing recombinant cortactin-B was loaded to the resulting H32K-Sepharose affinity column in the presence of 1 mM Ca2+, and eluted with 10 mM EDTA. M: molecular weight standard; Lane 1: cell extract containing over-expressed cortactin-B; Lane 2: eluent after washing; Lane 3–6: fractions eluted with 10 mM EDTA, 20 mM Tris-Cl pH7.5, 100 mM NaCl; Lane 7–10: fractions eluted with 50 mM glycine pH2.3, 250 mM NaCl. (B): Western blotting result by Odyssey of all cortactin-containing fractions eluted from the H32K-Sepharose column. The fractions in Panel A were transferred onto PVDF membrane and incubated with mouse anti-cortactin monoclonal antibody followed by fluorescently labeled anti-mouse secondary antibody. All lanes correspond to the lanes of the same number in Panel A.
4. ELISA: CaD and H32K bind to immobilized cortactin
Binding of CaD to cortactin was also demonstrated by ELISA. This method allows for testing of the CaD-cortactin interaction in their native conformations. CaD and H32K were first incubated in the presence of 1 mM CaCl2 with cortactin attached to an ELISA plate, followed by detection with polyclonal anti-CaD antibodies. As shown in Fig. 5, the antibody reaction increased linearly as a function of CaD or H32K concentration, indicating that both CaD and its C-terminal fragment are retained to the cortactin-coated plate.
Fig. 5.
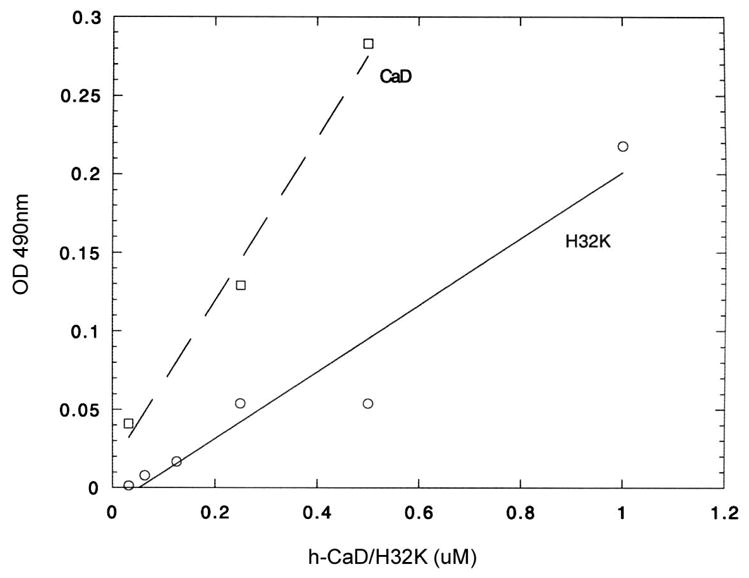
ELISA results showing cortactin interacts with both full-length gizzard CaD (squares, dash line) and H32K (circles, solid line) in the presence of Ca2+. The 96-well plate was coated with cortactin-B. After incubated with different ratio of h-CaD or H32K, followed with polyclonal anti-CaD antibodies (H-C) and anti-rabbit secondary antibody, the OD of each well was measured at 490 nm.
5. The N-terminal region of cortactin binds the C-terminal region of CaD
We have then tried to identify the interaction sites by mass spectrometry. Cortactin was first cleaved with trypsin; the resulting digest was mixed with H32K-Sepharose beads and centrifuged after incubation. The pellet fractions were washed and subjected to MALDI-TOF mass spectrometric analysis. Based on the cleavage specificity and obtained apparent masses (Table 1), we have tentatively assigned the binding fragments to the N-terminal sequences of cortactin (residues 4–147, which encompasses the NTA region and the first two repeats of the kelch motif).
Table 1.
Cortactin binds H32k via its N-terminal region. Trypsin-protease digested Cortactin peptides identified in the peptide fractions by mass spectrometric analysis after pull-down with H32K-coupled beads.
| Protein | Protease | m/z (calculated) | m/z (observed) | Amino acids |
|---|---|---|---|---|
| Cortactin | Trypsin | 1684 | 1684 | T43-K57 |
| 2261 | 2261 | F125-K144 | ||
| 2619 | 2619 | F125-K147/E71-R94 | ||
| 3065 | 3065 | G120-K147 | ||
| 3758 | 3762 | A4-R38 |
Reciprocally, by co-sedimenting a mixture of V8 protease-cleaved H32K with cortactin-Sepharose beads, we have determined that it is the C-terminal region of H32K (and therefore, of CaD), residues 693–771 (chicken sequence), that contains the cortactin-binding site (Table 2).
Table 2.
H32K binds Cortactin via its C-terminal region. V8-protease digested H32K peptides identified in the peptide fractions by mass spectrometric analysis after pulled-down with cortactin-coupled beads.
| Protein | Protease | m/z (calculated) | m/z (observed) | Amino acids |
|---|---|---|---|---|
| H32K | V8 | 1318 | 1318 | T176-P186(761–771) |
| 1431 | 1430 | T108-E121(693–706) | ||
| 3564 | 3564 | W122-E153(707–738) |
6. Cortactin alleviates the inhibitory effect of CaD on actomyosin ATPase activity
To test the effect of cortactin on the biological function of CaD we have performed actomyosin ATPase activity assays in the presence and absence of these proteins. We found that cortactin was able to partially reverse the inhibition of acto-S1 ATPase activity caused by either H32K (from 56% to 31% inhibition) or full-length CaD (from 45% to 35% inhibition; Table 3). Interestingly, cortactin alone also resulted in some inhibition (about 16%), although to a lesser extent compared to the same concentration of CaD (45–56%), in the ATPase activity. Presumably, cortactin binds to actin at the same site as that of myosin, which the C-terminal actin-binding cluster of CaD also binds (Foster et al., 2004).
Table 3.
Effect of cortactin on the CaD-mediated inhibition of acto-S1 ATPase activity*
| Relative ATPase activity | ||
|---|---|---|
| Inhibitor | S1+actin | S1 + actin + cortactin |
| None | 100 | 89 ± 3 |
| H32K | 44 ± 7 | 69 ± 9 |
| CaD | 55 ± 6 | 65 ± 7 |
The Mg2+-activated ATPase activities were measured according to Huang et al. (2003) using 3 μM actin, 0.3 μM S1, 0.5 μM H32K or CaD and 0.5 μM cortactin-B.
7. Competition between Cortactin and CaD for Actin Binding
Both cortactin and CaD bind F-actin. To test whether the binding of one is affected by the other, we have carried out competition experiments by co-sedimentation. When cortactin (1 μM) was mixed with F-actin (6 μM) in the presence of added H32K (2 μM) and centrifuged, the amount of cortactin in the pellet fraction was clearly less than in the absence of H32K, and the displaced cortactin was detected in the corresponding supernatant (Fig. 6A). Full-length smooth muscle CaD behaved in a similar manner as did H32K (data not shown). Thus it seemed that H32K (or CaD) was able to compete off some of the bound cortactin from F-actin. However, when the relative amounts of proteins were reversed, i.e., in a mixture of 1 μM H32K, 3 μM F-actin was incubated with an excess amount (5 μM) of cortactin, cortactin failed to displace H32K as effectively from actin filaments (Fig. 6B). Therefore, the affinity of H32K for actin appeared to be higher than that of cortactin. As H32K is known to bind F-actin via a two-pronged attachment (Huang et al., 2003; Foster et al., 2004), this observation suggested that cortactin at most competes with only one of the actin-binding sites of H32K.
Fig. 6.

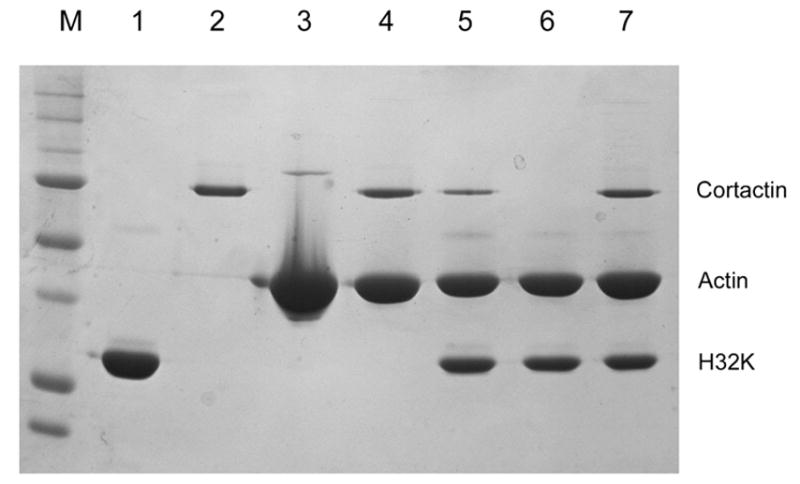
Competition between cortactin and H32K for F-actin binding. (A) Cortactin-B (1 μM) or H32K (2 μM), or both, mixed with F-actin (6 μM) in F-buffer, then centrifuged at 80K rpm for 20 min. Both the supernatant (Lanes 2, 4, 6) and pellet (Lanes 3, 5, 7) fractions were run through 10% SDS-PAGE after washing. M: molecular weight standard; Lane 1: cortactin-B; Lanes 2 and 3: cortactin-B and actin; Lanes 4 and 5: H32K and actin; Lanes 6 and 7: cortactin, H32K and actin. (B) Cortactin-B mixed with F-actin and H32K in F-buffer, then centrifuged at 80K rpm for 20 min, the pellets were run through 10% SDS-PAGE after washing. M: molecular weight standard; Lane 1: H32K; Lane 2: cortactin-B; Lane3: actin; Lane 4: 1 μM cortactin-B and 3 μM actin; Lane 5: 1 μM cortactin-B mixed with 3 μM actin and then added 5 μM H32K; Lane 6: 5 μM H32K mixed with 3 μM actin; Lane 7: 1 μM H32K mixed with 3 μM actin, and then added 5 μM cortactin-B.
When trypsin-digested H32K co-sedimented with F-actin was centrifuged again in the presence of cortactin, there were indeed several peptides being displaced from the pellet fraction to the supernatant. Mass spectrometric analysis revealed that these peptides corresponded to the actin-binding cluster near the C-terminus of CaD (residues 734–771). Thus it seems that it is this site having a lower affinity for actin and being competitive with cortactin. Interestingly, this peptide segment of CaD also contains the cortactin-binding elements (see above). Whether this segment enables a ternary complex of CaD-cortactin-actin is not clear at present and remains to be investigated. As shown previously (Weed et al., 2000), cortactin binds F-actin via its NTA domain, repeating domain, and the Pro-rich region (amino acid residues from 4 to 451 or 414 in smooth muscle sequence). Since only the NTA and part of the repeating domain is involved in CaD-binding, other parts of the cortactin molecule should still bind actin in the presence of CaD. The fact that CaD (or even H32K) is able to displace cortactin from F-actin may indicate the downstream actin binding sites are only of weak affinities.
8. Effect of Phosphorylation by ERK on the Competition between Cortactin and CaD
Cortactin is phosphorylated by ERK at Ser405 and Ser418. Using sedimentation experiments we found that both ERK-treated and untreated cortactin bind equally well to F-actin (data not shown). In either case cortactin could be displaced by H32K from F-actin. However, both cortactin and phosphocortactin failed to compete effectively with H32K. This is consistent with the finding that the CaD-binding sequence does not involve the ERK phosphorylation sites and there is no long-range effect between the two.
Discussion
Cortactin has been previously shown to exist at sites where CaD is also found, e.g., podosomes, focal adhesions, stress fibers, etc. (Tanaka et al., 1993; Webb et al., 2005). In agreement with these earlier reports, we also found that cortactin and CaD are co-localized at the cell cortex (Fig. 1). Co-localization alone, however, does not necessarily prove direct physical interaction, because it could happen through a third protein or more proteins. To demonstrate direct binding, additional lines of independence evidence must also exist. In this case, we have shown that CaD was able to adhere to cortactin band adsorbed on membranes (overlay experiments) or plastic surface (ELISA test), CaD could also be pulled down by cortactin-Sepharose beads, and reciprocally, cortactin was retained to CaD-Sepharose column. It is therefore quite clear that cortactin and CaD indeed interact with each other directly in vitro. Since the affinity column chromatography was performed in 100 mM NaCl and required mM levels of divalent metal ions such as Mg2+, it is conceivable that binding can occur under physiological conditions. What functional roles, if any, such binding plays can only be speculated at present.
Since the CaD’s C-terminal fragment, H32K, but not its N-terminal fragment, N240, interacts with cortactin in a similar manner as the full-length CaD, it is most likely that the C-terminal region of the CaD molecule contains the major interacting sites for cortactin. Reciprocally, the CaD-binding site was found to reside in the N-terminal part of cortactin, which also interacts with Arp2/3 and actin (Weed et al., 2000). This may explain the antagonistic effect of CaD on Arp2/3-mediated actin nucleation (Yamakita et al., 2003). It is also consistent with the observation that H32K could displace cortactin from F-actin in the competition experiments (Fig. 6). On the other hand, we found that the reverse was not true: Cortactin was unable to diminish binding of CaD or H32K to F-actin, but only displaced certain peptide segments from actin when CaD was first fragmented enzymatically. Based on the apparent masses of these displaced peptides, it appears that only one of the two actin-binding clusters of CaD near its C-terminus was competed off by added cortactin, such that CaD remains bound to F-actin via the other actin-binding cluster. Such an assignment is consistent with the results of the ATPase activity assays (Table 3), because it is this actin-binding cluster sits on the actin surface of subdomain 1 and 2, and inhibits myosin binding (Foster et al., 2004). This cortactin-sensitive actin-binding cluster of CaD also harbors the ERK-phosphorylation site(s). Interestingly, in those rat aorta fibroblast cells where CaD was found to be co-localized with cortactin, CaD also happened to be phosphorylated at ERK sites (Kordowska et al., 2006). The significance of this is still not clear and awaits further investigation.
It is particularly curious that the CaD-cortactin interaction is stronger in the presence of divalent metal ions, since neither CaD nor cortactin is known to bind Ca2+ or Mg2+. The involvement of other Me2+-binding proteins was ruled out, because Me2+-dependence was observed in both overlay assays and H32K-Sepharose affinity column chromatography, where any contaminating proteins should have been separated. We have also carried out titration studies in the presence and absence of Ca2+ using fluorescently labeled H32K. Although cortactin induced a small decrease in the emission intensity of labeled H32K, the spectral change was not reversible (data not shown). More detailed studies are underway to characterize this binding.
Cortactin redistributes itself from cytoplasm to membranes upon Rac stimulation (Weed et al., 1998). While it is evident that cortactin is involved in the dynamic assembly of cortical actin, the mechanism of the observed movement toward cell peripheries is not clearly understood. It is not known whether translocation occurs via a specific carrier protein or the identity thereof. Several other proteins also undergo similar cytosol-to-membrane translocation as a result of Rac activation, including p67-phox in neutrophils (Leusen et al., 1996) and c-Src (Fincham et al., 1996). c-Src, in particular, has been shown to interact directly with cortactin. It is therefore possible the two proteins may travel together as a complex. However, although cortactin is a well-known substrate of Src (Wu and Parsons, 1993), the observed translocation does not seem to be dependent on tyrosine phosphorylation (Lopez et al., 2001). The Rac-dependent translocation of cortactin appears to be more closely linked to Ser/Thr phosphorylation by kinases such as PAK (Vidal et al., 2002). Whether the complex involves PAK is an interesting question. Cortactin has also been shown to interact with a number of other proteins, such as CortBP1 (Du et al., 1998), ZO-1 (Katsube et al., 1998), dynamin (McNiven et al., 2000) and MLCK (Dudek et al., 2002). It is not clear any of these proteins would serve as carriers for cortactin, because they are not known to exhibit Rac-dependent translocation. CaD may be another candidate for such a role.
This is a first report on the interaction between CaD and cortactin. In unstimulated cells CaD binds to and stabilizes actin filaments. Upon activation, CaD is phosphorylated and moves to cell edges and becomes co-localized with cortactin. At the same time, the cytoplasmic actin cytoskeleton undergoes disassembly, whereas cortical actin is being assembled. The dynamic process must involve multiple protein components. For example, phosphorylation-induced dissociation of one of the actin-binding clusters of CaD allows for severing proteins to bind and thus facilitating actin cytoskeleton remodeling. The fact that cortactin binds to the C-terminal extreme of CaD, which contains such phosphorylation sites, may suggest a mechanism for cortactin to target a subset of actin filaments that are to be transported to the cell cortex upon activation. The in vitro evidence of direct interaction between CaD and cortactin, and the in vivo co-localization provides a potential player to more precisely regulate this complicated process. There are obviously many more questions to be answered before the biological significance can be assessed. This may be just an example that cytoskeleton proteins are interacting with each other to synergistically achieve the dynamic remodeling of the actin cytoskeleton.
Acknowledgments
The authors wish to thank Ms. Lijia Zhu for her help in protein preparation. This work was supported by a grant from NIH (P01-AR41637).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bursac P, Lenormand G, Fabry B, Oliver M, Weitz DA, Viasnoff V, Butler JP, Fredberg JJ. Cytoskeletal remodelling and slow dynamics in the living cell. Nat Mater. 2005;4(7):557–61. doi: 10.1038/nmat1404. [DOI] [PubMed] [Google Scholar]
- Clarkson MR, Murphy M, Gupta S, Lambe T, Mackenzie HS, Godson C, Martin F, Brady HR. High glucose-altered gene expression in mesangial cells. Actin-regulatory protein gene expression is triggered by oxidative stress and cytoskeletal disassembly. J Biol Chem. 2002;277(12):9707–12. doi: 10.1074/jbc.M109172200. [DOI] [PubMed] [Google Scholar]
- Cortes P, Mendez M, Riser BL, Guerin CJ, Rodriguez-Barbero A, Hassett C, Yee J. F-actin fiber distribution in glomerular cells: structural and functional implications. Kidney Int. 2000;58(6):2452–61. doi: 10.1046/j.1523-1755.2000.00428.x. [DOI] [PubMed] [Google Scholar]
- D’Angelo G, Graceffa P, Wang CLA, Wrangle J, Adam LP. Mammal-specific, ERK-dependent, caldesmon phosphorylation in smooth muscle. Quantitation using novel anti-phosphopeptide antibodies. J Biol Chem. 1999;274(42):30115–21. doi: 10.1074/jbc.274.42.30115. [DOI] [PubMed] [Google Scholar]
- Daly RJ. Cortactin signalling and dynamic actin networks. Biochem J Pt. 2004 doi: 10.1042/BJ20040737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Weed SA, Xiong WC, Marshall TD, Parsons JT. Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol Cell Biol. 1998;18(10):5838–51. doi: 10.1128/mcb.18.10.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Birukov KG, Zhan X, Garcia JG. Novel interaction of cortactin with endothelial cell myosin light chain kinase. Biochem Biophys Res Commun. 2002;298(4):511–9. doi: 10.1016/s0006-291x(02)02492-0. [DOI] [PubMed] [Google Scholar]
- Fincham VJ, Unlu M, Brunton VG, Pitts JD, Wyke JA, Frame MC. Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J Cell Biol. 1996;135(6):1551–64. doi: 10.1083/jcb.135.6.1551. Pt 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DB, Huang R, Hatch V, Craig R, Graceffa P, Lehman W, Wang CLA. Modes of caldesmon binding to actin: sites of caldesmon contact and modulation of interactions by phosphorylation. J Biol Chem. 2004;279(51):53387–94. doi: 10.1074/jbc.M410109200. [DOI] [PubMed] [Google Scholar]
- Huang R, Li L, Guo H, Wang CLA. Caldesmon binding to actin is regulated by calmodulin and phosphorylation via different mechanisms. Biochemistry. 2003;42(9):2513–23. doi: 10.1021/bi0268605. [DOI] [PubMed] [Google Scholar]
- Huber PA. Caldesmon. Int J Biochem Cell Biol. 1997;29(8–9):1047–51. doi: 10.1016/s1357-2725(97)00004-6. [DOI] [PubMed] [Google Scholar]
- Katsube T, Takahisa M, Ueda R, Hashimoto N, Kobayashi M, Togashi S. Cortactin associates with the cell-cell junction protein ZO-1 in both Drosophila and mouse. J Biol Chem. 1998;273(45):29672–7. doi: 10.1074/jbc.273.45.29672. [DOI] [PubMed] [Google Scholar]
- Kordowska J, Hetrick T, Adam LP, Wang CLA. Phosphorylated l-caldesmon is involved in disassembly of actin stress fibers and postmitotic spreading. Exp Cell Res. 2006;312(2):95–110. doi: 10.1016/j.yexcr.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Leusen JH, de Klein A, Hilarius PM, Ahlin A, Palmblad J, Smith CI, Diekmann D, Hall A, Verhoeven AJ, Roos D. Disturbed interaction of p21-rac with mutated p67-phox causes chronic granulomatous disease. J Exp Med. 1996;184(4):1243–9. doi: 10.1084/jem.184.4.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhuang S, Guo H, Mabuchi K, Lu RC, Wang CLA. The major myosin-binding site of caldesmon resides near its N-terminal extreme. J Biol Chem. 2000;275(15):10989–94. doi: 10.1074/jbc.275.15.10989. [DOI] [PubMed] [Google Scholar]
- Lopez I, Duprez V, Melle J, Dreyfus F, Levy-Toledano S, Fontenay-Roupie M. Thrombopoietin stimulates cortactin translocation to the cytoskeleton independently of tyrosine phosphorylation. Biochem J. 2001;356:875–81. doi: 10.1042/0264-6021:3560875. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuchi K, Li Y, Tao T, Wang CLA. Immunocytochemical localization of caldesmon and calponin in chicken gizzard smooth muscle. J Muscle Res Cell Motil. 1996;17(2):243–60. doi: 10.1007/BF00124246. [DOI] [PubMed] [Google Scholar]
- McNiven MA, Kim L, Krueger EW, Orth JD, Cao H, Wong TW. Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. Journal of Cell Biology. 2000;151(1):187–198. doi: 10.1083/jcb.151.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seow CY. Biophysics: fashionable cells. Nature. 2005;435(7046):1172–3. doi: 10.1038/4351172a. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Watanabe T, Nakamura N, Sobue K. Morphological and biochemical analyses of contractile proteins (actin, myosin, caldesmon and tropomyosin) in normal and transformed cells. J Cell Sci. 1993;104:595–606. doi: 10.1242/jcs.104.2.595. Pt 2. [DOI] [PubMed] [Google Scholar]
- Vidal C, Geny B, Melle J, Jandrot-Perrus M, Fontenay-Roupie M. Cdc42/Rac1-dependent activation of the p21-activated kinase (PAK) regulates human platelet lamellipodia spreading: implication of the cortical-actin binding protein cortactin. Blood. 2002;100(13):4462–9. doi: 10.1182/blood.V100.13.4462. [DOI] [PubMed] [Google Scholar]
- Wang CLA. Caldesmon and smooth-muscle regulation. Cell Biochem Biophys. 2001;35(3):275–88. doi: 10.1385/cbb:35:3:275. [DOI] [PubMed] [Google Scholar]
- Webb BA, Eves R, Crawley SW, Zhou S, Cote GP, Mak AS. PAK1 Induces Podosome Formation in A7r5 Vascular Smooth Muscle Cells in a PIX-dependent Manner. Am J Physiol Cell Physiol. 2005;8:8. doi: 10.1152/ajpcell.00095.2005. [DOI] [PubMed] [Google Scholar]
- Weed SA, Du Y, Parsons JT. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J Cell Sci. 1998;111 :2433–43. doi: 10.1242/jcs.111.16.2433. Pt 16. [DOI] [PubMed] [Google Scholar]
- Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, Parsons JT. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol. 2000;151(1):29–40. doi: 10.1083/jcb.151.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weed SA, Parsons JT. Cortactin: coupling, membrane dynamics to cortical actin assembly. Oncogene. 2001;20(44):6418–6434. doi: 10.1038/sj.onc.1204783. [DOI] [PubMed] [Google Scholar]
- Wu H, Parsons JT. Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol. 1993;120(6):1417–26. doi: 10.1083/jcb.120.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakita Y, Oosawa F, Yamashiro S, Matsumura F. Caldesmon inhibits Arp2/3-mediated actin nucleation. Journal of Biological Chemistry. 2003;278(20):17937–17944. doi: 10.1074/jbc.M208739200. [DOI] [PubMed] [Google Scholar]
- Zhou X, Hurst RD, Templeton D, Whiteside CI. High glucose alters actin assembly in glomerular mesangial and epithelial cells. Lab Invest. 1995;73(3):372–83. [PubMed] [Google Scholar]
- Zhuang S, Mabuchi K, Wang CLA. Heat treatment could affect the biochemical properties of caldesmon. J Biol Chem. 1996;271(47):30242–8. doi: 10.1074/jbc.271.47.30242. [DOI] [PubMed] [Google Scholar]