

Abstract
The regulation of myogenic progenitor cells during muscle regeneration is not clearly understood. We have previously shown that the Foxk1 gene, a member of the forkhead/winged helix family of transcription factors, is expressed in myogenic progenitor cells in adult skeletal muscle. In the present study, we utilize transgenic technology and demonstrate that the 4.6 kb upstream fragment of the Foxk1 gene directs β-galactosidase expression to the myogenic progenitor cell population. We further establish that Sox15 directs Foxk1 expression to the myogenic progenitor cell population, as it binds to an evolutionarily conserved site and recruits Fhl3 to transcriptionally coactivate Foxk1 gene expression. Knockdown of endogenous Sox15 results in perturbed cell cycle kinetics and decreased Foxk1 expression. Furthermore, Sox15 mutant mice display perturbed skeletal muscle regeneration, due in part to decreased numbers of satellite cells and decreased Foxk1 expression. These studies demonstrate that Sox15, Fhl3 and Foxk1 function to coordinately regulate the myogenic progenitor cell population and skeletal muscle regeneration.
Keywords: Foxk1, muscle regeneration, myogenic progenitors, Sox15, transgenic technologies
Introduction
Skeletal muscle has the capacity for self-repair. Resident within adult skeletal muscle is a pool of undifferentiated myogenic progenitor cells (MPCs) that includes satellite cells (Miller et al, 1999; Mourkioti and Rosenthal, 2005). Satellite cells are small mononuclear cells that share a common basal lamina with the larger multinucleated myocytes, and are located in a peripheral (or satellite) position with respect to the multinucleated myofiber (Mauro, 1961). Following muscle injury, the MPCs are activated, they proliferate and withdraw from the cell cycle to form multinucleated myofibers in a manner that recapitulates the fundamental events of muscle development in the fetus (Sheehan and Allen, 1999; Seale and Rudnicki, 2000; Snider and Tapscott, 2003; Shi and Garry, 2006). The MPCs are self-renewing and replenish a pool of quiescent progenitors; however, this capacity for self-renewal is finite and repeated injuries ultimately lead to loss of muscle mass and myopathies (Conboy and Rando, 2002; Olguin and Olwin, 2004; Collins et al, 2005). MPCs are arrested at an early stage of the myogenic program such that they do not express any of the bHLH proteins of the MyoD family (Shi and Garry, 2006). Recent studies have begun to identify factors that are expressed in the satellite cell population, including Foxk1, Pax7, C-met, syndecan3/4 and Pax3, although the molecular regulation of this cell population remains ill-defined (Garry et al, 1997; Borycki et al. 1999; Seale et al. 2000; Cornelison et al, 2001; Cornelison et al, 2004; Deasy et al, 2005; Montarras et al, 2005).
Members of the forkhead/winged helix (Fox) transcription factor family, which has more than 100 constituents, have been identified in a number of vertebrate cell lineages and are known to exert important regulatory functions during development in the control of cell fate, patterning, proliferation, differentiation and tissue morphogenesis (Lai et al, 1993; Ang and Rossant 1994). With respect to stem cells and/or tissue repair, a mammalian forkhead/winged helix protein termed Foxd3 (Genesis) is expressed selectively in embryonic stem cells and Foxa2 has been identified in regenerating hepatocytes (Sutton et al, 1996; Ye et al, 1997). We have previously shown that Foxk1 is expressed in the satellite cell population that is resident in adult skeletal muscle (Garry et al, 1997). Using a gene disruption strategy, we have observed that mice that lack Foxk1 have impaired muscle regeneration, dysregulation of the cyclin-dependent kinase inhibitor, p21 (Cdkn1a), and perturbed cell cycle progression of the MPC population (Garry et al, 2000; Hawke et al, 2003a).
Sox transcription factors are found in all metazoan species and, like forkhead transcription factors, play key roles in embryonic development. Satellite cells also express Sox transcription factors (Béranger et al, 2000; Schmidt et al, 2003). Members of the Sox transcription factor family have relative homology of the high-mobility group (HMG) DNA binding domain (Wilson and Koopman, 2002). For example, Sox2 is an essential regulator of ES cells, whereas Sox4 and Sox5 are key regulators of cardiac development (Koopman et al, 2004). Sox transcription factors interact with protein partners and function to either activate or repress downsteam target gene expression (Koopman et al, 2004). Sox15, a member of this family, is expressed in satellite cells, and mice-lacking Sox15 are viable but appear to have impaired skeletal muscle regeneration (Lee et al, 2004). Overall, the functional role of Sox15 in the MPC population is unclear.
Transcriptional regulators of gene expression interact with adaptor proteins to form a regulatory complex of nuclear proteins. The Four and a half LIM domain family of proteins are regulators of growth and discrete stages of development and cellular differentiation (Chu et al, 2000; Müller et al, 2002). The LIM domain is characterized by a double zinc-finger motif that mediates protein–protein interactions. Fhl3 has previously been described to be expressed in skeletal muscle and is localized to the cytoplasmic and the nuclear compartments, although the functional role of this factor is unclear (Morgan and Madgwick, 1998).
In the present study, we pursued a strategy to decipher the regulatory mechanisms that govern cell cycle re-entry of quiescent satellite cells. We demonstrate that Sox15 recruits Fhl3 to form a complex that coactivates the Foxk1 gene. We further establish that Sox15 directs Foxk1 expression to the MPC population. These studies support the notion that Foxk1 is a direct downstream target of Sox15. This Sox15–Fhl3 interaction and subsequent regulation of Foxk1 provides a mechanism whereby quiescent satellite cells re-enter the cell cycle and regenerate injured skeletal muscle. These studies enhance our understanding of the molecular regulation of the MPC population and will provide a platform for therapeutic applications for the treatment of debilitating myopathies.
Results
We have previously reported that the forkhead/winged helix transcription factor, Foxk1, is expressed in MPCs that are resident in adult skeletal muscle using light microscopic and ultrastructural immunohistochemical techniques (Garry et al, 1997). We have further defined Foxk1 as an important cell cycle regulator of the MPC population (Garry et al, 2000; Hawke et al, 2003a). To examine the regulation of the Foxk1 gene, we characterized a 4.6 kb Foxk1 promoter fragment that contained evolutionarily conserved regions between mouse and human. We generated transgenic mice using this 4.6 kb Foxk1 promoter fragment to drive the lacZ reporter gene (Figure 1A). Expression was observed in three transgenic lines and in all three lines β-galactosidase staining was specific to the muscle precursor cells of the developing limbs (E11.5, E13.5 and E14.5) (Figure 1B–E). These results indicate that the 4.6 kb promoter of Foxk1 contains regulatory elements that control expression in a cell lineage that are destined to become skeletal muscle (the MPC population) in the developing limbs (Figures 1E–K). During midgestational stages of embryogenesis (E13.5), expression of the 4.6 kb Foxk1 promoter was colocalized with Foxk1 in developing muscle using immunohistochemical techniques, a Foxk1 antiserum and a β-galactosidase antiserum (Figure 1G and H). Whereas the 4.6 kb Foxk1-lacZ expression largely recapitulated endogenous Foxk1 expression in the developing limbs, trunk muscles (low level) and heart (low level), β-galactosidase expression was not observed in the developing somites (Supplementary Figure 1).
Figure 1.

Transgenic embryonic expression of the Foxk1 promoter fused to lacZ. (A) Schematic outlining the transgenic construct that consists of a 4.6 kb upstream fragment of the Foxk1 gene fused to the lacZ reporter. (B) Whole-mount β-galactosidase expression in limbs, diaphragm and branchial arch derivatives in E11.5 (B), E13.5 (C, E) and E14.5 (D) 4.6 kb Foxk1–lacZ transgenic embryos. (F) Transverse section of the E13.5 embryo reveals β-galactosidase expression in muscle precursor cells of the limb. (G, H) Coexpression of β-galactosidase (α-bgal) and Foxk1 in intercostal muscles of 4.6 kb Foxk1–lacZ transgenic embryos. Low magnification (I) and high magnification (J) of an E13.5 parasaggital section of the Foxk1 promoter-lacZ expression (blue) and desmin immunolocalization (brown). Desmin is an intermediate filament protein expressed early during myogenesis. Desmin staining is observed in the heart and is coexpressed in a small subpopulation of the Foxk1 promoter-lacZ-positive mesenchymal percursor cells of the limb. h, heart.
Following birth, expression of the 4.6 kb Foxk1 promoter fragment is limited to the quiescent MPC population. In response to cardiotoxin-induced injury to hindlimb skeletal muscle, we have shown that the MPC population undergoes activation (D1 following injury) and a tremendous proliferative expansion (D2–D5) with the formation of newly regenerated myotubes (D5–D10) following cardiotoxin injury (Hawke et al, 2003a; Goetsch et al, 2003). In the present study, we observed that the expression of the 4.6 kb Foxk1 promoter recapitulates Foxk1 expression during muscle regeneration and identifies both quiescent satellite cells and proliferating myogenic progenitor cells, but is not expressed in differentiated myotubes using whole-mount (Figure 2A) and histological sections of 5 day injured (Figure 2B), 10 day injured (Figure 2C), 4.6 kb Foxk1:mdx (Figure 2D) and uninjured skeletal muscle (Figure 2E and F). The mdx mouse lacks dystrophin and is characterized by cycles of degeneration and regeneration of skeletal muscle.
Figure 2.
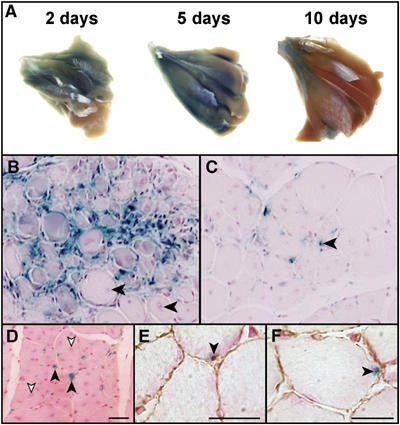
Foxk1 transgene expression is observed in MPCs of cardiotoxin-injured and uninjured adult skeletal muscle. (A) β-Galactosidase expression in 4.6 kb Foxk1 promoter-lacZ transgenic whole-mount stained gastrocnemius muscles at defined time periods following cardiotoxin-induced muscle injury. Note robust β-galactosidase expression in transgenic gastrocnemius muscles 2 days following injury (corresponding to satellite cell activation) and 5 days following injury (associated with satellite cell proliferation), with decreased expression within 10 days following injury (newly regenerated myofibers). (B) Transverse histological sections of transgenic (4.6 kb Foxk1 promoter-lacZ) hindlimb skeletal muscle 5 days following injury. Note robust expression in activated satellite cells (blue) but no expression in newly regenerated centronucleated myofibers (arrowhead). (C) The 4.6 kb Foxk1 promoter-lacZ expression (blue) is expressed in MPCs in skeletal muscle 10 days following injury (arrowhead). (D) Transverse section of the 4.6 kb Foxk1-lacZ transgenic:mdx tibialis anterior skeletal muscle reveals β-galactosidase expression in activated satellite cells (black arrowheads) and not newly regenerated centronucleated myofibers (white arrowheads). Scale bar, 40 μm. (E, F) Transverse sections of the 4.6 kb Foxk1 promoter-lacZ skeletal muscle examined for β-galasctosidase (blue) and laminin (brown) expression in uninjured skeletal muscle (arrowhead). Note that the β-galactosidase-expressing cells are sublaminar, consistent with expression in satellite cells (arrowheads). Scale bars, 20 μm.
A detailed analysis of the Foxk1 upstream fragment has been undertaken using a transgenic strategy. We generated mice harboring either 1.6 kb Foxk1 promoter-lacZ or 0.6 kb Foxk1 promoter-LacZ transgenes and we observed that a distal 3 kb fragment contains an enhancer(s) that directs expression in the MPC population (Supplementary Table 1).
Sox15 binds to the Foxk1 promoter
Database analysis of this 3 kb upstream fragment of the Foxk1 gene revealed the presence of a Sox binding element (SBE) that was evolutionarily conserved between mouse and human (Figure 3A). Using electrophoretic mobility shift assays (EMSA) and a 23-oligonucleotide probe of the Foxk1 promoter that contains the SBE, we examined the capacity of a myc-Sox15 fusion protein to bind to this DNA probe. As outlined in Figure 3B (mouse) and Supplementary Figure 2 (human), we demonstrated that Sox15 binds to this site (i.e. SBE) in the Foxk1 promoter, and that the myc-Sox15 fusion protein–oligonucleotide complex could be competed with cold competitor. We further demonstrated that mutagenesis of three nucleotides within the SBE of the radiolabeled oligonucleotide probe precluded formation of the protein–DNA complex (Figure 3B). Additionally, the incubation of the wild-type oligonucleotide probe that contains the SBE with the myc-Sox15 fusion protein and anti-myc serum resulted in the supershift of the protein–DNA complex (Figure 3B).
Figure 3.
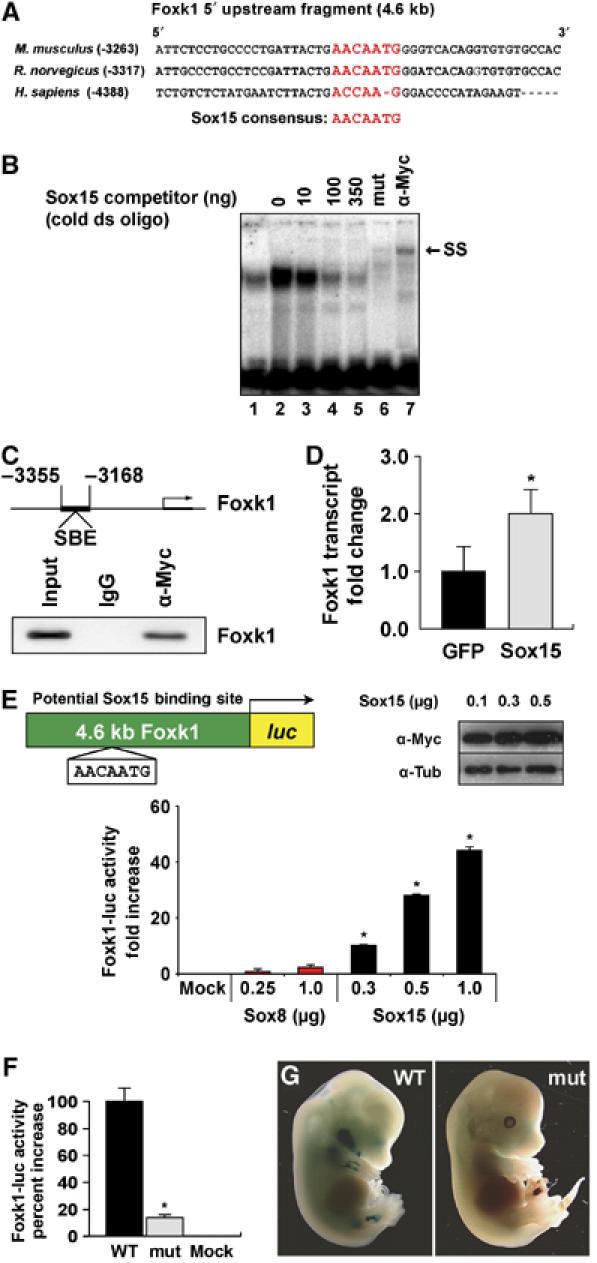
Sox15 binds to a conserved region of the Foxk1 promoter. (A) Alignment of the human, mouse and rat Foxk1 promoter sequences revealing the evolutionary conservation of the Sox binding element. (B) EMSA of the 32P-labeled SBE from the mouse Foxk1 promoter with in vitro-synthesized myc-Sox15 protein. Competition studies were performed with 0–350 ng of unlabeled Sox15 binding site DNA (lanes 2–5). Using the same probe that has a mutated Sox binding site, no protein–DNA complex is formed (lane 6; mut). Using a myc antibody that recognizes the myc-Sox15 fusion protein, the protein–DNA complex is supershifted (lane 7; SS). (C) ChIP assay was undertaken following the overexpression of the myc-Sox15 fusion protein in C2C12 myoblasts, crosslinking and IP of the myc-Sox15 fusion protein–DNA complexes. Co-IP of the targeted promoter DNA was analyzed by PCR amplification. DNA was also isolated without IP and used as input. IP of the myc-Sox15 fusion protein pulled down the Foxk1 promoter. Note that no DNA was immunoprecipitated with the control IgG. (D) Forced overexpression of Sox15 in C2C12 cells compared with overexpression of a GFP expression plasmid (control) reveals a two-fold increase in endogenous Foxk1 transcript expression (n=4 for each sample; *P<0.001). (E) Sox15 specifically transactivates the Foxk1 promoter. Schematic of the 4.6 kb Foxk1 promoter-luc plasmid used in these assays containing the Sox binding motif is shown. Overexpression of increasing amounts of Sox8 or Sox15 demonstrates a dose-dependent transcriptional activation of the Foxk1 promoter by Sox15 (n=9 for each sample; *P<0.001). In contrast, Sox8 does not activate the Foxk1 promoter. Western blot analysis of extracts isolated from C2C12 myoblasts following overexpression of myc-Sox15 reveals overexpression of the fusion protein in a dose-dependent manner in C2C12 myoblasts (α-tubulin is used as a loading control). (F) Transfection of Sox15 expression plasmids with wild-type (WT) or mutant (mut) Sox binding site reveals a 90% decrease in luciferase activity following transfection with the mutated Foxk1 vector (n=3 for each sample; *P<0.01). (G) Mutagenesis of the SBE in the 4.6 kb Foxk1 promoter-lacZ transgene extinguishes β-galactosidase expression in the limbs of E13.5 embryos compared with age-matched controls (a total of 15 mutant transgenics were evaluated and all had significantly decreased or no expression in the developing limbs).
To further investigate whether Sox15 interacts and binds to the Foxk1 promoter, we performed chromatin immunoprecipitation (ChIP) assays. Chromatin fragments from lysates of C2C12 cells transfected with a plasmid expressing myc-Sox15 were precipitated with a myc antibody or control IgG (Figure 3C). DNA from the immunoprecipitates was subjected to PCR analysis using primers specific to the Foxk1 promoter harboring the SBE. As shown in Figure 3C, chromatin fragments containing the Sox15 binding site in the Foxk1 gene were specifically immunoprecipitated by the myc antibody in cells expressing the myc-Sox15 fusion protein. No chromatin fragments were precipitated by control IgG that further established the specificity of Sox15 binding to the Foxk1 promoter (Figure 3C).
Sox15 transcriptionally activates Foxk1 gene expression
We performed transcriptional assays to assess the specificity of Sox transcription factors as regulators of Foxk1 gene expression. We fused the 4.6 kb Foxk1 promoter to the luciferase reporter (Figure 3E). We transfected C2C12 myoblasts (a cell line generated from satellite cells) with the Foxk1 promoter–reporter construct and Sox factors that have been reported to be expressed in the MPC population (i.e. Sox8 and Sox15) (Schmidt et al, 2003; Lee et al, 2004). We observed that Sox15, in a dose-dependent manner, was a potent transcriptional regulator of Foxk1 gene expression as it resulted in more than a 40-fold activation (Figure 3E). In contrast, Sox8 did not transcriptionally activate the Foxk1 gene (Figure 3E). To further examine the specificity of Sox15 transcriptional activity, we evaluated whether Sox15 was capable of activation of the myoglobin gene (a well-characterized muscle-specific promoter in our laboratory) (Garry et al, 1996, 1998; Grange et al, 2002). We observed that Sox15 is not a transcriptional activator of the 2.0 kb myoglobin promoter-luc construct (Supplementary Figure 3).
To further evaluate the capacity of Sox15 to transcriptionally activate Foxk1, we overexpressed Sox15 (pcDNA-GFP-Sox15) in C2C12 myoblasts and assayed for endogenous Foxk1 transcript expression using QRT–PCR analysis. We observed that 24 h following overexpression of Sox15, Foxk1 mRNA was significantly increased more than two-fold compared with the control (n=4 for each sample; *P<0.001) (Figure 3D). The induction of endogenous Foxk1 expression is consistent with the hypothesis that Sox15 is an upstream regulator of the Foxk1 gene.
Mutagenesis of the Sox binding site attenuates Foxk1 gene expression
We undertook in vitro and in vivo studies to further examine whether Sox15 is an upstream regulator of the Foxk1 gene. We mutagenized three nucleotides within the SBE located in the 4.6 kb Foxk1 promoter and performed transcriptional and transgenic analyses. Mutagenesis of the SBE in the 4.6 kb Foxk1-luc plasmid and cotransfection with a Sox15 expression plasmid resulted in a 90% reduction of luciferase activity in C2C12 cells compared with the wild-type control (n=3 for each group; *P<0.01) (Figure 3F). We further examined the endogenous expression of the 4.6 kb (ΔSBE)Foxk1-lacZ construct using transgenic technologies. Mutant transgenic embryos were harvested in parallel with controls, fixed, stained with X-gal and examined for β-galactosidase expression. Expression was observed in 15 ΔSBE mutant-Foxk1 transgenic embryos at E13.5 and all had a severe reduction of β-galactosidase expression in the fore and hindlimbs (Figure 3G and Supplementary Figure 4). These results further establish that Sox15 is a direct upstream transcriptional regulator of Foxk1 expression.
Sox15 physically interacts with Fhl3
Having established that Sox15 is an essential upstream regulator of Foxk1, we undertook a yeast two-hybrid assay to define the proteins that interact with Sox15 in the regulation of Foxk1 and the MPC population. We utilized the longest Sox15 construct (1–181) that lacked autoactivation as bait and screened a commercially available adult skeletal muscle library (Figure 4A). We identified the LIM-only protein, Fhl3, as a Sox15 interacting factor, which was confirmed using full-length Fhl3 in the yeast two-hybrid assay (Figure 4B). Fhl3 did not interact with the GAL4 DNA binding domain alone and there was no interaction between the bait and the GAL4 activation domain (Figure 4B). This interaction was further confirmed in mammalian cells using co-immunoprecipitation (co-IP) assays after myc-tagged Sox15 and HA-tagged Fhl3 or HA-tagged Sox15 and myc-tagged Fhl3 were coexpressed in C2C12 cells (Figure 4C).
Figure 4.
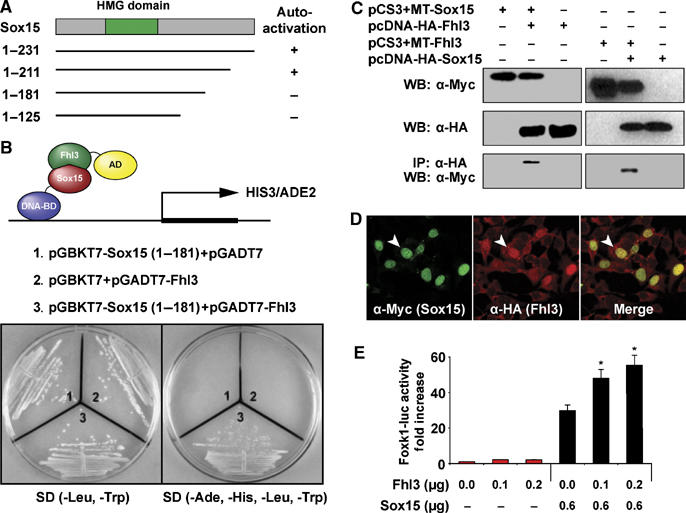
Sox15 physically interacts with Fhl3 and synergistically transctivates Foxk1 gene expression. (A) Selected Sox15 constructs were tested for autoactivation. The Sox (1–181) construct lacked autoactivation and was used as bait in the yeast two-hybrid assay. (B) Two-hybrid results, following the screening of a skeletal muscle library, confirmed the interaction of full-length Sox15 and Fhl3. (C) Co-IP assay following overexpression of myc-Sox15 and HA-Fhl3 or HA-Sox15 and myc-Fhl3 confirmed the interaction. (D) Overexpression of the respective tagged proteins reveals colocalization (yellow nuclei in the merge panel) in the nuclear compartment of C2C12 cells (arrowhead). (E) Cotransfection of Fhl3, Sox15 and 4.6 kb Foxk1-luc in C2C12 cells reveals that Fhl3 by itself is unable to transactivate Foxk1 expression, but in tandem with Sox15 there is a synergistic coactivation of Foxk1 gene expression (n=6; *P<0.001).
To further investigate the interaction between Fhl3 and Sox15, we transfected C2C12 myoblasts with HA-Fhl3 and myc-Sox15 and examined the localization of these fusion proteins using immunohistochemical and confocal microscopic techniques. Sox15 was restricted to the nuclear compartment and colocalized with Fhl3 (Figure 4D). In addition, Fhl3 was abundantly expressed in myoblasts in the cytoplasmic and nuclear compartment and decreased in expression following differentiation using immunohistochemical and Western blot analyses (Figure 4D and Supplementary Figure 5). Future studies may further examine the interacting partners for Sox15 using an array of in vitro and in vivo (i.e. FRET) techniques.
Having established that Fhl3 interacts with Sox15 and is coexpressed in C2C12 myoblasts, we examined the role of Fhl3 as a transcriptional regulator of Foxk1. We found that Fhl3 alone had no transcriptional activity, but in combination with Sox15 there was a dose-dependent transcriptional synergy in the activation of Foxk1 gene expression (Figure 4E). These studies support the hypothesis that Sox15 interacts with a complex that includes Fhl3 to regulate Foxk1 gene expression and the MPC population.
Identification of the Sox15–Fhl3 interacting domains
To enhance our understanding regarding the molecular basis of the Sox15–Fhl3 interaction, we generated deletional constructs to identify the interaction domains for each protein. We mapped the interaction domains using yeast two-hybrid and GST pull-down assays. The Fhl3 binding domain of Sox15 was mapped using a series of Sox15 deletion mutants. Sox15 deletional mutants (1–181 and 136–181) strongly interacted with Fhl3 using a yeast two-hybrid assay (Figure 5A). These results were confirmed using GST pull-down assays, as full length (1–231) and the deletion mutant (136–181) interacted with Fhl3 (Figure 5B). In contrast, deletion constructs that contained the N-terminal (1–46) or the HMG domain (47–135) did not interact with Fhl3 (Figure 5A and B). We conclude that residues between 136 and 181 of Sox15 are sufficient to bind Fhl3. We further emphasize that unlike other Sox factors, where the HMG domain serves a dual role as a DNA binding domain and an interaction domain to partner with other nuclear factors, the Fhl3 interacting domain is a non-HMG domain (136–181).
Figure 5.
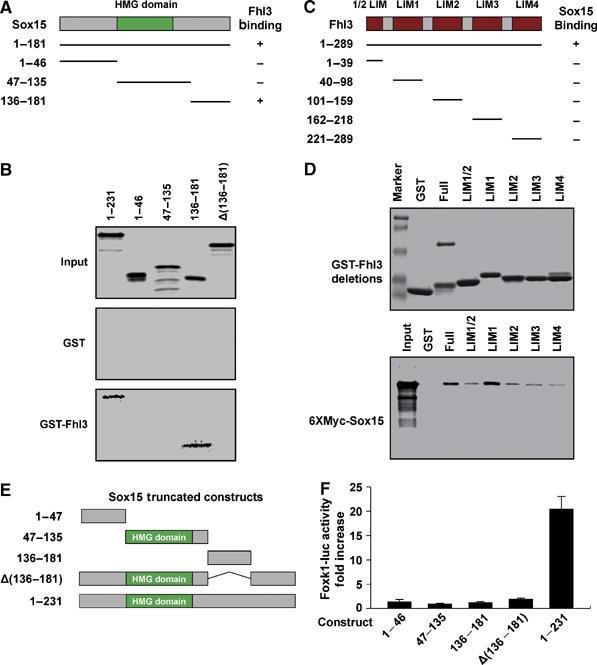
Interaction of Sox15 and Fhl3 requires specific domains. (A) Schematic of Sox15 and the deletion constructs used in the yeast two-hybrid assay and GST pull-down assays. Identification of the Sox15 binding domain that interacts with Fhl3 as determined by the yeast two-hybrid assay. (B) In vitro-translated Sox15 deletion constructs were used for GST pull-down assays. As shown in (B), the Sox15 construct (136–181) harbors the Fhl3 binding domain. This is further established, as full-length Sox15 lacking the 136–181 domain (Δ136–181) is unable to bind to Fhl3. (C) Schematic representation of Fhl3 full-length and deletional constructs used in the yeast two-hybrid and GST pull-down assays. The ability of the respective constructs to bind Sox15 using yeast two-hybrid assays is noted. (D) In vitro-translated Fhl3 deletion constructs were used for GST pull-down assays. Although all the LIM domains are able to interact with Sox15, LIM1 domain demonstrates the strongest affinity. These results support the conclusion that Fhl3 has multiple interacting domains with Sox15. (E) Schematic of full-length and deletional Sox15 constructs containing or lacking the Fhl3 binding site used in the transcriptional assays presented in (F). (F) Cotransfection of the respective Sox15 constructs and the 4.6 kb Foxk1-luc plasmid reveals that the Fhl3 binding domain and the carboxy-terminal region are essential for transcriptional activation of the Foxk1 gene (n=3 for each sample).
To map the Sox15 binding region of Fhl3, we performed yeast two-hybrid and GST pull-down assays using Fhl3 deletional constructs and in vitro-translated Sox15. As shown in Figure 5C, only full-length (1–289) Fhl3 interacted with Sox15 using a yeast two-hybrid assay. Furthermore, GST pull-down assays revealed that full-length Fhl3 and each individual LIM domain interacted with Sox15 but with different affinities (Figure 5D). We observed that the LIM1 domain had the highest affinity for Sox15 (Figure 5D). These data support the conclusion that Fhl3 forms multiple specific contacts with Sox15, which are consistent with previous reports of protein–protein interactions of other LIM-only proteins (Müller et al, 2002; Turner et al, 2003).
To further examine the functional role of Sox15 and Fhl3 interaction, we examined the transcriptional activity of Sox15 deletional constructs that differed in the carboxy-terminal region and the Fhl3 interacting domain. Following transfection of the respective Sox15 constructs with the 4.6 kb Foxk1 reporter plasmid in C2C12 myoblasts, we observed that only the full-length Sox15 displays transcriptional regulation of the Foxk1 gene (Figure 5E and F). Deletion of the Fhl3 interacting domain (construct designated as Δ136–181) resulted in a lack of transcriptional activity, further supporting the notion that the transcriptional activation domain for Sox15 is also, in part, harbored within the 136–231 region (Figure 5E and F).
Inactivation of Sox15 results in perturbed cell cycle kinetics
Our initial efforts to further define the functional role of Sox15 in the MPC population utilized three distinct Sox15 small interfering RNA (siRNA) double-stranded (ds) oligomers (Figure 6 and Supplementary Figure 6). Sox15-specific siRNA were transfected into asynchronously dividing C2C12 myoblasts to knock down Sox15 transcript expression. Forty-eight hours following transfection, we analyzed the experimental and control cells for phenotypic differences and analyzed the respective cell populations for their cell cycle kinetics and gene expression (Figure 6 and Supplementary Figure 6). Transfection with the RISC control (nonfunctional, nontargeting siRNAs) had no effect on cellular proliferation, cell cycle kinetics or gene expression (Figure 6). In contrast, transfection of Sox15 siRNA resulted in a severe decrease of Sox15 mRNA (Figure 6E). The knockdown of Sox15 expression resulted in decreased cellular proliferation as measured by BrdU incorporation (which labels cells in the S-phase of the cell cycle as a measure of cellular proliferation), impaired cell cycle kinetics (i.e. G0/G1 cell cycle arrest) and decreased Foxk1 transcript expression (Figure 6A–E). No evidence of increased apoptosis was observed in the experimental versus the control samples using FACS analysis (n=3 for each sample). The impaired cell cycle kinetics (i.e. G0/G1 cell cycle arrest) and decreased Foxk1 transcript expression were observed using three separate double-stranded oligos directed against different regions of the Sox15 transcript (Figure 6 and Supplementary Figure 6). The G0/G1 cell cycle arrest following Sox15 knockdown (Figures 6C and D) was associated with an induction of Cdkn1a (p21), but no change in Cdkn1b (p27) transcript levels was measured by QRT–PCR analysis (Figure 6E). These results are similar to those observed in the Foxk1 mutant satellite cells (Hawke et al, 2003a). In contrast, knockdown of Fhl3 in C2C12 cells had no effect on cell cycle kinetics (Supplementary Figure 7). The lack of an effect of Fhl3 knockdown on cell cycle kinetics may be due to redundancy of other LIM only family members as Fhl2 is capable of synergistic coactivation of Foxk1 gene expression (Supplementary Figure 8) but does not physically interact with Sox15 using co-IP assays (data not shown).
Figure 6.
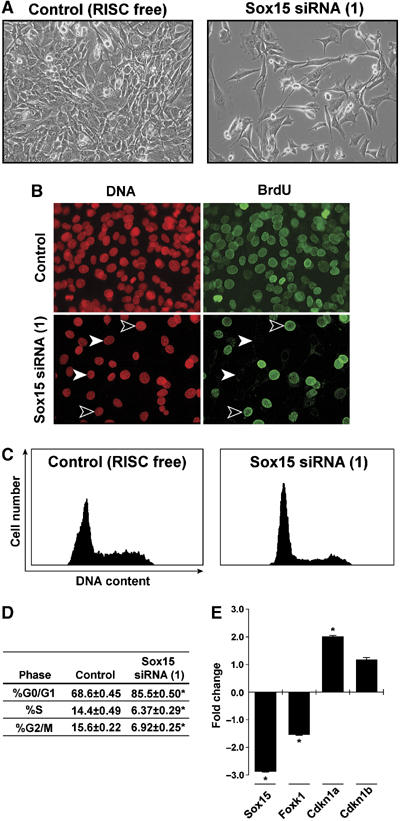
Inactivation of Sox15 by siRNA results in perturbed cell cycle kinetics and decreased Foxk1 gene expression. (A) siRNA primer pairs directed against Sox15 were transfected into C2C12 cells. The control consisted of transfection of a nonsense ds-oligo (RISC). Forty-eight hours following transfection, decreased numbers of cells were observed in the siRNA(1) samples compared with the control samples. There were no differences in cell death between the respective samples. (B) Decreased BrdU incorporation was observed in the C2C12 cells 48 h following transfection with siRNA Sox15 versus transfection of RISC. All nuclei are labeled with propidium iodide staining. Arrowheads represent BrdU-positive (black arrowheads) and -negative (white arrowheads) cells. (C) Perturbed cell cycle kinetic profile of the siRNA cells (i.e. G0/G1 arrest) versus the control. (D) Quantitation of the cell cycle kinetics for each phase is designated as mean±s.e.m. Note the cell cycle arrest with a significantly greater percentage of the siRNA-treated cells in the G0/G1 phase. Results represent triplicate analyses (*P<0.001). (E) QRT–PCR analysis reveals the knockdown (decreased expression) of Sox15 transcript in the siRNA samples compared to control. Note decreased Foxk1 expression and increased expression of Cdkn1a (p21) without any changes in p27 (Cdknb) transcript levels with the siRNA samples. *P<0.005. Data are mean±s.e.m. (n=3 for each sample).
Sox15-null mice have perturbed muscle regeneration
To further examine the role of Sox15 in the MPC population, we analyzed the Sox15-deficient mice. In contrast to previous reports, which analyzed the Sox15 mutant phenotype in a mixed 129/C57BL/6 strain (Lee et al, 2004), we undertook our studies in the pure C57BL/6 background. Two weeks following an intramuscular delivery of cardiotoxin, wild-type mice had evidence (centronucleated myotubes, which are a hallmark of regeneration) of complete skeletal muscle regeneration, whereas the Sox15 mutant mice had a modest impairment with evidence of persistent necrosis and replacement of myofibers with adipose tissue (Figure 7A and B). Using electron microscopy, we observed approximately an 80% reduction in the number of satellite cells located within the Sox15-null tibialis anterior muscles compared with the wild-type controls (n=3 for each sample; Figure 7C–E). Sox15 mutant MPCs displayed impaired proliferation, as measured by Ki67 immunohistochemical staining (Supplementary Figure 9), and a three-fold decrease in Foxk1 expression without evidence of impaired differentiation (Figure 7F–J). These data further establish a role for Sox15 as a regulator of Foxk1 expression and the myogenic progenitor cell population.
Figure 7.
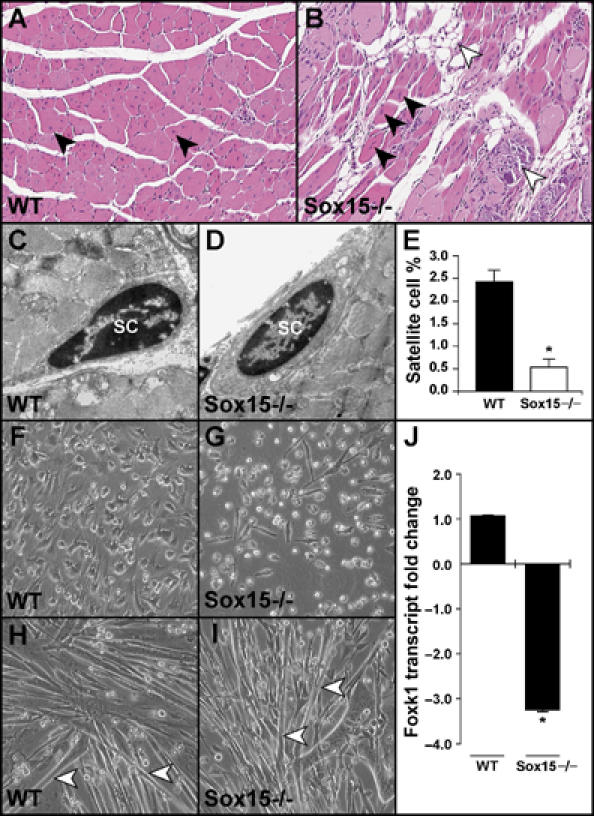
Sox15-deficient skeletal muscle has perturbed regeneration. (A, B) Adult male WT and Sox15−/− mice received intramuscular delivery of cardiotoxin into the tibialis anterior muscles and were killed 2 weeks following injury. Note the centronucleated myofibers (arrowheads) throughout the WT muscle. While the Sox15−/− muscle has evidence of centronucleated myofibers (black arrowheads), there is evidence of impaired regeneration with the presence of fibrous, necrotic tissue (white arrowheads) and adipose tissue replacing myofibers (B; white arrowhead). (C–E) Using electron microscopy, both WT and Sox15−/− tibialis anterior (TA) muscles have evidence of satellite cells (SC), but quantitation (E) reveals a severe decrease in the number of satellite cells in the absence of Sox15. Note the total of nuclei evaluated in WT (1400 nuclei) and Sox15−/− (1096 nuclei) TA muscles (n=3 for each sample; *P<0.005). (F–I) Primary MPCs (including satellite cells) were isolated from postnatal day 2 littermates (offspring from Sox15 heterozygous matings), genotyped and equal numbers of myoblasts were plated. Note decreased numbers of Sox15-null MPCs (G) compared with the WT control (F). Once the respective myoblast cultures were 90% confluent, they were cultured in differentiation media for an additional 3 days. Both the WT and Sox15-null myoblasts differentiated at the same rate and formed equal numbers of myotubes (H and I; white arrowheads) (performed in triplicate). (J) QRT–PCR analysis for Foxk1 expression in WT and Sox15−/− primary MPCs. Note more than a three-fold decrease in Foxk1 expression in the absence of Sox15 (n=3 for each sample; *P<0.005).
Coexpression of Sox15, Fhl3 and Foxk1
Previous studies by our laboratory and others have demonstrated that Foxk1 and Sox15 are expressed in satellite cells that are resident in adult skeletal muscle (Garry et al, 1997; Lee et al, 2004). To further examine Fhl3 biology, we used Northern blot analysis to examine transcript expression in a spectrum of adult murine tissues. We observed that the Fhl3 transcript was approximately 1.8 kb in size and abundantly expressed in striated muscle (heart and skeletal muscle) (Supplementary Figure 10A). Using semiquantitative RT–PCR analysis, we observed that Sox15, Fhl3 and Foxk1 are coexpressed in C2C12 cells (Supplementary Figure 10B). Moreover, using immunohistochemical techniques, we examined the endogenous Sox15 and Fhl3 expression in unperturbed adult murine skeletal muscle. Using polyclonal antisera and immunohistochemical techniques, we observed that both Sox15 and Fhl3 were expressed in a sublaminar cell population that is likely to be the MPC population (Supplementary Figure 10C–K). It is likely that Fhl3 is expressed more broadly and is not limited to the satellite cell pool in skeletal muscle as the other LIM-only proteins such as Fhl1 and Fhl2 are expressed in a variety of lineages. These results further support the conclusion that Foxk1, Sox15 and Fhl3 are expressed in the MPC population of adult skeletal muscle.
Discussion
In response to a severe injury that destroys a majority of the tissue, skeletal muscle is capable of complete regeneration and restoration of the cytoarchitecture within a 2-week period (Miller et al, 1999; Seale and Rudnicki 2000; Shi and Garry 2006). This remarkable regenerative capacity is due to a rare population of cells termed myogenic progenitor cells that are resident in adult skeletal muscle. Although intense interest has been directed toward muscle regeneration, the molecular mechanisms that direct the MPC population are incompletely defined. In the present study, we have begun to decipher the MPC regulatory program by making three principal findings that enhance our mechanistic understanding of the MPC population. First, we utilized transgenic technology to define modular sequences of the Foxk1 gene that directs lacZ reporter expression to the MPC population. We have previously demonstrated that Foxk1 is restricted to the MPC population in adult skeletal muscle (Garry et al, 1997, 2000; Hawke et al, 2003a). Using a gene disruption strategy, we have observed that mice that lack Foxk1 have impaired muscle regeneration, dysregulation of the cyclin-dependent kinase inhibitor, p21, and perturbed cell cycle progression of the MPC population (Garry et al, 2000; Hawke et al, 2003a). The analysis of several transgenic constructs of the Foxk1 promoter revealed a modular region that contained an evolutionarily conserved SBE that directed expression to the MPC population during embryogenesis and in adult skeletal muscle.
The second major finding of the present study is that Sox15 is a potent transcriptional activator of the Foxk1 gene. The Sox transcription factor family has a number of members that function as key regulators of stem cell and progenitor cell function (Wilson and Koopman, 2002; Koopman et al, 2004). For example, Sox2 forms a transcriptional complex with Oct3/4 to regulate gene expression and maintain the pluripotent state of ES cells (Wilson and Koopman, 2002). In a similar manner, Sox2 has been shown to be an important transcriptional regulator in neural stem cells. Moreover, Sox17, Sox5, Sox6, Sox8, Sox9 and Sox10 are important transcriptional regulators in endodermal, neural, cardiac, neural crest and oligodendrocyte precursor cells, respectively (Wilson and Koopman, 2002). Sox8 and Sox15 are expressed in the MPC population that resides in adult skeletal muscle (Béranger et al, 2000; Schmidt et al, 2003; Lee et al, 2004). Recent studies suggested that the overexpression of either Sox8 or Sox15 resulted in an inhibition of the C2C12 differentiation program (Béranger et al, 2000; Schmidt et al, 2003). Furthermore, mice (in a mixed 129/C57BL/6 strain) lacking Sox15 were viable but appeared to have impaired muscle regeneration in response to a crush injury and appeared to have a differentiation defect, although the mechanisms underlying this phenotype are unknown (Lee et al, 2004). In the present study, we provide molecular biological and biochemical data supporting the role of Sox15 as an upstream regulator of Foxk1. Furthermore, using a knockdown strategy to decrease endogenous Sox15 expression, we observed impaired cell cycle kinetics and decreased Foxk1 expression. These studies have been extended through our analysis of Sox15-deficient skeletal muscle in the C57BL/6 strain. Sox15 mutant skeletal muscle in the C57Bl/6 strain had impaired regeneration following the delivery of a myonecrotic agent, severely decreased numbers of MPCs (satellite cells) resident in adult skeletal muscle, impaired proliferation of the MPCs (without impaired differentiation) and decreased levels of Foxk1 transcript expression. One explanation for the differences observed in the present study versus the original report (Lee et al, 2004) characterizing the Sox15-null phenotype is most likely due to strain differences and the presence of modifier genes. Collectively, the knockdown and gene disruption strategies to inactivate Sox15 outlined in the present study support an important role for Sox15 in the regulation of Foxk1 and the MPC population.
Previous studies have demonstrated that the functional role of Sox transcription factors is in part determined by their collaborating or interacting factors (i.e. Sox factors have been shown to interact with Oct3/4, Mef2, Wnt/β-catenin, etc.) (Wilson and Koopman, 2002). Our third principal observation is that Sox15 physically interacts with Fhl3 to synergistically coactivate Foxk1 gene expression. FHL proteins have similarly been shown to function as critical regulators of progenitor cell populations (Chu et al, 2000; Fimia et al, 2000; Kong et al, 2001; Müller et al, 2002; Morlon and Sassone-Corsi, 2003; Günther et al, 2005). One member of this family, Fhl2 (also called DRAL for ‘downregulated in rhabdomyosarcoma), is expressed in cardiac progenitors and has been shown to function as a coactivator of CREB/CREM (Kong et al, 2001; Fimia et al, 2000). In the present study, we showed that Fhl3 has multiple specific contacts with the non-HMG domain of Sox15 (136–185) and this complex coactivates the Foxk1 gene in the nuclear compartment. These adaptor proteins (non-DNA binding proteins) presumably recruit other factors that further facilitate gene expression. While knockdown studies of Fhl3 revealed no differences in the MPC cell cycle kinetics, redundancy by other family members (i.e. Fhl2) may compensate for decreased expression of Fhl3. Future strategies to produce a complete absence of Fhl3 (i.e. the production of mice lacking Fhl3) may be necessary to comprehensively analyze the role of Fhl3 and MPC proliferation.
In summary, adult skeletal muscle has a tremendous regenerative capacity due to resident MPCs including satellite cells. Intense efforts have been directed toward an enhanced understanding of the molecular regulation of the quiescent, activated and proliferating states of the satellite cell pool. Although the role of Sox and Fox factors has been shown to regulate essential functions in stem cell and progenitor cells, the interaction and coordinated regulation of Sox and Fox factors have not been described previously. Therefore, our results not only define Sox15 as the first transcriptional coactivator of Foxk1 but also integrate cofactors such as Fhl3 in a novel pathway linking the molecular regulation of the MPC population and regeneration. Definition of these molecular cascades in the MPC population will serve as a platform for the design of cell-based therapies for the future treatment of myopathic diseases.
Materials and methods
Transgenic mice
The transgene constructs (4.6, 1.6 and 0.6 kb Foxk1 fragments) were subcloned into a lacZ reporter cassette (generously provided by E Olson) (Cheng et al, 1993; Masino et al, 2004). Transgenic mice were generated following microinjection of the respective constructs into fertilized F2 eggs (B6SJLFF1; Jackson Laboratories), which were reimplanted into pseudopregnant F1 foster ICR mothers (Harlan). Identification of transgenic mice was undertaken using PCR for lacZ, and β-galactosidase expression was assessed using whole-mount and histological/histochemical techniques (Cheng et al, 1993; Masino et al, 2004).
Combinatorial matings were used to generate 4.6 kb Foxk1 promoter:mdx mice. C57BL/10ScSn mdx mice were obtained from Jackson Laboratories (Bar Harbor, ME). Cardiotoxin-induced muscle injury was performed as previously described and as outlined in Supplementary data (Garry et al, 1997, 2000; Hawke et al. 2003b).
Founder analyses were undertaken for the mutagenesis of the SBE within the 4.6 kb Foxk1 upstream fragment. The SBE was mutated from the sequence AACAATG to AATCCTG in the 4.6 kb Foxk1 promoter-lacZ (wild type) vector using PCR-based site-directed mutagenesis and ligated into the 4.6 kb Foxk1–lacZ plasmid using BglII and AflII restriction enzyme cuts that flanked the Sox mutation. As described above, embryos were harvested at E13.5 and processed for β-galactosidase staining. The yolk sacs from each embryo were genotyped as outlined above. Whole-mount preparations of the embryos are further described in Supplementary data.
Sox15-null mice
Sox15-null mice were engineered as previously described (Lee et al, 2004) and mated into the C57BL/6 strain.
Immunohistochemistry of tissue sections, C2C12 cells and primary MPCs
Immunohistochemistry was performed as previously described (Garry et al, 1996, 1997) and outlined in greater detail in Supplementary data. Primary antisera utilized in this study included an affinity-purified Foxk1 rabbit polyclonal antibody (1:200 dilution), a rabbit Sox15 polyclonal antibody (1:50 dilution) and a chicken anti-Fhl3 polyclonal serum (1:500 dilution; Abcam), a monoclonal laminin antibody (1:200; Dako), a monoclonal β-galactosidase antibody (1:50; Hybridoma Bank) and a monoclonal desmin antibody (1:250; Dako). Following incubation with the respective fluorophore-conjugated secondary antisera, the sections were coverslipped with Vectashield and imaged using a Zeiss LSM510 META confocal microscope. Detailed immunohistochemical colocalization assays undertaken in C2C12 cells and primary MPCs are outlined in the Supplementary data.
Electrophoretic mobility shift assay
Sox15 in vitro-translated protein was generated using the pCS3+MT-Sox15 vector and the Promega TNT kit (Molkentin et al, 1996; Yang et al, 2000). A 100 μM length of a 23-bp oligonucleotide (5′-ATTACTGAACAATGGGGTCACAG-3′) corresponding to a region in the Foxk1 5′ upstream region and its reverse compliment, and the Sox mutated binding element oligonucleotide 5′ATTACTGAATCCTGGGGTCACAG3′ and its reverse compliment were used as DNA probes. Further details of the EMSA are described in Supplementary data.
ChIP and GST pull-down assays
ChIP assays were performed following the transfection of the myc-Sox15 plasmid into C2C12 myoblasts, formaldehyde fixation and neutralization with glycine (Liu et al, 2005). Briefly, the nuclear extracts were sonicated and the chromatin complex was immunoprecipitated with anti-myc conjugated with agarose (Sigma Chemical) or anti-rabbit IgG conjugated to agarose (Sigma Chemical). The immunoprecipitation product was washed, eluted, reverse crosslinked, treated with protease K, extracted with phenol/chloroform/isoamyl alcohol, precipitated with ethanol and finally resuspended in H2O. Standard PCR was performed to analyze the DNA fragment.
GST pull-down assays utilized Escherichia coli BL21 expressing GST–Fhl3 fusion proteins, which were extracted with B-PER bacterical protein extraction reagent (Pierce Biochemicals) and then purified with glutathione–Sepharose 4B (GE Healthcare) (Liu et al, 2005). Sox15 constructs were cloned into the pCS3+MT vector and in vitro translated using TNT-coupled reticulocyte lysate systems (Promega). GST fusion proteins bound to Sepharose beads were incubated with 35S-Met-labeled in vitro-translated protein product and the BL21 cell extract. The beads pull-down complex was washed four times and resuspended in loading buffer, analyzed using a 12% gel and imaged.
Transcriptional assays
C2C12 myoblasts and COS-7 cells were cultured in 35 mm dishes containing DMEM (Gibco) supplemented with 20 and 10% fetal bovine serum respectively. Approximately 1.2 × 105 of cells were transfected with lipofectamine (Invitrogen) and assayed for both luciferase and β-galactosidase activity (Molkentin et al, 1996; Yang et al, 2000). Cells were transfected with 0.5 μg of the 4.6 kb Foxk1 promoter fused to the luciferase reporter, along with 0.25 μg β-actin-LacZ (internal control), increasing amounts of transcription factor and an empty vector (pcDNA3.1) to standardize the total amount of transfected DNA at 2 μg. Cells were incubated in transfected media supplemented with Opti-MEM (Gibco) for 4 h and then incubated overnight with normal growth media. Luciferase assays were performed using the Promega luciferase assay system. All fold changes in luciferase activity were normalized to β-galactosidase activity and to the vector alone (Molkentin et al, 1996; Yang et al, 2000). All transfection experiments were performed in triplicate and replicated.
Yeast two-hybrid and co-IP assays
The yeast two-hybrid screen was performed following the standard procedure as described (Yeast Protocols Handbook). Sox15 (amino acids 1–181) was cloned into pGBKT7 and used as bait to screen human skeletal muscle cDNA library (Clontech). For interacting domain mapping experiments, the Sox15 deletion constructs were cloned by PCR into pGBKT7, and the Fhl3 deletion constructs were cloned into pGADT7. The interaction was analyzed on selection medium plates (Ade–His–Leu–Trp) (Wang et al, 2001). Co-IP assays were undertaken as previously described and are outlined in Supplementary data.
RNA interference
siRNA ds oligos (SMARTpool, Dharmacon) were transfected in C2C12 myoblasts with modifications as described (Ma et al, 2005). Sequences are available upon request. Additional methodological information is available in Supplementary data.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Figure 10
Supplementary Table 1
Supplementary data
Acknowledgments
We acknowledge the contributions of Justin Kallhoff, John Shelton and Dr James A Richardson (UT Southwestern Medical Center) in FACS and histological analyses. Funding support was obtained from the National Institutes of Health (AR47850), the Muscular Dystrophy Association, the Donald W Reynolds Foundation and the Gail Griffiths Hill Foundation. DJG is an Established Investigator of the AHA.
References
- Ang SL, Rossant J (1994) HNF-3-beta is essential for node and notochord formation in mouse development. Cell 78: 561–574 [DOI] [PubMed] [Google Scholar]
- Béranger F, Mejean C, Moniot B, Berta P, Vandromme M (2000) Muscle differentiation is antagonized by SOX15, a new member of the SOX protein family. J Biol Chem 275: 16103–16109 [DOI] [PubMed] [Google Scholar]
- Borycki AG, Li J, Jin F, Emerson CP, Epstein JA (1999) Pax3 functions in cell survival and in pax7 regulation. Development 126: 1665–1674 [DOI] [PubMed] [Google Scholar]
- Cheng TC, Wallace MC, Merlie JP, Olson EN (1993) Separable regulatory elements governing myogenin transcription in mouse embryogenesis. Science 261: 215–217 [DOI] [PubMed] [Google Scholar]
- Chu PH, Ruiz-Lozano P, Zhou Q, Cai C, Chen J (2000) Expression patterns of FHL/SLIM family members suggest important functional roles in skeletal muscle and cardiovascular system. Mech Dev 95: 259–265 [DOI] [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, Morgan JE (2005) Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122: 289–301 [DOI] [PubMed] [Google Scholar]
- Conboy IM, Rando TA (2002) The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell 3: 397–409 [DOI] [PubMed] [Google Scholar]
- Cornelison DD, Filla MS, Stanley HM, Rapraeger AC, Olwin BB (2001) Syndecan-3 and syndecan-4 specifically mark skeletal muscle satellite cells and are implicated in satellite cell maintenance and muscle regeneration. Dev Biol 239: 79–94 [DOI] [PubMed] [Google Scholar]
- Cornelison DD, Wilcox-Adelman SA, Goetinck PF, Rauvala H, Rapraeger AC, Olwin BB (2004) Essential and separable roles for Syndecan-3 and Syndecan-4 in skeletal muscle development and regeneration. Genes Dev 18: 2231–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deasy BM, Gharaibeh BM, Pollett JB, Jones MM, Lucas MA, Kanda Y, Huard J (2005) Long-term self-renewal of postnatal muscle-derived stem cells. Mol Biol Cell 16: 3323–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Cesare DD, Sassone-Corsi P (2000) A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol Cell Biol 20: 8613–8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry DJ, Bassel-Duby R, Richardson J, Grayson J, Neufer P, Williams RS (1996) Postnatal development and plasticity of specialized muscle fiber characteristics in the hindlimb. Dev Gen 19: 146–156 [DOI] [PubMed] [Google Scholar]
- Garry DJ, Meeson A, Elterman J, Zhao Y, Yang P, Bassel-Duby R, Williams RS (2000) Myogenic stem cell function is impaired in mice lacking the forkhead/winged helix protein MNF. Proc Natl Acad Sci USA 97: 5416–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garry DJ, Ordway GA, Lorenz JN, Radford NB, Chin ER, Grange RW, Bassel-Duby R, Williams RS (1998) Mice without myoglobin. Nature 395: 905–908 [DOI] [PubMed] [Google Scholar]
- Garry DJ, Yang Q, Bassel-Duby R, Williams RS (1997) Persistent expression of MNF identifies myogenic stem cells in postnatal muscles. Dev Biol 188: 280–294 [DOI] [PubMed] [Google Scholar]
- Goetsch SC, Hawke TJ, Gallardo TD, Richardson JA, Garry DJ (2003) Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol Genomics 14: 261–271 [DOI] [PubMed] [Google Scholar]
- Grange RW, Meeson A, Chin E, Lau K, Stull J, Shelton J, Williams RS, Garry DJ (2002) Functional and molecular adaptations in skeletal muscle of myoglobin mutant mice. Am J Physiol 282: H726–H733 [DOI] [PubMed] [Google Scholar]
- Günther T, Poli C, Muller JM, Catala-Lehnen P, Schinke T, Yin N, Vomstein S, Amling M, Schule R (2005) Fhl2 deficiency results in osteopenia due to decreased activity of osteoblasts. EMBO J 24: 3049–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawke TJ, Jiang N, Garry DJ (2003a) Absence of p21 rescues myogenic progenitor cell proliferative and regenerative capacity in Foxk1 null mice. J Biol Chem 278: 4015–4020 [DOI] [PubMed] [Google Scholar]
- Hawke TJ, Meeson A, Jiang N, Graham S, Hutcheson K, DiMaio M, Garry DJ (2003b) p21 is essential for normal myogenic progenitor cell function in regenerating skeletal muscle. Am J Physiol 285: C1019–C1027 [DOI] [PubMed] [Google Scholar]
- Kong Y, Shelton JM, Rothermel B, Li Z, Richardson JA, Bassel-Duby R, Williams RS (2001) Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation 103: 2731–2738 [DOI] [PubMed] [Google Scholar]
- Koopman P, Schepers G, Brenner S, Venkatesh B (2004) Origin and diversity of the Sox transcription factor gene family: genome-wide analysis in Fugu rubripes. Gene 328: 177–186 [DOI] [PubMed] [Google Scholar]
- Lai E, Clark KL, Burley S, Darnell JE (1993) Hepatocyte nuclear factor 3/forkhead or ‘winged helix' proteins: A family of transcription factors of diverse biological function. Proc Natl Acad Sci USA 90: 10421–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Goring W, Ochs M, Muhlfeld C, Steding G, Paprotta I, Engel W, Adham IM (2004) Sox15 is required for skeletal muscle regeneration. Mol Cell Biol 24: 8428–8436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZP, Wang Z, Yanagisawa H, Olson EN (2005) Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev Cell 9: 261–270 [DOI] [PubMed] [Google Scholar]
- Ma K, Chan JK, Zhu G, Wu Z (2005) Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol Cell Biol 25: 3575–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino AM, Gallardo TD, Wilcox CA, Olson EN, Williams RS, Garry DJ (2004) Transcriptional regulation of cardiac progenitor cell populations. Circ Res 95: 389–397 [DOI] [PubMed] [Google Scholar]
- Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9: 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JB, Schaefer L, Dominov JA (1999) Seeking muscle stem cells. Curr Top Dev Biol 43: 191–219 [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN (1996) Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol Cell Biol 16: 2627–2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarras D, Morgan J, Collins C, Relaix F, Zaffran S, Cumano A, Partridge T, Buckingham M (2005) Direct isolation of satellite cells for skeletal muscle regeneration. Science 309: 2064–2067 [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Madgwick AJA (1998) The LIM proteins FHL1 and FHL3 are expressed differently in skeletal muscle. Biochem Biophys Res Commun 255: 245–250 [DOI] [PubMed] [Google Scholar]
- Morlon A, Sassone-Corsi P (2003) The LIM-only protein FHL2 is a serum-inducible transcriptional coactivator of AP-1. Proc Natl Acad Sci USA 100: 3977–3982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourkioti F, Rosenthal N (2005) IGF-1, inflammation and stem cells: interactions during muscle regeneration. Trends Immunol 26: 535–542 [DOI] [PubMed] [Google Scholar]
- Müller JM, Metzger E, Greschik H, Bosserhoff A-K, Mercep L, Buettner R, Schule R (2002) The transcriptional coactivator FHL2 transmits Rho signals from the cell membrane into the nucleus. EMBO J 21: 736–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olguin HC, Olwin BB (2004) Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev Biol 275: 375–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Glaser G, Wernig A, Wegner M, Rosorius O (2003) Sox8 is a specific marker for muscle satellite cells and inhibits myogenesis. J Biol Chem 278: 29769–29775 [DOI] [PubMed] [Google Scholar]
- Seale P, Rudnicki MA (2000) A new look at the origin, function, and stem cell status of muscle satellite cells. Dev Biol 218: 115–124 [DOI] [PubMed] [Google Scholar]
- Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, Rudnicki MA (2000) Pax7 is required for the specification of myogenic satellite cells. Cell 102: 777–786 [DOI] [PubMed] [Google Scholar]
- Sheehan SM, Allen RE (1999) Skeletal muscle satellite cell proliferation in response to members of the fibroblast growth factor family and hepatocyte growth factor. J Cell Physiol 181: 499–506 [DOI] [PubMed] [Google Scholar]
- Shi X, Garry DJ (2006) Muscle stem cells in development, regeneration, and disease. Genes Dev 20: 1692–1708 [DOI] [PubMed] [Google Scholar]
- Snider L, Tapscott SJ (2003) Emerging parallels in the generation and regeneration of skeletal muscle. Cell 113: 811–812 [DOI] [PubMed] [Google Scholar]
- Sutton J, Costa R, Klug M, Field L, Xu D, Largaespada D, Fletcher C, Jenkins N, Copeland N, Klemsz M, Hromas R (1996) Genesis, a winged helix transcriptional repressor with expression restricted to embryonic stem cells. J Biol Chem 271: 23126–23133 [DOI] [PubMed] [Google Scholar]
- Turner J, Nicholas H, Bishop D, Matthews J, Crossley M (2003) The LIM protein FHL3 binds basic kruppel-like factor/kruppel-like factor 3 and its co-repressor C-terminal-binding protein 2. J Biol Chem 278: 12786–12795 [DOI] [PubMed] [Google Scholar]
- Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN (2001) Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 105: 851–862 [DOI] [PubMed] [Google Scholar]
- Wilson M, Koopman P (2002) Matching SOX: partner proteins and co-factors of the SOX family of transcriptional regulators. Curr Opin Genet Dev 12: 441–446 [DOI] [PubMed] [Google Scholar]
- Yang Q, Kong Y, Rothermel B, Garry DJ, Bassel-Duby R, Williams RS (2000) The winged-helix/forkhead protein myocyte nuclear factor beta (MNF-beta) forms a co-repressor complex with mammalian sin3B. Biochem J 345: 335–343 [PMC free article] [PubMed] [Google Scholar]
- Ye H, Kelly T, Samadani U, Lim L, Rubio S, Overdier D, Roebuck K, Costa R (1997) Hepatocyte nuclear factor3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol 17: 1626–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Supplementary Figure 9
Supplementary Figure 10
Supplementary Table 1
Supplementary data