

Abstract
In mammals, maintenance of energy and nutrient homeostasis during food deprivation is accomplished through an increase in mitochondrial fatty acid oxidation in peripheral tissues. An important component that drives this cellular oxidative process is the transcriptional coactivator PGC-1α. Here, we show that fasting induced PGC-1α deacetylation in skeletal muscle and that SIRT1 deacetylation of PGC-1α is required for activation of mitochondrial fatty acid oxidation genes. Moreover, expression of the acetyltransferase, GCN5, or the SIRT1 inhibitor, nicotinamide, induces PGC-1α acetylation and decreases expression of PGC-1α target genes in myotubes. Consistent with a switch from glucose to fatty acid oxidation that occurs in nutrient deprivation states, SIRT1 is required for induction and maintenance of fatty acid oxidation in response to low glucose concentrations. Thus, we have identified SIRT1 as a functional regulator of PGC-1α that induces a metabolic gene transcription program of mitochondrial fatty acid oxidation. These results have implications for understanding selective nutrient adaptation and how it might impact lifespan or metabolic diseases such as obesity and diabetes.
Keywords: caloric restriction, gene transcription, lipid metabolism, mitochondrial oxidation, Sirtuins
Introduction
Mammals have adapted to different environmental conditions and food by coordinating changes in tissue-specific metabolic pathways to maintain energy and nutrient homeostasis. Among the changes in food availability, nutrient deprivation triggers a whole rearrangement of glucose and lipid metabolism in key metabolic tissues such as skeletal muscle and liver. As glucose is the main fuel substrate for neuronal and red blood cells, peripheral tissues undergo a major shift from glucose to fatty acid oxidation, and gluconeogenic precursors such as lactate and alanine are delivered from muscle to the liver to synthesize glucose (Storlien et al, 2004). In muscle cells, a major regulatory metabolic enzyme that allows this nutrient shift is pyruvate dehydrogenase kinase-4 (PDK4) that inactivates pyruvate dehydrogenase (PDH) by phosphorylation and prevents the entry of pyruvate into the TCA cycle. PDK4 enzymatic activity is regulated by nutrient deprivation (Sugden et al, 1993). At the transcriptional level, several transcription factors including FOXO1, ERRα and PPARα are activators of PDK4 gene expression (Pilegaard and Neufer, 2004). Interestingly, PGC-1α, a common transcriptional coactivator of these factors, also induces PDK4, consistent with a key role of PGC-1α in fatty acid utilization (Wende et al, 2005). Furthermore, as PGC-1α induces OXPHOS genes involved in the final step of electron transport chain and ATP synthesis, it allows complete mitochondrial oxidation of fatty acids (Wu et al, 1999; Koves et al, 2005).
Nutrient or caloric restriction results in major tissue-specific metabolic changes that ultimately cause an increase of longevity in many different organisms (Bordone and Guarente, 2005; Sinclair, 2005). A key component of this response is Sir2, an NAD+-dependent protein deacetylase that mediates increases of lifespan in response to restriction of nutrients (Bordone and Guarente, 2005). Genetic evidence for a role of Sir2 in this nutrient/aging pathway has been provided in model organisms such as yeast (Kaeberlein et al, 1999), worms (Tissenbaum and Guarente, 2001) and flies (Rogina and Helfand, 2004). Interestingly, in yeast, caloric restriction by lowering glucose levels has been associated with increased rates of respiration in an Sir2-dependent pathway to allow a more efficient use of fuel substrates (Lin et al, 2000). In mammals, SIRT1 controls different tissue-specific metabolic processes. For example, the ability of SIRT1 to repress PPARγ confers increases of lipolytic rates in white adipose tissue (Picard et al, 2004) and insulin secretion in β-pancreatic cells (Bordone et al, 2006). Moreover, we have previously shown that SIRT1 is increased in fasted liver to deacetylate and activate PGC-1α to promote hepatic glucose output (Rodgers et al, 2005). Together, these metabolic adaptations transcriptionally controlled by SIRT1 might be important for the effects of nutrient restriction in organismal survival and longevity.
PGC-1α is a metabolic coactivator that by interacting with transcription factors induces mitochondrial biogenesis and respiration (Lin et al, 2005; Finck and Kelly, 2006). In skeletal muscle cells, PGC-1α is sufficient to convert to type I fibers that are rich in mitochondria and highly oxidative (Lin et al, 2002). In addition, interaction of PGC-1α with PPARα and ERRα transcription factors potently induces fatty acid oxidation genes such as MCAD and CPT-1b (Vega et al, 2000; Huss et al, 2002; Mootha et al, 2004; Schreiber et al, 2004). Interestingly, PGC-1α and mitochondrial OXPHOS targets are downregulated in human skeletal muscle of type II diabetic patients that might be linked to increased intramyocellular triglyceride accumulation and insulin resistance (Mootha et al, 2003; Patti et al, 2003). In fact, there is a strong correlation between the presence of intramyocellular lipid in skeletal muscle and liver and progression of type II diabetes (Guan et al, 2002; Kelley et al, 2002; Petersen et al, 2004). Therefore, a knowledge of the molecular mechanisms by which cells increase the rates of fatty acid oxidation in response to low nutrients is important to understand the pathophysiology of these metabolic diseases and their connection to aging biology.
We report here the identification of SIRT1 as the main deacetylase of PGC-1α that positively regulates mitochondrial and fatty acid utilization genes. Moreover, SIRT1 is required for increased rates of fatty acid oxidation in response to low glucose, providing a new metabolic regulator that allows mammalian cells to switch from glucose to fatty acid oxidation in nutrient deprivation conditions.
Results
SIRT1 deacetylates PGC-1α in skeletal muscle cells
Skeletal muscle undergoes a major metabolic reprogramming under nutrient deprivation conditions (Shuldiner and McLenithan, 2004). PGC-1α has been previously shown to regulate different metabolic pathways in muscle cells, leading to a more oxidative capacity towards lipid utilization (Lin et al, 2005). In liver cells, PGC-1α acetylation decreases in fasting conditions mainly through activation of SIRT1 deacetylase (Rodgers et al, 2005). In order to determine whether changes in PGC-1α acetylation also occurred in skeletal muscle, PGC-1α was immunoprecipitated from muscle tissue in fed and fasted conditions. Figure 1A shows that fasting strongly induced PGC-1α deacetylation. To investigate whether SIRT1 could target PGC-1α in cultured muscle cells, we treated C2C12 with nicotinamide, an SIRT1 inhibitor. As shown in Figure 1B, nicotinamide induced PGC-1α acetylation and was blocked with ectopic expression of SIRT1. GCN5 acetyltransferase is part of a major protein complex associated with PGC-1α and acetylates PGC-1α (Lerin et al, 2006). Consistent with these results, PGC-1α was also acetylated by GCN5 in muscle cells (Figure 1B). Interestingly, treatment with nicotinamide and GCN5 strongly synergized to acetylate PGC-1α and again expression of SIRT1 largely decreased PGC-1α acetylation. To demonstrate further that SIRT1 is the main PGC-1α deacetylase, we used SIRT1−/− mouse embryonic fibroblasts (MEFs) (Chua et al, 2005). As shown in Figure 1C, in SIRT1+/+ MEFs, PGC-1α was deacetylated and treatment with nicotinamide induced its acetylation. In contrast, in SIRT1−/− MEFs, PGC-1α was constitutively acetylated and nicotinamide did not further increase it. Importantly, in these cells, deacetylation of PGC-1α was totally rescued by expression of SIRT1 (Figure 1D).
Figure 1.
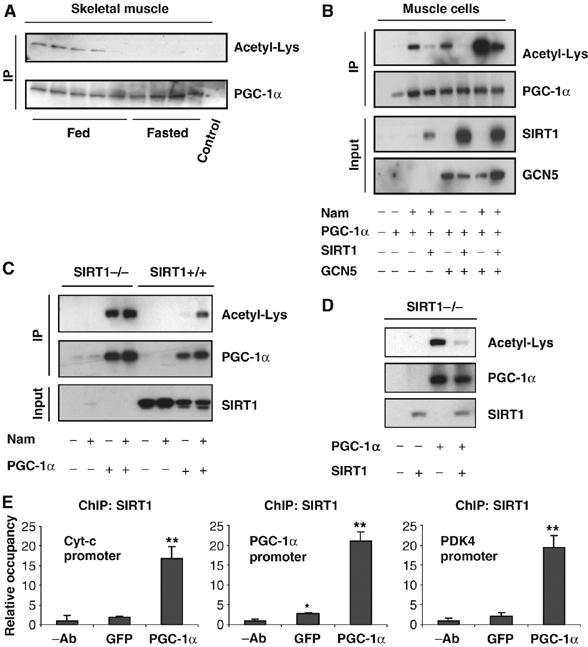
SIRT1 deacetylates PGC-1α in skeletal muscle cells. (A) Fasting-induced PGC-1α deacetylation in skeletal muscle. Mice were either fed or fasted for 16 h and skeletal muscle was used to immunoprecipitate PGC-1α and Western blot was performed to detect PGC-1α levels and acetylation. (B) Nicotinamide and GCN5 induce PGC-1α acetylation. C2C12 myotubes were treated with 5 mM nicotinamide for 12 h and/or infected with the indicated adenoviruses for 48 h. After treatment, cells were harvested and protein cell extracts were used for PGC-1α immunoprecipitation and Western blot analysis using the specified antibodies. (C) PGC-1α is constitutively acetylated in SIRT1−/− MEFs. MEFs were treated with nicotinamide and/or infected with adenoviruses encoding PGC-1α as described in (B). (D) SIRT1 rescues PGC-1α deacetylation in SIRT1−/− MEFs. Adenoviruses expressing PGC-1α and SIRT1 were infected in MEFs as described in (B). (E) SIRT1 is bound to PGC-1α-targeted gene promoters. C2C12 myotubes were infected with adenoviruses encoding either GFP or PGC-1α. Forty-eight hours after infection, cells were harvested and ChIP analysis using SIRT1 antibody was performed as described in Materials and methods. Values in (E) represent the mean of two experiments performed in triplicate. Error bars represent s.e.m. Statistical analyses were performed using Student's t-test. *P<0.05 and **P<0.005.
To demonstrate that SIRT1 localized to PGC-1α target genes in skeletal muscle cells, we performed ChIP assays (Figure 1E). SIRT1 was bound to promoters of PGC-1α targets such as cytochrome c, PDK4 and PGC-1α. Ectopic expression of PGC-1α induced a 7–10-fold recruitment of SIRT1 to these promoters. Together, these results indicate that PGC-1α is deacetylated by SIRT1 in skeletal muscle cells and SIRT1 localized to promoters that are controlled by PGC-1α, suggesting a regulatory role of SIRT1 on the expression of these genes.
SIRT1 regulates mitochondrial and fatty acid metabolism
In skeletal muscle cells, PGC-1α activates mitochondrial gene expression in response to energy demands (Wu et al, 1999; Lin et al, 2002). As SIRT1 was localized to promoters controlled by PGC-1α, we investigated whether SIRT1 regulated PGC-1α-induced mitochondrial gene expression in myotubes infected with adenoviruses expressing PGC-1α and SIRT1 shRNA (Figure 2G). As expected, PGC-1α increased a repertoire of mitochondrial gene expression, including regulatory genes ERRα, genes of the TCA cycle (IDH3α), respiratory chain (cyt-c, COXVa) and fatty acid utilization (MCAD, CPT-1b and PDK4). Remarkably, PGC-1α-increased mitochondrial and fatty acid utilization gene expression was largely prevented by knocking down SIRT1 (Figure 2A and B). Mitochondrial transcriptional regulator ERRα, but not NRF-1, followed exactly the same pattern (Figure 2C). However, the glycolytic gene pyruvate kinase did not change under these conditions (Figure 2D). These fluctuations in gene expression translated to an increase in fatty acid oxidation induced by PGC-1α that was prevented by SIRT1 knockdown (Figure 2E). Interestingly, in these conditions, shRNA SIRT1 prevented PGC-1α-decreased glucose oxidation (Figure 2F). These data indicate that SIRT1 is required, at least in part, for maximum expression of a large set of PGC-1α target genes and increased fatty acid oxidation.
Figure 2.
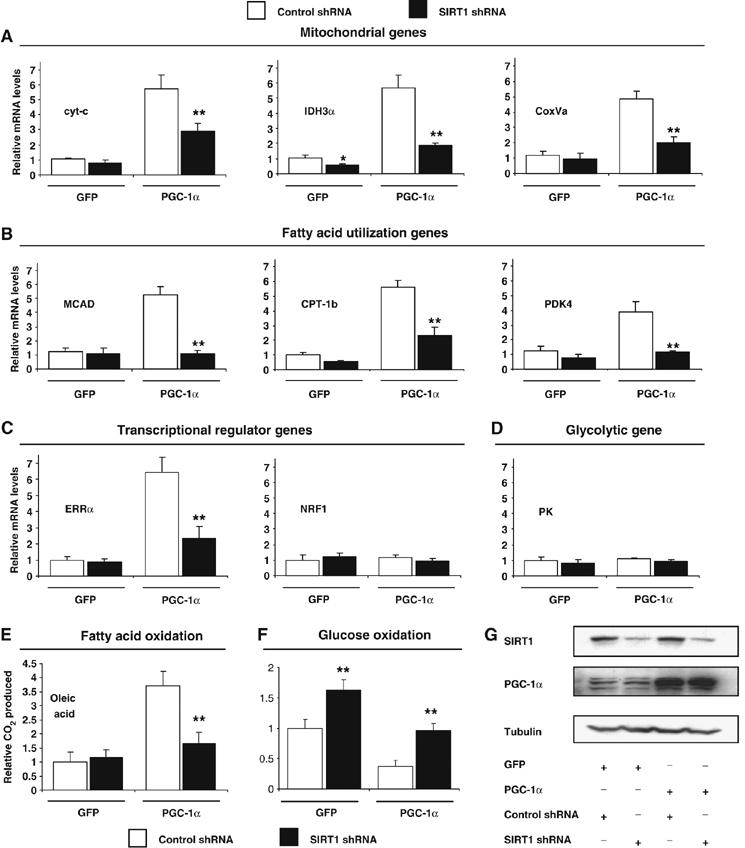
SIRT1 regulates PGC-1α-mediated mitochondrial and fatty acid metabolism in C2C12 skeletal muscle cells. (A) SIRT1 knockdown decreases PGC-1α-induced mitochondrial, (B) fatty acid utilization, (C) ERRα, but not (D) pyruvate kinase gene expression. C2C12 myotubes were infected for 3 days with the indicated adenoviruses. After harvesting, RNA was extracted and used for measuring indicated gene expression using quantitative RT–PCR analysis. (E) PGC-1α-induced oxidation rates of fatty acids are reduced by SIRT1 knockdown. C2C12 myotubes were treated as in (A). Oleic acid oxidation rates were measured as described in Materials and methods. (F) PGC-1α-decreased oxidation rates of glucose are prevented by SIRT1 knockdown. Glucose oxidation rates were measured in C2C12 myotubes as described in Materials and methods. (G) Knockdown of SIRT1 in C2C12 myotubes. Cells were infected with the different adenoviruses as in (A–F). Protein cell extracts were used for Western blot analysis that was performed with the indicated antibodies. Values represent the mean of 2–3 experiments performed in duplicate. Error bars represent s.e.m. Statistical analyses were performed using Student's t-test. *P<0.05 and **P<0.005, shRNA control versus shRNA SIRT1.
The effects of knocking down SIRT1 on gene expression without ectopic PGC-1α expression were very minor in C2C12 muscle cells (Figure 2). As these cells express very low levels of PGC-1α, we used primary skeletal muscle cells that express a significant level of PGC-1α. We tested whether SIRT1 was required to maintain mitochondrial and fatty acid oxidation gene expression. Primary myotubes transduced with an adenovirus expressing SIRT1 shRNA resulted in an efficient knockdown of SIRT1 (Figure 3E). The expression levels of mitochondrial and fatty acid utilization genes (Figure 3A and B) were significantly reduced in the cells transduced with SIRT1 shRNA. Interestingly, PGC-1α and its target mitochondrial transcriptional regulators, ERRα and mtTFA, were also downregulated (Figure 3C). These gene expression changes functionally translated to decreases in TCA cycle measured by citrate synthase activity (Figure 3D). These results indicate that SIRT1 controls a broad set of genes involved in mitochondrial oxidative function in muscle cells.
Figure 3.
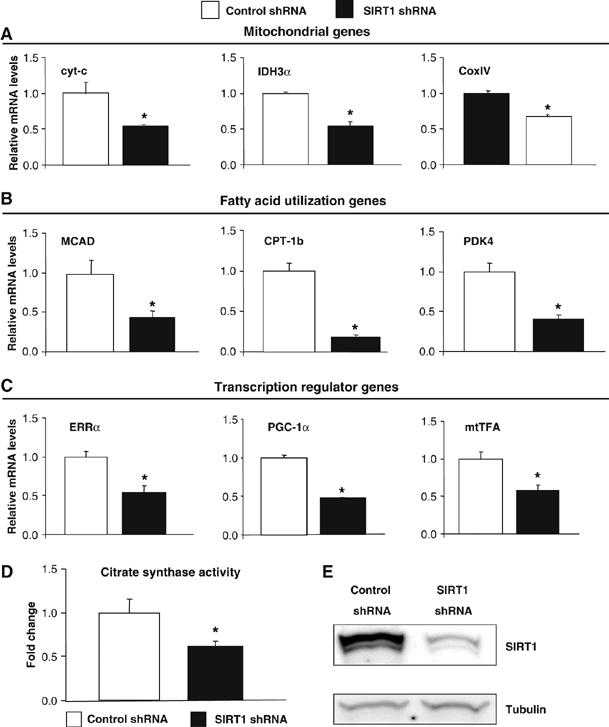
SIRT1 regulates mitochondrial and fatty acid oxidation genes in primary skeletal muscle cells. (A) SIRT1 knockdown decreases mitochondrial genes, (B) fatty acid utilization genes and (C) transcriptional regulator gene expresson in primary myotubes. Mouse primary myotubes were transduced with adenovirus expressing either SIRT1 shRNA or control shRNA for 71 h. Total RNA was extracted and used for measuring indicated gene expression using quantitative RT–PCR. (D) SIRT1 knockdown decreases citrate synthase enzymatic activity in primary myotubes. (E) Knockdown of SIRT1 in primary skeletal myotubes. Cells were infected with the indicated adenoviruses and protein cell extracts were used to perform Western blot analysis for SIRT1 and tubulin as a control. Values represent the mean of 2–3 experiments performed in duplicate. Error bars represent s.e.m. Statistical analyses were performed using Student's t-test. *P<0.05. shRNA control versus shRNA SIRT1.
To have genetic evidence to further support the role of SIRT1 regulating mitochondrial and fatty acid oxidation functions, we analyzed the same set of genes in MEFs lacking SIRT1. SIRT1−/− MEFs displayed a decrease in expression of genes associated with mitochondrial (Figure 4A), fatty acid utilization (Figure 4B) and mitochondrial transcriptional regulators compared with wild-type cells (Figure 4C). To directly show that these effects were entirely dependent on SIRT1, we ectopically expressed SIRT1 in SIRT1−/− MEFs. Importantly, this decreased gene expression pattern was entirely rescued by SIRT1 in a dose-dependent manner (Figure 4D). Moreover, these changes in gene expression resulted in a decreased rate of fatty acid oxidation in SIRT1−/− cells (Figure 4E). Taken together, these results indicate that SIRT1 positively regulates complete mitochondrial and fatty acid oxidation, and at least in skeletal muscle cells, SIRT1 largely contributes to the effects of PGC-1α in regulating expression of genes linked to these metabolic pathways.
Figure 4.

SIRT1 regulates mitochondrial and fatty acid metabolism in MEFs. Decreased mitochondrial (A), fatty acid utilization (B) and mitochondrial transcriptional regulator (C) gene expression in SIRT1−/− MEFs. MEFs were cultured until confluence, harvested and RNA was extracted to measure mitochondrial genes by quantitative RT–PCR. (D) Ectopic expression of SIRT1 rescues mitochondrial and fatty acid utilization gene expression. SIRT1−/− MEFs were infected with different amounts of adenoviruses encoding for SIRT1. After 3 days of infection, total RNA was isolated and indicated gene expressions were measured by quantitative RT–PCR. (E) Decreased rates of fatty acid oxidation in SIRT1−/− MEFs. Cells were treated as described in (A), but were used to measure oleic acid oxidation rates as described in Materials and methods. Values represent the mean of 2–3 experiments performed in duplicate. Error bars represent s.e.m. Statistical analyses were performed using Student's t-test. *P<0.05 and **P<0.005.
GCN5 and nicotinamide regulate mitochondrial and fatty acid metabolism in skeletal muscle cells
We have recently identified GCN5 as the main PGC-1α acetyltransferase and a negative regulator of PGC-1α biological functions (Lerin et al, 2006). We therefore investigated whether GCN5, by opposing SIRT1-positive effects, would negatively regulate PGC-1α function on mitochondrial and fatty acid utilization genes. Indeed, in C2C12 myotubes, GCN5 largely abolished PGC-1α-induced mitochondrial, fatty acid utilization and mitochondrial transcriptional regulator gene expression (Figure 5A). As C2C12 muscle cells express low levels of endogenous PGC-1α, we again used primary skeletal myotubes to analyze the effects of GCN5 expression. The same pattern of gene expression was observed in these cells, but without ectopically expressing PGC-1α (Supplementary Figure S1). To support that these effects of GCN5 were mediated through acetylation of PGC-1α, we used the SIRT1 inhibitor, nicotinamide. Consistent with the effects of GCN5, treatment of C2C12 or primary skeletal muscle cells with nicotinamide caused a decrease in expression of PGC-1α targeted mitochondrial and fatty acid utilization genes (Figure 5B and Supplementary Figure S2). To demonstrate that these effects were dependent on SIRT1, we again used SIRT1−/− MEFs. As shown in Figure 5C, nicotinamide decreased expression of cyt-c and MCAD by approximately three-fold in SIRT1+/+ MEFs; however cells that lack SIRT1 did not decrease these genes in response to nicotinamide. Moreover, and consistent with the effects of transcription on these genes, PGC-1α potently induced fatty acid oxidation that was blocked by expression of GCN5 or nicotinamide treatment (Figure 5D). Together, these results suggest that acetylation of PGC-1α is a regulatory chemical modification that controls the oxidative function of this transcriptional coactivator.
Figure 5.
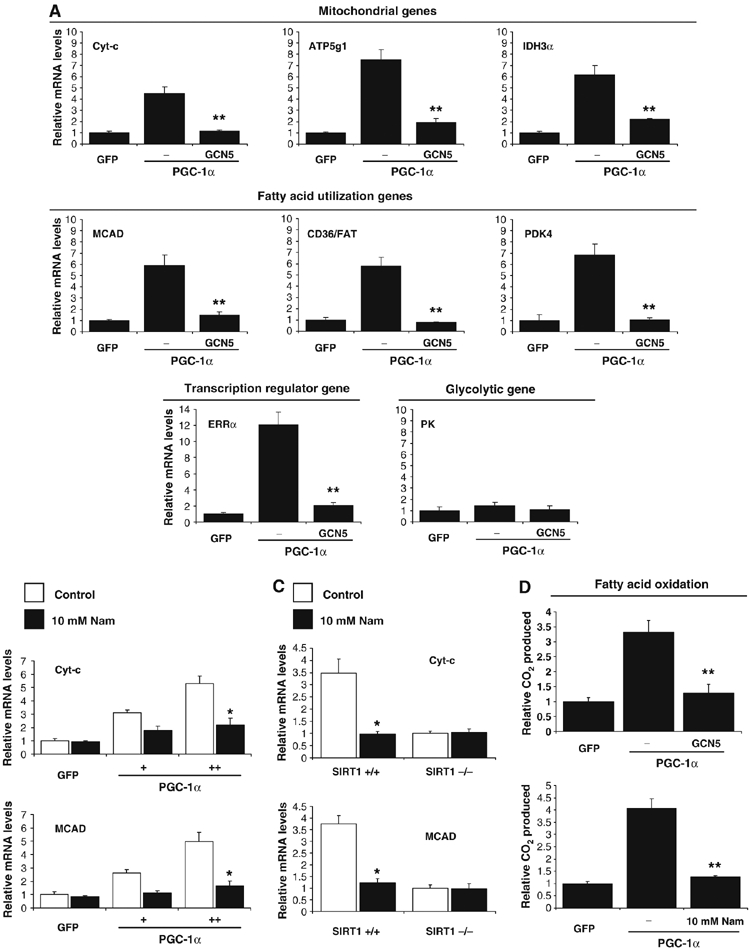
GCN5 regulates mitochondrial and fatty acid metabolism in skeletal muscle cells. (A) GCN5 downregulates PGC-1α-induced mitochondrial gene expression. C2C12 myotubes were infected for 3 days with the indicated adenoviruses. Total RNA was extracted and analyzed for mitochondrial, fatty acid utilization and transcriptional regulator gene expression using quantitative RT–PCR analysis. (B) Nicotinamide downregulates PGC-1α-induced mitochondrial and fatty acid oxidation gene expression in C2C12 myotubes and (C) MEFs. C2C12 cells were infected with the indicated adenoviruses. At day 3 after infection, cells were treated for 12 h with 10 mM nicotinamide. Confluent MEFs were also treated for 12 h with nicotinamide and harvested for RNA analysis. (D) GCN5 and nicotinamide prevented PGC-1α-increased fatty acid oxidation rates. Cells were treated as in (A–C), but oleic acid oxidation assays were performed as described in Materials and methods. Values represent the mean of 2–3 experiments performed in duplicate. Error bars represent s.e.m. Statistical analyses were performed using Student's t-test. *P<0.05 and **P<0.005.
SIRT1 is required to switch on fatty acid oxidation in response to low glucose concentrations
SIRT1 and GCN5 are modulators of PGC-1α and changes of amounts and/or activities of these two enzymes will define the ability of PGC-1α to regulate metabolic genes. However, an important question is what are the stimuli or signals that are involved in this physiological process. In hepatocytes, we have shown that SIRT1 is regulated through a nutrient pathway with changes in NAD+ and pyruvate in food-deprivation conditions (Rodgers et al, 2005). Moreover, Figure 1A shows fasting-induced PGC-1α deacetylation in skeletal muscle. In order to investigate whether SIRT1 was a target of a nutrient response in skeletal muscle cells and regulated fatty acid oxidation—a metabolic pathway that is highly induced during nutrient deprivation—we first analyzed whether changes in glucose concentration might control fatty acid oxidation rates. As shown in Figure 6A, decreases in glucose concentrations induced fatty acid oxidation up to three-fold in C2C12 myotubes. Interestingly, this decrease in glucose concentration resulted in an increase of NAD+, a substrate and activator of SIRT1, as well as an increase in the ratio of free NAD+/NADH measured by lactate and pyruvate concentrations (Figure 6B and Supplementary Figure S3). These data suggest that this change in NAD+ levels could be sensed via SIRT1 and activate its enzymatic activity. To test this, we determined acetylation levels of PGC-1α in high and low glucose concentrations. Figure 6C shows that PGC-1α acetylation is largely decreased by lowering levels of glucose, which is consistent with an increase in NAD+ and SIRT1 deacetylase activity. To demonstrate that SIRT1 mediated low glucose-induced fatty acid oxidation through regulation of gene expression, we used SIRT1−/− cells (Figure 6D). In SIRT1+/+ cells, low glucose induced expression of mitochondrial, fatty acid utilization and mitochondrial transcriptional regulator gene expression. Notably, this induction was totally blunted in SIRT1−/− MEFs. Furthermore, we tested the requirement of SIRT1 in C2C12 myotubes expressing PGC-1α (Figure 6E). As expected, low glucose induced expression of the same set of PGC-1α-targeted genes; however knockdown of SIRT1 blocked this induction. As a control, PGC-1β did not change with these treatments. Consistent with this gene expression pattern, Figure 6F shows that SIRT1−/− MEFs completely lack the ability to induce fatty acid oxidation in response to low glucose. Furthermore, knockdown of SIRT1 in C2C12 myotubes also prevented the induction of fatty acid oxidation under low glucose concentrations, both in GFP- and PGC-1α-expressing cells (Figure 6F). Taken together, these results indicate that changes in glucose concentrations that occur in physiological situations such as starvation or caloric restriction induce an autonomous switch to increase oxidation of fatty acids. Importantly, this metabolic response entirely required SIRT1 to efficiently trigger fatty acid oxidation.
Figure 6.
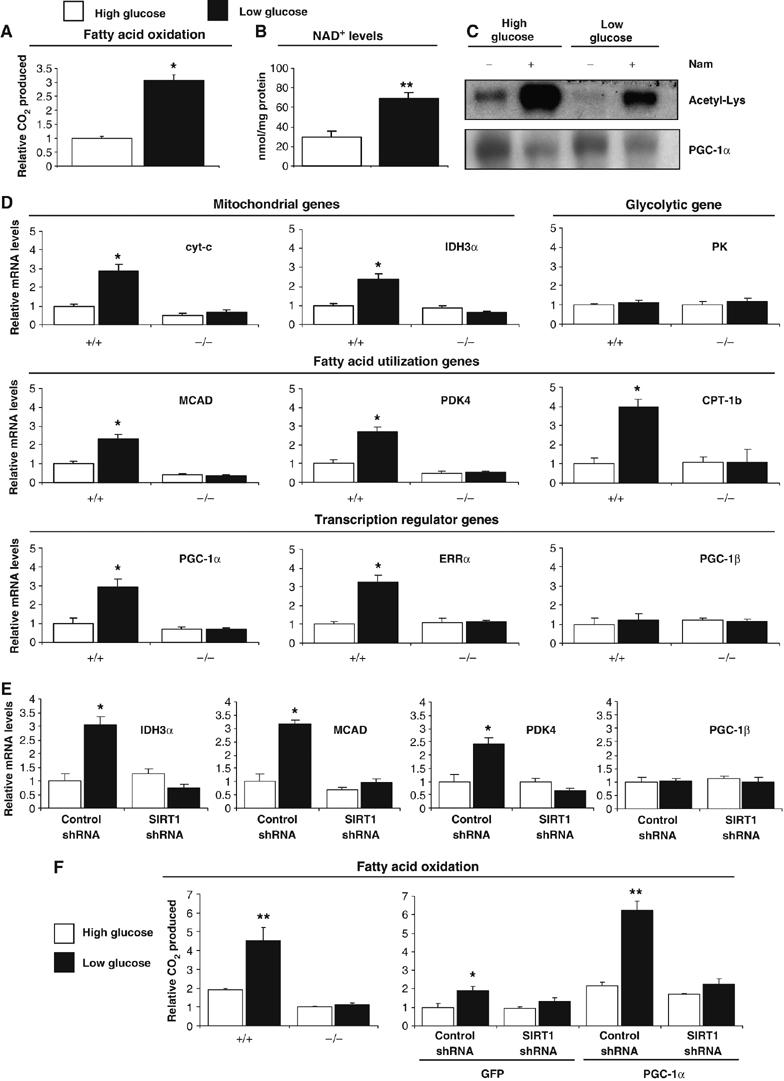
SIRT1 is required for the switch to fatty acid oxidation in response to low glucose concentrations. (A) Low glucose concentration induced increase in rates of fatty acid oxidation and (B) increased in cellular NAD+ levels. C2C12 myotubes were treated with 25 mM (open bars) or 5 mM (filled bars) glucose for 12 h. Palmitic acid oxidation was measured and in another set of experiments, deproteinized cell extracts were used to determine NAD+ levels, as described in Materials and methods. (C) Low glucose concentration induces deacetylation of PGC-1α. C2C12 myotubes were infected with PGC-1α adenoviruses and treated for 12 h with 25 and 5 mM of glucose with or without nicotinamide (10 mM). (D) SIRT1 is required to increase mitochondrial and fatty acid utilization genes in response to low glucose in MEFs and (E) C2C12 myotubes. C2C12 cells were infected with the adenoviruses encoding PGC-1α and shRNA control or shRNA SIRT1. Twelve hours before harvesting, MEFs or C2C12 cells were treated with different concentrations of glucose and indicated gene expression was analyzed. (F) SIRT1 is required for induction of mitochondrial fatty acid oxidation in response to low glucose. C2C12 myotubes and MEFs were treated as in (D). Oxidation rates of oleic acid were analyzed as described in Materials and methods. Values represent the mean of 2–3 experiments performed in duplicate. Error bars represent s.e.m. Statistical analyses were performed using Student's t-test. *P<0.05 and **P<0.005.
Discussion
Here we show the requirement of SIRT1 to induce a complete metabolic program of mitochondrial fatty acid oxidation in response to nutrient deprivation. We propose the following model of how nutrients can be sensed in cells to trigger a transcriptional metabolic response. In low levels of glucose, mammalian cells rearrange metabolic pathways that cause increases in NAD+ levels, which are sensed by the protein deacetylase SIRT1. Once activated, SIRT1 targets and deacetylates the transcriptional coactivator PGC-1α at promoter regions to induce gene expression of mitochondrial and fatty acid oxidation to maintain the bioenergetic state of the cell. This model provides a molecular mechanism by which fluctuations in nutrients target two key transcriptional regulators, SIRT1 and PGC-1α that control expression of mitochondrial oxidative genes, allowing cells to survive and adapt in periods of nutrient deprivation (Figure 7).
Figure 7.
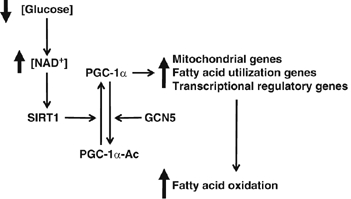
Model of glucose-dependent control of fatty acid oxidation through SIRT1/PGC-1α. Decreases in glucose elevate intracellular levels of NAD+ that will activate SIRT1deacetylase enzymatic activity. SIRT1 interacts and deacetylates PGC-1α, which induces an upregulation of genes linked to mitochondrial function and fatty acid utilization. Finally, this change in PGC-1α-targeted gene expression results in the activation of complete oxidation of fatty acids. See text for further details.
The ability of cells to switch from glucose to fatty acid oxidation in response to low concentrations of glucose correlates with changes in NAD+ levels. Absence or decreased levels of SIRT1 prevent this metabolic response. We propose that this is mainly due to a reduction in expression of genes involved in mitochondrial and fatty acid utilization, however the essential and required target genes are unknown. A key enzyme in this switch is PDK4, which acts by inhibition of PDH activity, thereby preventing the entry of pyruvate into the TCA cycle (Sugden et al, 1993). In this scenario, oxidation of free fatty acids is the main supply of acetyl-CoA and NADH to the TCA and respiratory electron transport chain respectively. The fact that PDK4 is tightly controlled by SIRT1 and PGC-1α indicates that it might be a crucial target through which these transcriptional regulators control this nutrient response. In addition to PDK4 and genes of fatty acid oxidation such as CPT1b and MCAD, SIRT1 and PGC-1α also coordinate increases of genes of mitochondrial respiration. This increase is important because if the rate of fatty acid oxidation is not coupled to the use of NADH by mitochondrial respiration, it would inhibit the TCA cycle and compromise synthesis of ATP as energetic substrate for the cell. In fact, uncompleted rates of fatty acid oxidation in skeletal muscle could generate lipid intermediates that are linked to insulin resistance states (Koves et al, 2005). In this situation, increases in NADH levels could also potentially inhibit the activity of SIRT1 as has been previously proposed (Lin et al, 2004). Interestingly, this might suggest the existence of a negative feedback loop by increases of NADH to slow down rates of fatty acid oxidation through inhibition of SIRT1. Therefore, it is crucial in conditions of nutrient deprivation that cells maintain the ability to shift to oxidation of fatty acids and to couple it to mitochondrial respiration. Nicotinamide, an inhibitor and product of SIRT1 catalytic activity, also affected gene expression regulated by PGC-1α and prevented the low glucose response to induce oxidation of fatty acids. As nicotinamide is an endogenous inhibitor of SIRT1, it could also play a role in this metabolic response. In fact, in yeast, Pnc1p, an enzyme that synthesizes NAD+ from nicotinamide and ADP-ribose, is induced by caloric restriction, suggesting that cellular nicotinamide concentrations might modulate lifespan (Anderson et al, 2003). In addition, in neuronal cells, caloric restriction also regulates endogenous levels of nicotinamide (Qin et al, 2006).
The levels of PGC-1α acetylation are controlled by the acetyl transferase GCN5 and SIRT1 deacetylase. It is currently not clear how or whether GCN5 is regulated or if it is constitutively active. We have shown previously that SIRT1 protein levels are increased in hepatocytes during starvation, ultimately leading to deacetylation of PGC-1α, which correlated with increases in NAD+ and pyruvate (Rodgers et al, 2005). Interestingly, in myotubes, we did not observe any effect of pyruvate on SIRT1 protein levels (data not shown). It is conceivable that selective tissue transcription factors that interact with PGC-1α might account for these differences. However, in response to low concentrations of glucose, increased NAD+ levels correlated with PGC-1α deacetylation suggesting that SIRT1 enzymatic activity was increased. The extent to which the observed changes in NAD+ directly affect SIRT1 enzymatic activity in cells is unknown. It is possible that in addition to NAD+, other alternative regulatory mechanisms or signaling pathways that are activated by low glucose concentrations might also regulate the activity of SIRT1 on PGC-1α. Such putative mechanisms are currently under exploration. We have previously mapped up to 13 acetylation sites on different structural domains of PGC-1α (Rodgers et al, 2005). At this point, it is not entirely clear which acetylation residues are required for the metabolic effects. Interestingly, acetylation of PGC-1α by GCN5 drives PGC-1α to nuclear foci that are transcriptionally inactive (Lerin et al, 2006), which could conceivably be the same mechanism that functions in myotubes.
In summary, we show here that SIRT1 is an important functional regulator of PGC-1α, and that under conditions of nutrient restriction, SIRT1 regulates PGC-1α target genes and is essential in responding to increases of complete fatty acid oxidation. This is a crucial metabolic adaptation that will not only allow cells to survive periods of low nutrients, but will also integrate muscle cells in the physiological response of food deprivation to spare glucose for neuronal and red blood cells. It is also possible that this metabolic adaptation might impact the molecular mechanisms by which caloric restriction modulates lifespan. As SIRT1 is an enzyme, it encourages the possibility of screening drugs that would modulate its activity on PGC-1α and activate mitochondrial oxidative genes that are dysregulated in diseases such as diabetes and neurodegeneration.
Materials and methods
Plasmids
PGC-1α, GCN5, SIRT1, control and SIRT1 shRNA adenoviruses were constructed using the pAd-Easy system as previously described (Rodgers et al, 2005).
Animal experiments
C57B1/6 mice were fed ad libitum or fasted for 16 h. Skeletal muscle whole-cell homogenates were prepared with RIPA buffer and used for Western blot analysis as previously described (Rodgers et al, 2005).
Cell culture and adenoviral infections
C2C12 skeletal muscle cells were cultured in DMEM with 10% calf serum. Differentiation was induced at ∼90–100% confluency by switching the cell media to DMEM supplemented with 2% horse serum (HS) (differentiation media) for 72 h. Following differentiation into myotubes, C2C12 cells was infected with adenovirus for 4 h in differentiation media. The media was then replaced with fresh DMEM/2%HS for an additional 48 h. The cells were then washed with PBS and incubated with indicated treatment for 12 h before collection in RIPA buffer (protein analysis) or Trizol (RNA analysis).
MEFs were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). Upon reaching 70% confluency, cells were infected for 24 h in DMEM with 10% FBS. The media was then replaced with fresh DMEM/10% FBS for an additional 48 h. The cells were washed with PBS and incubated with the indicated treatment for 12 h before collection in RIPA buffer (protein analysis) or Trizol (RNA analysis).
Primary muscle cells were isolated and cultured from 2 to 3-week-old FVB mice as described previously (Sabourin et al, 1999). To induce differentiation, myoblasts were grown to 80% confluence and then switched to the differentiation medium, DMEM, containing 5% HS. Myotubes were transduced with adenovirus for 24 h and media were then replaced with fresh DMEM with 5% horse serum for an additional 48 h. Cells were washed with PBS and lysed in RIPA buffer. Protein lysates were subjected to Western blot analysis to determine the expression level of SIRT1 protein. Total RNA was extracted from the cells and subjected to quantitative RT–PCR analysis to determine gene expression. The Taqman primer/probe sets were purchased from ABI (assay on demand) and 18S was used for normalization. Citrate synthase activity was measured in the cells as described previously (Moyes et al, 1997).
Gene expression analysis
Total RNA prepared from either C2C12 cells or from MEFs was extracted with Trizol (Invitrogen). Complementary DNA generated by Superscript II enzyme (Invitrogen) was analyzed by quantitative reverse transcriptase-mediated PCR using an iQ SYBR Green Supermix (Bio-Rad). All data were normalized to tubulin expression. The oligonucleotide primers used are available upon request.
PGC-1α acetylation assays
PGC-1α lysine acetylation was analyzed by immunoprecipitation of PGC-1α followed by Western blot using acetyl-lysine antibodies (Cell Signalling and Technology) as previously described (Rodgers et al, 2005). C2C12 cells or MEFs were infected with adenovirus expressing Flag-tagged PGC-1α as described above. After the indicated 6 h or 12 h treatment, PGC-1α was immunoprecipitated using anti-Flag beads (Sigma) and examined for acetylation.
Fatty acid and glucose oxidation and NAD+ measurements assays
Cellular oleic and palmitic acid oxidation rates were determined in C2C12 cells and MEFs using modifications of protocols previously described (Garcia-Martinez et al, 2005; Wende et al, 2005). C2C12 cells were cultured in 12-well dishes, then differentiated and infected as described above. The cells were rinsed with PBS and incubated with MEM supplemented with 0.5% HS and either 500 μM oleic or palmitic acid for 12 h. Cells were then incubated for an additional 3 h with fresh DMEM/0.5% HS that was supplemented with [1-14C]oleic or [1-14C]palmitic acid (3.0 mCi/mmol). The oxidation reactions were terminated and CO2 was released from the media by the addition of 3 M perchloric acid. Filter paper saturated with phenylethylamine was placed over each well to capture CO2. Following a 3 h incubation with gentle shaking at 25°C, 14CO2 resulting from oxidized fatty acid was quantified by scintillation counting of the filter paper. Each experiment was performed in triplicate and the results were normalized to total protein.
Glucose oxidation rates were measured using methods previously described (Wende et al, 2005).
NAD+ nucleotide concentration was directly measured as described (Lin et al, 2001). In brief, a confluent 6 cm dish of C2C12 myotubes was homogenized in 400 μl of acid extraction buffer to obtain the NAD+ concentration. Homogenates were neutralized with 200 μl of 0.4 M Tris buffer. The concentration of NAD+ was measured fluorometrically after an enzymatic reaction using 1.5 μl of sample. For NAD+/NADH measurements, C2C12 myotubes were deproteinized and concentrations of lactate and pyruvate were determined as previously described (Rodgers et al, 2005).
Statistical analysis
Data are the means±s.e.m. Statistical analysis was performed by a two-tailed unpaired Student's t-test. P<0.05 was considered to be statistically significant.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank members of the Puigserver laboratory for insightful discussions on the manuscript. CL and JTR were supported by fellowships from the American Heart Association. These studies were supported in part by an Ellison Medical Foundation New Scholar Award, American Diabetes Association, US Department of Defense and NIH RO1, DK069966 (to PP).
References
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423: 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordone L, Guarente L (2005) Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol 6: 298–305 [DOI] [PubMed] [Google Scholar]
- Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J, McDonagh T, Lemieux M, McBurney M, Szilvasi A, Easlon EJ, Lin SJ, Guarente L (2006) Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol 4: e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KF, Mostoslavsky R, Lombard DB, Pang WW, Saito S, Franco S, Kaushal D, Cheng HL, Fischer MR, Stokes N, Murphy MM, Appella E, Alt FW (2005) Mammalian SIRT1 limits replicative lifespan in response to chronic genotoxic stress. Cell Metab 2: 67–76 [DOI] [PubMed] [Google Scholar]
- Finck BN, Kelly DP (2006) PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 116: 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez C, Marotta M, Moore-Carrasco R, Guitart M, Camps M, Busquets S, Montell E, Gomez-Foix AM (2005) Impact on fatty acid metabolism and differential localization of FATP1 and FAT/CD36 proteins delivered in cultured human muscle cells. Am J Physiol Cell Physiol 288: C1264–C1272 [DOI] [PubMed] [Google Scholar]
- Guan HP, Li Y, Jensen MV, Newgard CB, Steppan CM, Lazar MA (2002) A futile metabolic cycle activated in adipocytes by antidiabetic agents. Nat Med 8: 1122–1128 [DOI] [PubMed] [Google Scholar]
- Huss JM, Kopp RP, Kelly DP (2002) Peroxisome proliferator-activated receptor coactivator-1alpha (PGC-1alpha) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-alpha and -gamma. Identification of novel leucine-rich interaction motif within PGC-1alpha. J Biol Chem 277: 40265–40274 [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13: 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, Goodpaster BH, Storlien L (2002) Muscle triglyceride and insulin resistance. Annu Rev Nutr 22: 325–346 [DOI] [PubMed] [Google Scholar]
- Koves TR, Li P, An J, Akimoto T, Slentz D, Ilkayeva O, Dohm GL, Yan Z, Newgard CB, Muoio DM (2005) Peroxisome proliferator-activated receptor-gamma co-activator 1alpha-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J Biol Chem 280: 33588–33598 [DOI] [PubMed] [Google Scholar]
- Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab 3: 429–438 [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 1: 361–370 [DOI] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418: 797–801 [DOI] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L (2000) Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289: 2126–2128 [DOI] [PubMed] [Google Scholar]
- Lin SJ, Ford E, Haigis M, Liszt G, Guarente L (2004) Calorie restriction extends yeast lifespan by lowering the level of NADH. Genes Dev 18: 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SS, Manchester JK, Gordon JI (2001) Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J Biol Chem 276: 36000–36007 [DOI] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, Bolouri MS, Ray HN, Sihag S, Kamal M, Patterson N, Lander ES, Mann M (2003) Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115: 629–640 [DOI] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM (2004) Err{alpha} and Gabpa/b specify PGC-1{alpha}-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci USA 101: 6570–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes CD, Mathieu-Costello OA, Tsuchiya N, Filburn C, Hansford RG (1997) Mitochondrial biogenesis during cellular differentiation. Am J Physiol 272: C1345–C1351 [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci USA 100: 8466–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type II diabetes. N Engl J Med 350: 664–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 429: 771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilegaard H, Neufer PD (2004) Transcriptional regulation of pyruvate dehydrogenase kinase 4 in skeletal muscle during and after exercise. Proc Nutr Soc 63: 221–226 [DOI] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Thiyagarajan M, Macgrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng HH, Pedrini S, Gandy S, Sauve A, Pasinetti GM (2006) Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer's disease amyloid neuropathology by calorie restriction. J Biol Chem 281: 21745–21754 [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434: 113–118 [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL (2004) Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA 101: 15998–16003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabourin LA, Girgis-Gabardo A, Seale P, Asakura A, Rudnicki MA (1999) Reduced differentiation potential of primary MyoD−/− myogenic cells derived from adult skeletal muscle. J Cell Biol 144: 631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A (2004) The estrogen-related receptor {alpha} (ERR{alpha}) functions in PPAR{gamma} coactivator 1{alpha} (PGC-1{alpha})-induced mitochondrial biogenesis. Proc Natl Acad Sci USA 101: 6472–6477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuldiner AR, McLenithan JC (2004) Genes and pathophysiology of type II diabetes: more than just the Randle cycle all over again. J Clin Invest 114: 1414–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA (2005) Toward a unified theory of caloric restriction and longevity regulation. Mech Ageing Dev 126: 987–1002 [DOI] [PubMed] [Google Scholar]
- Storlien L, Oakes ND, Kelley DE (2004) Metabolic flexibility. Proc Nutr Soc 63: 363–368 [DOI] [PubMed] [Google Scholar]
- Sugden MC, Howard RM, Munday MR, Holness MJ (1993) Mechanisms involved in the coordinate regulation of strategic enzymes of glucose metabolism. Adv Enzyme Regul 33: 71–95 [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L (2001) Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410: 227–230 [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20: 1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Huss JM, Schaeffer PJ, Giguere V, Kelly DP (2005) PGC-1alpha coactivates PDK4 gene expression via the orphan nuclear receptor ERRalpha: a mechanism for transcriptional control of muscle glucose metabolism. Mol Cell Biol 25: 10684–10694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures