

Abstract
Functional crosstalk between cell-surface and intracellular ion channels plays important roles in excitable cells and is structurally supported by junctophilins (JPs) in muscle cells. Here, we report a novel form of channel crosstalk in cerebellar Purkinje cells (PCs). The generation of slow afterhyperpolarization (sAHP) following complex spikes in PCs required ryanodine receptor (RyR)-mediated Ca2+-induced Ca2+ release and the subsequent opening of small-conductance Ca2+-activated K+ (SK) channels in somatodendritic regions. Despite the normal expression levels of these channels, sAHP was abolished in PCs from mutant mice lacking neural JP subtypes (JP-DKO), and this defect was restored by exogenously expressing JPs or enhancing SK channel activation. The stimulation paradigm for inducing long-term depression (LTD) at parallel fiber–PC synapses adversely established long-term potentiation in the JP-DKO cerebellum, primarily due to the sAHP deficiency. Furthermore, JP-DKO mice exhibited impairments of motor coordination and learning, although normal cerebellar histology was retained. Therefore, JPs support the Ca2+-mediated communication between voltage-gated Ca2+ channels, RyRs and SK channels, which modulates the excitability of PCs and is fundamental to cerebellar LTD and motor functions.
Keywords: afterhyperpolarization, long-term depression, Purkinje cell, ryanodine receptor, SK channel
Introduction
Ca2+ is an indispensable messenger coupling the activation of cell-surface receptors and ionic channels with intracellular signaling, and regulates a large variety of cellular processes. To establish Ca2+-signaling systems, neurons contain various types of Ca2+-related channels, such as cell-surface Ca2+ channels, intracellular Ca2+ release channels and Ca2+-activated ionic channels, and further form integrated networks of functional communications among the channels (Berridge, 2002; Verkhratsky, 2005). A few types of Ca2+-mediated channel communications have been electrophysiologically identified from neurons, and their contributions to cell excitability were extensively studied at the single-cell level. On the other hand, Ca2+ signaling in neurons is temporally restricted to a very narrow range by their characteristic cell structures and intracellular Ca2+-buffering effects generated by specific and nonspecific Ca2+-binding cytosolic proteins (Goldberg et al, 2003; Bloodgood and Sabatini, 2005). Therefore, effective Ca2+-mediated channel communication requires their assembly in specific subcellular compartments. The junctional membrane complex between the cell membrane and the endoplasmic/sarcoplasmic reticulum (ER/SR) detected in electron microscopic observations has been thought to function as a structural platform for Ca2+-mediated crosstalk between cell-surface and intracellular channels (Berridge, 2002). However, molecular components and their physiological roles of the proposed channel crosstalk are largely unknown in neurons (Levitan, 2006).
In muscle excitation–contraction coupling, the activation of dihydropyridine receptor channels opens ryanodine receptor (RyR) channels and triggers Ca2+ release from the SR (Endo, 1985; Meissner, 1994). The functional coupling between the channels occurs in junctional membrane complexes designated as ‘triad junction' in skeletal muscle, ‘diad' in cardiac muscle and ‘peripheral coupling' in immature striated muscle (Flucher, 1992; FranziniArmstrong and Protasi, 1997). Our previous study indicates that junctophilin (JP) subtypes, namely JP1–4, contribute to the formation of junctional membrane complexes by spanning the ER/SR membrane and interacting with the plasma membrane (Takeshima et al, 2000). In JP1-knockout mice with perinatal lethality, mutant skeletal muscle shows deficiency in triad junctions and insufficient contraction due to impaired communication between dihydropyridine receptors and RyRs (Ito et al, 2001). JP2-knockout mice exhibit embryonic cardiac failure, and deficiency in peripheral couplings leads to functional uncoupling between dihydropyridine receptors and RyRs in mutant cardiomyocytes bearing arrhythmic Ca2+ signaling. In striated muscles, JPs are therefore associated with the physiological functioning of RyRs. In the central nervous system, both JP3 and JP4 are expressed throughout neural sites (Nishi et al, 2003). Mice lacking either JP3 or JP4 show no obvious abnormalities (Nishi et al, 2002), whereas mice lacking both JP subtypes (JP-DKO) seem to bear serious symptoms accompanied by abnormal nervous functions. Our current study has indicated that Ca2+-mediated communication between N-methyl-D-aspartate (NMDA) receptor channels, RyRs and Ca2+-activated K+ channels is disconnected in mutant hippocampal pyramidal neurons from JP-DKO mice with impaired learning and memory (Moriguchi et al, 2006). In this report, we propose that cerebellar Purkinje cells (PCs) lacking NMDA receptor channels possess another type of Ca2+-mediated crosstalk between cell-surface and intracellular channels. Moreover, we present evidence that this channel crosstalk is supported by JPs and required for integrated cerebellar motor functions.
Results
Impaired motor coordination and learning in JP-DKO mice
JP-DKO mice show severe growth retardation and die at the weaning stage under normal housing conditions, but can survive on wet paste food (Moriguchi et al, 2006). The rescued mature JP-DKO mice were healthy, but were smaller and had weaker muscle than control mice (Supplementary Figure 1). Because of the neuron-specific expression of both JP3 and JP4, the muscular weakness might result from an impaired function of the motor control system in JP-DKO mice. Therefore, we focused on abnormalities in the cerebellar motor function of JP-DKO mice, and the JP3 (+/−) and JP4 (+/−) (JP-DHE) and wild-type mice were used as controls in this study.
JP-DKO mice showed an abnormal behavior when placed on a narrow bar in the fixed-bar test. Control mice walked quickly and proficiently, whereas JP-DKO mice walked slowly and frequently crawled on the bar (Figure 1A). Moreover, the mutant mice frequently stopped and wound their tail around the bar and lost their balance. In the rota-rod test, JP3 or JP4 knockout mice showed mildly impaired performance but retained motor learning during the training period (Figure 1B). However, JP-DKO mice failed to stay on a rotating rod and could not improve their performance during the training.
Figure 1.
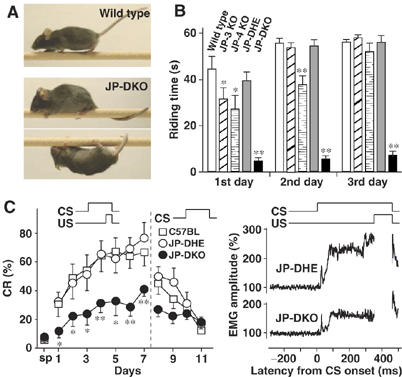
Poor performance of JP-DKO mice in cerebellar-dependent tasks. (A) In the fixed-bar test, JP-DKO mice showed abnormal behavior. The mouse was placed on a narrow bar and its behavior was photographed. (B) In the rota-rod test, JP-DKO mice failed to stay on a rotating rod. Mice were placed on a rod rotating at 16 r.p.m. for measuring their riding time in four trials for 3 days. A maximum of 60 s was allowed per trial. Mean±s.e.m. (wild type n=14, JP-3 KO n=7, JP-4 KO n=10, JP-DHE n=10, JP-DKO n=11; 8–9 weeks old). *P<0.05, **P<0.01, significantly different from the value for wild type (t-test). (C) In delay eyeblink conditioning, JP-DKO mice showed impaired performance. Conditioned response (CR) frequency of delay eyeblink conditioning (left panel). CS, intense sound as conditioned stimulus; US, electrical shock as unconditioned stimulus. After spontaneous eyeblink frequency (sp) was examined, mice were subjected to the acquisition session for 7 days and the extinction session for 4 days. Mean±s.e.m. (C57BL n=10, JP-DHE n=10, JP-DKO n=9; 7–10 weeks old). *P<0.05, **P<0.01, significantly different from the value for JP-DHE mice (t-test). No significant differences in performance were detected between C57BL and JP-DHE mice. The average amplitude of eyelid electromyography (EMG) on day 7 (right panel) is shown. The accumulated amplitude in JP-DKO mice is significantly lower than that in JP-DHE mice due to decreased CR%. The transient electromyographic response at ∼20 ms preceding the CR at ∼80–350 ms was derived from the auditory startle reflex elicited upon the sound and was retained in JP-DKO mice. Therefore, it is unlikely that the poor performance is caused by hearing inability in JP-DKO mice.
JP-DKO mice were made to experience delay eyeblink conditioning, a form of cerebellum-dependent discrete motor learning, in which an intense sound of a conditioned stimulus and an electrical shock of an unconditioned stimulus temporally overlap and co-terminate (Figure 1C). The unconditioned stimulus was delivered through paired electrodes implanted in the upper eyelid, and the percentage of conditioned responses (CR%) was monitored through electromyographic activity recorded with another pair of electrodes implanted. In the trials, animals learn the adaptive timing of eyeblinking. Therefore, CR% for JP-DHE mice progressively increased to >70% during the 7-day acquisition session. This performance was almost identical to that of wild-type mice in previous studies (Kishimoto et al, 2002). However, CR% for JP-DKO mice was less than 40% even on day 7, and there were significant differences in CR% during the development between JP-DHE and DKO mice. The normalized electromyographic amplitude on day 7 for JP-DKO mice was clearly lower than that for JP-DHE mice. Therefore, learning this task is greatly impaired in JP-DKO mice. Because delay eyeblink conditioning is a cerebellum-dependent task (McCormick and Thompson, 1984), we reasonably assumed cerebellar dysfunction in JP-DKO mice.
Apparently normal morphology and excitatory circuit in JP-DKO cerebellum
In morphological analyses, JP-DKO mice showed no abnormalities in basic cerebellar histology or cytology, including cerebellar size, foliation and trilaminar organization in the cerebellar cortex and monolayer alignment of PCs (Supplementary Figure 2). Furthermore, immunohistochemical studies showed that dendritic branching of PCs, distribution patterns of excitatory and inhibitory terminals, and palisade-like arrangement of Bergmann fibers were normal in JP-DKO mice.
In the adult cerebellum of wild-type animals, each PC is innervated by single climbing fiber (CF). However, in the majority of model animals bearing motor discoordination, PCs are innervated by multiple CFs from different origins (e.g., Kano et al, 1995, 1997; Kakizawa et al, 2000; Miyazaki et al, 2004; Tohgo et al, 2006). We therefore examined whether JP-DKO PCs had persistent multiple CF innervation. Excitatory postsynaptic currents (EPSCs) were recorded from PCs by the whole-cell voltage clamp technique using the pipette solution containing an excess amount of BAPTA (20 mM) to buffer cytosolic Ca2+. The stimulating pipette was systematically moved in the granule cell layer of cerebellar slices from 5- to 9-week-old mice, and EPSCs evoked with various stimulus intensities were monitored at each site. CF stimulation results in the simultaneous activation of hundreds of excitatory synaptic terminals, and hence induces EPSPs in an all-or-none manner when only single CFs innervate PCs. Most JP-DKO PCs showed large EPSCs elicited in an all-or-none manner, and we detected no difference in PC population innervated by two or more CFs between control and JP-DKO mice (Figure 2A).
Figure 2.
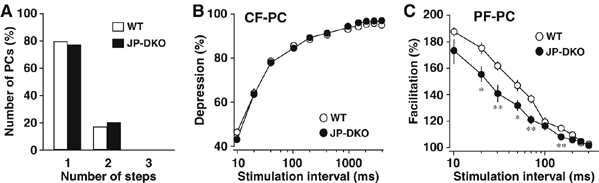
Excitatory inputs to PCs in JP-DKO cerebellum. (A) Frequency histograms of PCs in terms of the number of discrete CF-EPSC steps. Wild type (WT) n=39, JP-DKO n=38. (B) Paired-pulse depression of CF-EPSCs recorded from PC in WT (n=9) and JP-DKO (n=12) mice. Mean±s.e.m. The amplitude of the second response is expressed as a percentage of the first response and is plotted along the interpulse intervals. (C) Paired-pulse facilitation of PF-mediated EPSCs recorded from WT (n=11) and JP-DKO (n=12) mice. Mean±s.e.m. The PF-EPSCs were examined as in (B), and the data suggest that irregular transmission could occur at high-frequency PF stimuli. *P<0.05, **P<0.01, significantly different from the value for WT mice (t-test). Data are obtained from 5- to 9-week-old mice.
We next examined basic properties in excitatory inputs to JP-DKO PCs. The rise time and amplitude of mono-innervating CF-EPSCs were the same between control and JP-DKO mice, whereas the decay time constant was slightly larger in JP-DKO mice (Supplementary Table I). As for the paired-pulse depression of CF-EPSCs, there was no significant difference between control and mutant mice (Figure 2B). EPSCs elicited by the stimulation of parallel fibers (PFs), another excitatory input to PCs, showed similar kinetics in control and JP-DKO mice (Supplementary Table I). In paired-pulse facilitation of PF-EPSCs, paired-pulse ratios in JP-DKO mice were slightly lower than those in control mice only at shorter interpulse intervals (Figure 2C). Therefore, the JP-DKO cerebellum retains basic excitatory circuitry, although there may be subtle electrophysiological changes in synaptic transmission.
Reversed cerebellar plasticity in JP-DKO mice
The induction of long-term depression (LTD) at PF-PC synapses is considered to be a cellular basis for cerebellar motor learning (Linden and Connor, 1995; Ito, 2002). Therefore, we surveyed whether any abnormalities of LTD underlay motor deficits in JP-DKO mice. EPSCs elicited by PF stimulation (PF-EPSC) were recorded using the whole-cell voltage-clamp mode before and after conjunctive CF and PF stimulation (1 Hz for 5 min) in the current-clamp mode for LTD induction. After the conjunctive stimulation, the amplitudes of PF-EPSCs in control mice were consistently decreased by ∼25% from the baseline levels (Figure 3). The observed LTD is comparable to that in experiments using wild-type mice (Ito, 2002). However, the conjunctive stimulation induced a completely opposite change in PF-EPSCs of JP-DKO PCs. The averaged EPSC amplitudes during the 21–30 min period after the conjunctive stimulation were >150% of the baseline levels. Therefore, the LTD paradigm adversely induces long-term potentiation (LTP) in the JP-DKO cerebellum, and this reversed plasticity at PF-PC synapses may underlie the impaired motor learning in the mutant mice.
Figure 3.
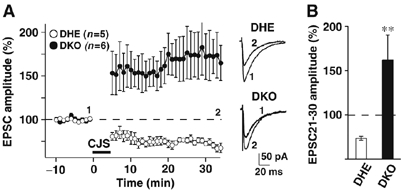
Reversed synaptic plasticity in JP-DKO cerebellum. LTP was induced in JP-DKO mice with conjunctive stimulation (stimuli of both PFs and CFs at 1 Hz for 5 min), which induces LTD in DHE mice. (A) (left) Changes in PF-EPSC amplitude before and after the conjunctive stimulation. The amplitude was normalized by the mean value observed for 10 min before the conjunctive stimulation. (right) Sample traces immediately before (1) and 30 min after the conjunctive stimulation (2). (B) Average PF-EPSC amplitude during the 21–30 min period after the conjunctive stimulation. Mean±s.e.m. **P<0.01, significantly different from the value for JP-DHE mice. Data were obtained from 8- to 10-week-old mice.
Deficiency of SK channel-mediated slow afterhyperpolarization in JP-DKO PCs
Because erratic activity of PCs results in cerebellar dysfunction (Sausbier et al, 2004), it is possible that PC excitability is abnormal in JP-DKO mice. We then performed whole-cell current-clamp recording of PCs using a K+-based pipette solution containing a low concentration of EGTA (1 mM) to examine voltage responses evoked by CF stimulation in cerebellar slices (Figure 4). In control PCs, CF stimulation elicited a voltage response consisting of a fast initial spike and several subsequent slower spikelets, which was followed by repolarization and slow afterhyperpolarization (sAHP) (Figure 4A, upper). In contrast, sAHP was absent in JP-DKO mice (Figure 4A, lower). Normal PCs have been shown to contain apamin-sensitive small-conductance (SK) and iberiotoxin-sensitive large-conductance (BK) Ca2+-dependent K+ channels. Both types of channel are activated after Ca2+ influx via major P/Q-type and minor T-type voltage-gated Ca2+ channels and likely contribute to action potential repolarization and afterhyperpolarization triggered by depolarizing pulses (Womack et al, 2004). The bath application of apamin (200 nM) completely abolished sAHP following complex spike recorded from control PCs, without changing the number and frequency of spikelets (Figure 4A and C). In contrast, apamin had no effect on the voltage response in JP-DKO PCs (Figure 4A and C). These results demonstrate that SK channels predominantly contribute to sAHP generation in PCs and suggest that SK channel-mediated sAHP generation is impaired in JP-DKO PCs.
Figure 4.
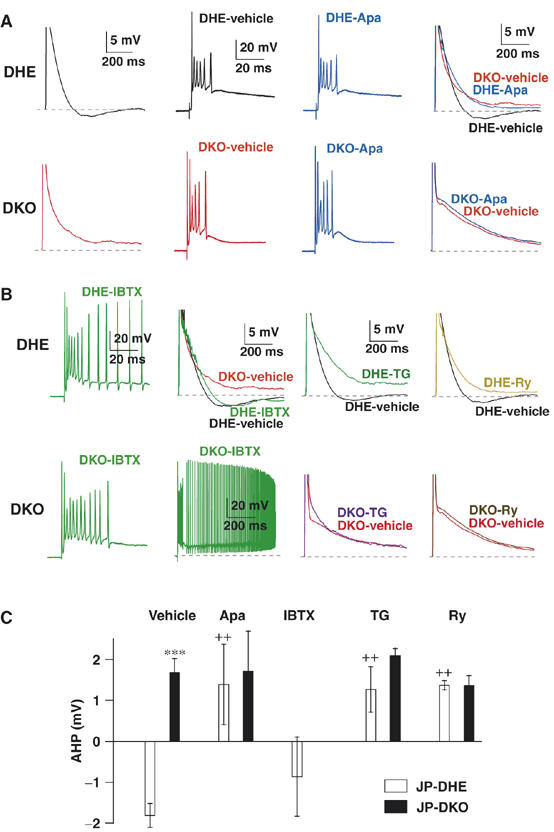
Abolished sAHP after complex spike in JP-DKO PCs. (A) Representative voltage response to CF stimulation recorded from control (DHE, upper) and JP-DKO (DKO, lower) PCs in current-clamp mode. Apamin (Apa; 200 nM), an SK channel blocker abolished sAHP in control PCs, but had no effect on sAHP in JP-DKO mice. (B) Effects of inhibitors for BK channel (iberiotoxin (IBTX); 100 nM), SR/ER Ca2+-ATPase (thapsigargin (TG); 2 μM) and RyRs (ryanodine (Ry); 100 μM) on voltage responses to CF stimuli in control (upper) and JP-DKO (lower) PCs. (C) Average amplitudes of sAHP in control and JP-DKO PCs treated with vehicle, Apa, IBTX, TG or Ry. The data represent mean±s.e.m. from at least five experiments. ***P<0.001, significantly different from the value for JP-DHE mice; ++P<0.01, significantly different from the value for vehicle-treated groups in JP-DHE mice (t-test). Data were obtained from 8- to 10-week-old mice.
There were no significant differences in the number and frequency of spikelets during complex spikes between control and JP-DKO mice in the presence and absence of apamin (Figure 4A). Therefore, SK channels seem to make no significant contribution to the early phase of the voltage response. The bath application of iberiotoxin (100 nM) produced no significant effects on sAHP, but increased the firing frequency and number of spikelets of complex spikes in control PCs (Figure 4B and C). In JP-DKO PCs, where SK channel activation is lacking, iberiotoxin induced sustained depolarization, presumably because of excessive repetitive firing under the lack of both SK and BK channel functions (Figure 4B). The iberiotoxin-induced responses, together with the regular decay phase after complex spikes under normal conditions, indicate that BK channels function normally in JP-DKO PCs.
Requirement of RyR-mediated Ca2+ release for SK channel opening
We examined the source of Ca2+ that activates SK channels in PCs. A possible signaling cascade involves Ca2+ release from the ER triggered by Ca2+ influx through voltage-gated Ca2+ channels, namely Ca2+-induced Ca2+ release (Endo, 1985). The pretreatment of cerebellar slices with thapsigargin (2 μM) for more than 60 min, which inhibits the SR/ER Ca2+-ATPase and induces Ca2+ store depletion, completely abolished sAHP in control PCs, but had no effect on JP-DKO PCs (Figure 4B and C). The bath application of ryanodine (100 μM), which induces use-dependent blockade of RyRs, impaired sAHP in control mice after repetitive CF stimuli (up to 100 times), but had no effect in JP-DKO mice (Figure 4B and C). Therefore, Ca2+ release from intracellular stores through RyRs seems to be essential for SK channel activation. This conclusion was further supported by the abolishment of sAHP with the bath application of dantrolene (30 μM) and the pipette-mediated application of ruthenium red (30 μM), both of which are RyR inhibitors (Supplementary Figure 3).
In addition to RyRs, inositol trisphosphate receptors (IP3Rs) are coexpressed on intracellular stores as Ca2+ release channels in PCs. The pipette application of heparin (4 mg/ml), an IP3R inhibitor, did not affect sAHP generation after CF-evoked complex spikes in control PCs (Supplementary Figure 3). The virus vector-mediated transfection of IP3 5-phosphatase, the enzyme for selective hydrolysis of IP3, resulted in the complete suppression of increase in IP3 level and weakening of PF synaptic connection to PCs owing to reduced presynaptic functions (Furutani et al, 2006). Although the activity of IP3 5-phosphatase overexpressed in PCs using the viral vector was confirmed by significant increases in the paired-pulse ratios of PF-EPSCs, we could not detect any effects on sAHP recorded from the same PCs (Supplementary Figure 3). These observations indicate that the IP3 signaling cascade is not involved in sAHP generation. Furthermore, thapsigargin and ryanodine had no effect on the iberiotoxin-sensitive responses during complex spikes (data not shown). Taken together, Ca2+-induced Ca2+ release through RyRs seems to trigger SK channel activation to generate sAHP in control PCs but not in JP-DKO PCs. In contrast, Ca2+ influx through voltage-gated channels likely leads to direct activation of BK channels and this mechanism is probably intact in JP-DKO PCs.
Proposed platform for crosstalk between RyRs and SK channels in PCs
Among the SK channel subtypes, both SK2 and SK3 produce apamin-sensitive currents and are expressed in neurons (Finlayson et al, 2001). In situ hybridization analyses have demonstrated that among RyR and SK channel subtypes, RyR1 and SK2 are predominantly expressed in mouse PCs (Mori et al, 2000; Bond et al, 2004). Because the JP-DKO cerebellum retained normal expression of these channels (Figure 5A), the sAHP deficiency is unlikely to result from decreased channel density. Alternatively, it may be possible that Ca2+ influx evoked by CF stimuli was weakened, and thus RyR-mediated Ca2+-induced Ca2+ release and the following SK channel activation were impaired in JP-DKO PCs. However, similar Ca2+ transients in response to CF stimuli were observed between control and JP-DKO PCs, and the effects of ryanodine on Ca2+ transient were somewhat less than obvious in both PCs (Supplementary Figure 4). The observations suggest that RyR-mediated Ca2+ release makes a minor contribution to Ca2+ transients during and after complex spikes and also that overall Ca2+ signaling, predominantly composed of P/Q-type channel-mediated Ca2+ influx, is apparently normal in the mutant PCs.
Figure 5.
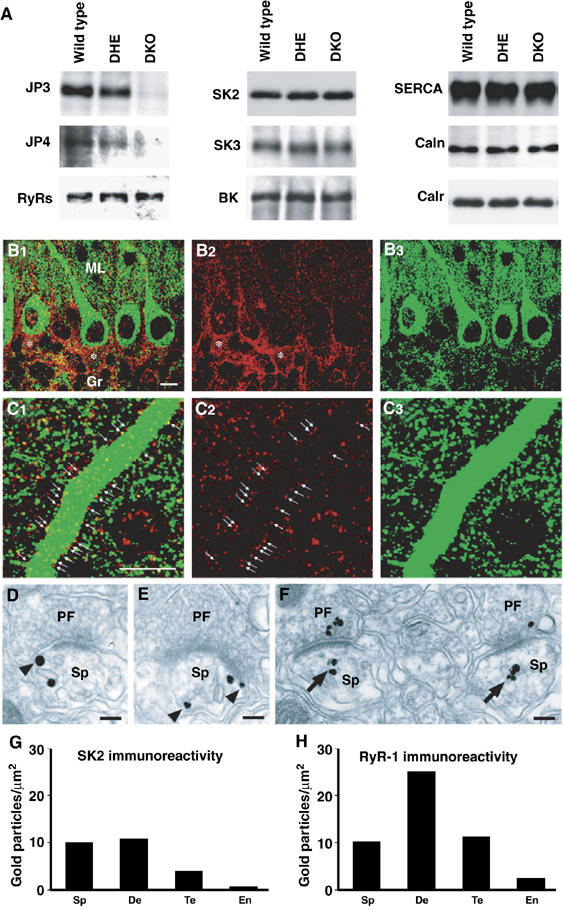
Expression of functional components for sAHP generation in PCs. (A) Normal contents of major Ca2+ store-related proteins in JP-DKO cerebellum. Total proteins were prepared from the cerebellum and analyzed by Western blotting. Antibodies detected junctophilin type 3 (JP3), junctophilin type 4 (JP4), ryanodine receptor subtypes (RyRs), SK channel type 2 (SK2) and type 3 (SK3), BK/MaxiK channel α subunit (BK), SR/ER Ca2+ ATPase type 2 (SERCA), calnexin (Caln) and calreticulin/calregulin (Calr). In quantitative analysis of immunoreactivities from at least five mice, no differences in protein expression level were observed between the genotypes. (B, C) Double immunofluorescence for SK2 (red) and RyR1 (green). Asterisks indicate SK2 in the pinceau formation, that is, clustered basket cell axons surrounding the initial segment of PC axons. Arrows indicate SK2 on the surface of PC dendritic shafts. Note the presence of dense punctate labeling for SK2 and RyR1 in the neuropil. Scale bars, 10 μm. (D–F) Silver-enhanced pre-embedding immunogold for SK2 (D, E) and RyR1 (F) at synapses between PF terminals (PF) and PC spines (Sp). Note that SK2 is associated with either the cell membrane or the smooth ER (sER, arrowheads), whereas RyR1 is preferentially distributed around the sER (arrows). Scale bars, 0.1 μm. (G, H) Quantitative measurement of the labeling density of SK2 (G) and RyR1 (H) in spines (Sp), dendritic shafts (De), PF synaptic terminals (Te) and capillary endothelial cells (En). Scores on the bars represent the number of immunogold particles per 1 μm2 of given elements. Labeling densities in all of these neuronal elements were much higher than those in capillary endothelial cells indicative of background immunogold decoration. Data were obtained from 8- to 10-week-old mice.
To locate the proposed platform for sAHP generation in PCs, we developed antibodies specific to RyR1 and SK2 (Supplementary Figures 5 and 6) for immunohistochemical analysis. Both antibodies produced reliable fluorescent signals detected as tiny puncta in the cerebellar cortex (Figure 5B and C). RyR1 was enriched inside dendritic shafts and perikarya of PCs, whereas SK2 was distributed both inside and along their surface (arrows in Figure 5C). In the neuropil, both the immunofluorescences were apposed to each other and often positioned side by side. By the silver-enhanced pre-embedding immunogold method, SK2 was distributed in association with either the cell membrane (arrowheads in Figure 5D and E) or the smooth ER, whereas RyR1 was preferentially around the smooth ER (arrows in Figure 5F). Based on the quantification of immunogold labeling, SK2 was distributed at higher levels in dendritic spines and shafts than nerve terminals (Figure 5G), whereas RyR1 was higher in dendritic shafts than dendritic spines and nerve terminals (Figure 5H). Therefore, SK2 is colocalized with RyR1 in the same somatodendritic compartments of PCs, showing different relative abundance depending on subcellular elements. This colocalization likely serves as the cellular machinery of sAHP generation in PCs. However, we could not detect any difference in the immunostaining of SK2 and RyR1 between control and JP-DKO PCs at the fluorescence-microscopic level (data not shown), suggesting that the expression and distribution of these channels are roughly normal under the lack of JPs.
Rescued sAHP in JP-DKO PCs
To test whether the ectopic expression of JP can restore sAHP in JP-DKO PCs, recombinant sindbis virus carrying two transgenes for JP4 and enhanced green fluorescence protein (EGFP) was injected into the cerebellar cortex of the mutant mice. The following day, clear EGFP signals were detected in infected PCs on the slice preparations (Figure 6A). CF stimulation elicited a complex spike followed by sAHP in the mutant PCs showing EGFP fluorescence, and the observed sAHP was sensitive to apamin (Figure 6A and B). On the other hand, sAHP was still absent in non-transfected PCs without EGFP fluorescence. We also examined the effects of two fluorescent fusion proteins, DsRed-JP4 and JP4-DsRed (Supplementary Figure 7). Although both fusion proteins were expressed in the somatodendritic regions of JP-DKO PCs, sAHP was restored in JP4-DsRed-expressing cells but not in DsRed-JP4-expressing cells. The fluorescence signals of JP4-DsRed were preferentially detected as puncta at the cell periphery in the dendritic shafts and soma regions of infected PCs, whereas DsRed-JP4 signals constantly diffused throughout the inside of cells. The N-terminal regions of JP subtypes share repeated MORN motif sequences essential for interacting with the plasma membrane toward the formation of junctional membrane complexes (Takeshima et al, 2000). In the N-terminal fusion of DsRed-JP4, the bulk effect of DsRed followed by the MORN motifs might inhibit its interaction with the plasma membrane for constructing the structural platform to generate sAHP. These observations demonstrate that sAHP deficiency in JP-DKO PCs is rescued by functional expression of exogenous JP4, and also suggest that the generation of sAHP requires JP-mediated close association between the cell membrane and the ER at somatodendritic regions in PCs.
Figure 6.
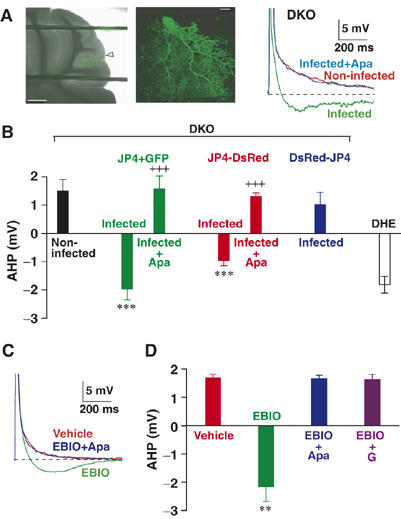
Rescued sAHP in JP-DKO PCs. (A) (left and middle) Fluorescent images of PCs expressing JP4 and EGFP in cerebellar slice at different magnifications. The arrowhead indicates the site of penetration for viral injection. Scale bars, (left) 500 μm, (middle) 20 μm. (right) Representative traces of voltage response to CF stimulation recorded from JP-DKO PCs. sAHP was not observed in noninfected PCs, whereas apamin (Apa)-sensitive sAHP was rescued in infected PCs expressing both JP4 and EGFP. (B) Average amplitudes of sAHP. The data represent mean±s.e.m. (n=5, except for JP4+GFP-infected (n=8) and DHE PCs (n=11)). ***P<0.001, significantly different from the value for noninfected PCs in JP-DKO mice; +++P<0.001, significantly different from the value for apamin-untreated PCs expressing the same proteins. (C) Representative traces of voltage response to CF stimulation recorded from JP-DKO PCs. The bath application of 1-EBIO (1 mM) rescued apamin-sensitive sAHP. (D) Average amplitudes of sAHP. The data represent mean±s.e.m. (vehicle n=8, EBIO n=3, EBIO+Apa n=3, EBIO+thapsigargin (TG) n=5). **P<0.01, significantly different from the value for vehicle-treated PCs in JP-DKO mice. Data were obtained from 8- to 10-week-old mice.
We further examined the acute effect of 1-ethyl-2-benzimidazolinone (EBIO) as an SK channel enhancer, which facilitates the Ca2+ sensitivity for channel gating through its direct interaction with the binding site composed of an SK channel subunit and calmodulin (Pedarzani et al, 2001; Stocker, 2004). The bath application of EBIO (1 mM) immediately resulted in the clear restoration of sAHP following complex spikes in JP-DKO PCs (Figure 6C and D), suggesting normal levels of physiologically active SK channels on the cell membrane in mutant PCs. As in normal sAHP observed in control mice, the rescued sAHP was certainly mediated by SK channels and required Ca2+ release from intracellular stores because both apamin and thapsigargin abolished EBIO-mediated effects (Figure 6C and D). Taken together, we conclude that the generation of sAHP requires Ca2+-mediated crosstalk between RyRs and SK channels at the microdomain level and that JPs physiologically contribute to establishing this channel communication in PCs.
Requirement of sAHP for LTD induction in cerebellum
To examine the relationship between sAHP deficiency and reversed synaptic plasticity in JP-DKO mice, we finally examined the effect of apamin on LTD induction in control mice. In the presence of apamin (200 nM), the same conjunctive stimulation reversibly induced PF-LTP (Figure 7). The LTP levels at 20–30 min after the conjunctive stimulation were similar to those observed in JP-DKO slices, although there was a significant difference in the time course for LTP induction between apamin-treated control PCs and JP-DKO PCs (see Figure 3). The observations likely suggest that sAHP deficiency is at least one of the major factors causing the reversed PF synapse plasticity in JP-DKO mice. Therefore, functional crosstalk between RyRs and SK channels in PCs is probably essential for cerebellar plasticity.
Figure 7.
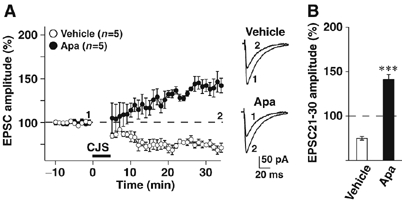
Contribution of sAHP to cerebellar LTD induction. (A) (left) Changes in PF-EPSC amplitude before and after conjunctive stimulation. The amplitude was normalized by the mean value observed for 10 min before the conjunctive stimulation. In the presence of apamin (Apa; 200 nM), LTP was induced by the conjunctive stimulation, which induced LTD in vehicle-treated group. (right) Sample traces immediately before (1) and 30 min after the conjunctive stimulation (2). (B) Average PF-EPSC amplitude during the 21–30 min period after the conjunctive stimulation. Mean±s.e.m. ***P<0.001, significantly different from the value for vehicle-treated group. Data were obtained from 8- to 10-week-old mice.
Discussion
Requirement of JPs for crosstalk between RyRs and SK channels
Previous studies indicated that BK and SK channels coexpressed on PCs collaboratively govern firing frequency to regulate membrane excitability. Although both channels are activated downstream of Ca2+ influx via P/Q-type voltage-gated Ca2+ channels (Edgerton and Reinhart, 2003; Womack et al, 2004), our study indicates that their activation mechanisms are definitely different from each other. The number and frequency of spikelets during CF-evoked PC excitation are mainly controlled by BK channels directly activated by Ca2+ influx via P/Q-type channels. On the other hand, SK channels are probably activated by RyR-mediated Ca2+-induced Ca2+ release after the Ca2+ influx to produce the sAHP phase in PCs (Figure 4). Our results also indicate that sAHP is impaired owing to insufficient communication between RyRs and SK channels in JP-DKO PCs (Figure 6). Therefore, we propose that neural JP subtypes contribute to the formation of structural platform essential for the proper functioning of Ca2+-mediated communication between P/Q-type channels, RyRs and SK channels to generate sAHP (Figure 8). This RyR-based channel communication likely requires close association between the channels on distinct membrane systems because Ca2+ mobility is highly restricted by strong cytosolic buffering effects. Although our electron microscopic observations could not detect any difference in junctional membrane complexes between control and JP-DKO PCs (data not shown), immunohistochemical analysis (Figure 5) and functional expression of fluorescence-labeled JPs (Supplementary Figure 7) clearly indicate that the platform for the channel communication is constructed at somatodendritic regions in PCs. Two possibilities can be assumed to understand sAHP deficiency in JP-DKO PCs at the molecular level; insufficient communication between P/Q-type channels and RyRs, and/or between RyRs and SK channels under platform-deficient conditions.
Figure 8.
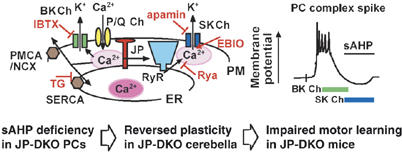
Role of JP-mediated channel crosstalk in PCs. Previous data have demonstrated that Ca2+ influx through predominant P/Q-type voltage-gated Ca2+ channels induces the activation of BK and SK channels in PCs. In our present study, JP-DKO PCs showed no sAHP produced by SK channels but probably retained normal BK channel currents. Moreover, JP-mediated channel crosstalk between P/Q-type channels, RyRs and SK channels was proposed as cellular machinery for sAHP generation in PCs (upper); Ca2+ influx via P/Q-type channels activates both RyRs on the ER and BK channels on the plasma membrane (PM). SK channels on the PM are subsequently activated by Ca2+-induced Ca2+ released through RyRs. Cytosolic Ca2+ is likely cleared by SR/ER and plasma membrane Ca2+-ATPase (SERCA/PMCA) and Na+/Ca2+ exchanger (NCX). JPs seem to prepare a structural platform for the Ca2+-mediated channel communication, and insufficient channel communication can be predicted at the microdomain level in JP-DKO PCs. The deficiency of sAHP likely induces reversed plasticity in the mutant cerebellum and motor dysfunction in the mutant mice. Rya, ryanodine; IBTX, iberiotoxin; TG, thapsigargin.
SK and BK channels coexpressed in PCs seem to serve as Ca2+ sensors at specific compartments underneath the cell membrane. BK channels probably function normally in JP-DKO PCs, suggesting normal Ca2+ signaling around BK channels. On the other hand, insufficient Ca2+ signaling is reasonably proposed at compartments surrounding SK channels in JP-DKO PCs, although even for a most advanced imaging system, it was impossible to capture abnormal Ca2+ transients predicted in JP-DKO PCs (Supplementary Figure 4). Therefore, delicate Ca2+ signaling within a narrow range, but not overall cytosolic Ca2+ increase, likely regulates SK channel opening in PCs. Moreover, different activation mechanisms between BK and SK channels would reflect their distinct subcellular distribution. BK channels may not be in the proposed platform constructed by JPs but may be distributed together with P/Q-type channels on PCs.
sAHP deficiency and reversed LTD in PCs
The conjunctive stimulation for LTD induction at PF synapses adversely induced LTP in JP-DKO PCs lacking sAHP after CF-evoked complex spikes (Figure 3). In wild-type PCs, bath application of apamin abolished sAHP and led to reversed plasticity similar to that of JP-DKO PCs (Figure 7). Therefore, sAHP generation in PCs during the conjunctive stimulation is essential for LTD induction. However, the temporal LTD profile in apamin-treated wild-type PCs was slightly different from that in JP-DKO PCs. In particular, relative PF-EPSC amplitudes were remarkably facilitated in JP-DKO PCs shortly after the conjunctive stimulation (compare Figures 3 and 7). In JP-DKO PCs, besides sAHP deficiency, chronic effects derived from poor Ca2+-mediated channel communication might develop ‘metabolic ailments', which further aggravate the reversed plasticity at the initial phase. Because protein kinase Cα (PKCα) plays an essential role in PCs during LTD induction (Chung et al, 2003; Leitges et al, 2004), irregular PKCα activity could be predicted in JP-DKO PCs. However, our immunoblotting experiments showed normal phosphorylation levels of PKCα and major PKC target proteins, myristoylated, alanine-rich protein kinase C substrate (MARCKS) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptor subunit GluR1, in the JP-DKO cerebellum (H Takeshima, unpublished observation). Moreover, DNA microarray analysis did not detect obvious alteration of the gene expression profile of JP-DKO mice, and the JP-DHE and JP-DKO cerebella showed similar mRNA expression patterns for signaling proteins bearing established or putative roles in LTD induction (A Ikeda and H Takeshima, unpublished observation). These results may suggest that as yet unrecognized cellular factors restrict the temporal profile of the reversed plasticity in JP-DKO PCs.
Role of RyRs in neuronal functions
Of the two types of Ca2+-release channels coexpressed in PCs, IP3Rs are essential for the induction of cerebellar LTD by contributing to the metabotropic glutamate receptor type 1 signaling of PCs triggered by PF synaptic stimuli (Ito, 2002). However, the role of RyRs in LTD induction was largely unknown, although it has been reported that the LTD induction in primary cultured PCs is inhibited by ruthenium red and ryanodine (Kohda et al, 1995). The present study has successfully demonstrated distinct roles of RyRs and IP3Rs in cerebellar LTD induction by establishing JP-mediated channel crosstalk between RyRs and SK channels in PCs. In addition to the major distribution of RyR1 in somatodendritic regions, our immunoelectron microscopic observations demonstrated the presence of RyR1 in PF synaptic terminals (Figure 5). Therefore, RyRs are likely expressed in both somatodendritic and presynaptic regions in neurons. The physiological function of presynaptic RyRs remains to be investigated, although the subcellular distribution implies their putative roles in Ca2+ signaling for transmitter release.
In the brain, RyR subtypes, namely RyR1–3, together with JP subtypes, are expressed in most neurons; however, their roles have not been established yet (Mori et al, 2000; Nishi et al, 2003). Our current data indicate that Ca2+-mediated functional communication between NMDA receptors, RyRs and SK channels is present in hippocampal CA1 pyramidal neurons for AHP generation, and that this mechanism is abolished in JP-DKO mice showing impaired CA1 LTP (Moriguchi et al, 2006). Results from hippocampal CA1 neurons and cerebellar PCs in JP-DKO mice indicate that neuronal JPs support the Ca2+-mediated functional crosstalk of RyRs with cell-surface ionic channels, and further suggest that RyR-mediated Ca2+ release in somatodendritic regions generally links with SK channel opening to produce the AHP phase after firing. Excitation–contraction coupling in striated muscle involves functional crosstalk between dihydropyridine-sensitive Ca2+ channels and RyRs, and requires junctional membrane structures produced by JP1 and JP2 (Takeshima et al, 2000; Ito et al, 2001). The JP-mediated channel crosstalk in skeletal muscle absolutely requires the accurate pair of channel subtypes, the combination of Cav1.1 and RyR1 (Meissner, 1994; FranziniArmstrong and Protasi, 1997). In contrast, neuronal channel crosstalk seems rather flexible, because NMDA receptors and P/Q-type channels likely activate RyRs in CA1 neurons and PCs, respectively. Moreover, the activation of SK channels may not require specific RyR subtypes. For example, RyR1 is preferentially expressed in PCs, whereas CA1 neurons contain both RyR2 and RyR3 (Mori et al, 2000). In both neurons, AHPs generated by JP-mediated channel crosstalk seem to contribute to the precise establishment of neuronal plasticity, which is fundamental for integrated brain functions. JP-DKO mice may be an ideal model system for unveiling physiological roles of functional crosstalk between RyRs and SK channels in other types of neurons.
Materials and methods
Knockout mice and morphological analysis
JP3-knockout and JP4-knockout mice (Nishi et al, 2002, 2003) were crossed to generate JP-DKO mice. As control mice, we used JP-DHE and wild-type mice in this study. JP-DKO and JP-DHE mice were fed paste food as described previously (Moriguchi et al, 2006), and wild-type (C57BL) mice were fed normal pellet food. Immunochemical analyses were conducted essentially as described previously (Nakagawa et al, 1998; Hashimoto et al, 2001; Miyazaki et al, 2003; Tohgo et al, 2006), and the methods are briefly described in the legends for Supplementary Figures 2, 5 and 6.
Behavioral tests
The fixed-bar and rota-rod tests were carried out as described previously (Kishimoto et al, 2002; Nishi et al, 2002; Tohgo et al, 2006). For delay eyeblink conditioning (Kishimoto et al, 2002), mice were surgically implanted with four Teflon-coated stainless steel wires (A-M Systems, WA, USA) under the left eyelid for recording electromyographic activity and delivering electric shock. A periorbital shock (100 Hz square pulses for 100 ms) was applied as unconditioned stimulus, and a tone (1.0 kHz, 80 dB for 450 ms) was used as conditioning stimulus; the conditioning stimulus preceded and coterminated with the unconditioned stimulus. The acquisition session consisted of 100 paired trials of the conditioned and unconditioned stimulus. The extinction sessions consisted of 100 conditioned stimulus trials. Spontaneous eyeblink frequency was measured in 100 nonstimulus trials. The electromyographic signal was band-pass filtered between 0.15 and 1.0 kHz and fed into a computer at a sampling rate of 10 kHz.
Electrophysiological measurements
Electrophysiological experiments were carried out essentially as described previously (Kakizawa et al, 2000, 2005; Hashimoto et al, 2001). The resistance of patch pipettes was 2.0–3.5 MΩ when filled with an intracellular solution composed of (in mM) 120 K-gluconate, 5 KCl, 5 NaCl, 1 EGTA, 4 ATP, 0.4 GTP and 10 HEPES (pH 7.3; adjusted with KOH). For the voltage-clamp recording of PCs (Figure 2), a pipette solution with the following composition was used (in mM) 60 CsCl, 10 Cs D-gluconate, 20 TEA-Cl, 20 BAPTA, 4 MgCl2, 4 ATP, 0.4 GTP and 30 HEPES (pH 7.3). The standard bathing solution was composed of (in mM) 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgSO4, 1.25 NaH2PO4, 26 NaHCO3 and 20 glucose, which was bubbled continuously with a mixture of 95% O2 and 5% CO2. Bicuculline (10 μM) was always present in the saline to block spontaneous inhibitory postsynaptic currents. Square pulses were applied for focal stimulation (duration, 0.1 ms; amplitude, 0–90 V for CF stimulation, 0–10 V for PF stimulation) through a glass pipette with a 5–10-μm-diameter tip and filled with the standard bath solution. The membrane potentials were held at −90 to −80 mV for recording PF-EPSCs, and at −20 to −10 mV for recording CF-EPSCs, after the compensation for liquid junction potential. Ionic currents were recorded using a patch-clamp amplifier (EPC-9, HEKA or Axopatch-1D, Axon Instruments). Stimulation and on-line data acquisition were performed using PULSE software (HEKA) on a Macintosh computer. Signals were filtered at 3 kHz and digitized at 20 kHz. The fitting of the decay phases of EPSCs was performed with PULSE-FIT software (HEKA). For the LTD experiments (Figures 3 and 7), the intensity of the stimulus was adjusted to evoke PF-EPSCs whose initial amplitudes were between 60 and 120 pA. After obtaining a stable initial recording for at least 10 min, conjunctive stimulus was applied to induce LTD. The conjunctive stimulation protocol is composed of 300 single PF stimuli in conjunction with single CF stimuli repeated at 1 Hz. Series resistance and membrane resistance were monitored throughout the experiments, and the data were discarded when either of these resistances varied by more than 10%. The data were also discarded when the slope of PF-EPSC amplitude averaged every minute during the initial recording for 10 min was larger than 2% or when the amplitude did not become stable within 30 min after the onset of whole-cell configuration (Namiki et al, 2005).
Sindbis virus infection
Mouse JP4 cDNA (Takeshima et al, 2000) was cloned into pIRES2-EGFP (Clontech) to generate the transgene encoding JP4-IRES-EGFP, or into pDsRed-Monomer (Clontech) to generate the transgenes encoding DsRed-JP4 and JP4-DsRed fusion proteins. The transgene for overexpressing IP3 5-phosphatase was constructed as described previously (Furutani et al, 2006). The transgenes were cloned into pSinRep5 (Invitrogen), and the resulting vector was used as the template for in vitro transcription using SP6 RNA polymerase (Ambion). The RNAs transcribed and helper RNA derived from a DH26 (S) cDNA template (Invitrogen) were cotransfected into BHK cells to prepare the sindbis viral vectors. For in vivo infection with viral particles, JP-DKO mice (8–10 weeks old) were anesthetized with pentobarbital, and through an incision in the scalp, a small piece of the occipital bone and the dura covering the surface of cerebellar lobule VII were removed (Kakizawa et al, 2000, 2005). Then, solutions containing viral particles carrying the transgenes were injected into lobule VII of the cerebellar cortex with a microglass needle (tip diameter of 20–40 μm) attached to a manipulator (Namiki et al, 2005). A volume of about 1 μl was delivered unilaterally within 5–10 min by air pressure. After the injection, the scalp was sutured.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Table I
Supplementary Information
Acknowledgments
We thank Drs John Adelman (Vollum Institute, Oregon Health and Science University, USA) and Rafael Lujan (Universidad de Castilla-La Mancha, Albacete Spain) for specificity verification of SK2 antibody by blank immunostaining in the SK2-knockout brain, and Ms Miyuki Kameyama for technical assistance in knockout mouse generation. This work was supported in part by grants from the Ministry of Education, Science, Sports and Culture of Japan (JSPS), the Ministry of Health and Welfare of Japan, the Sumitomo Foundation, the Naito Foundation, the Life Science Foundation, the Takeda Science Foundation and the Uehara Memorial Foundation.
References
- Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32: 235–249 [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL (2005) Neuronal activity regulates diffusion across the neck of dendritic spines. Science 310: 866–869 [DOI] [PubMed] [Google Scholar]
- Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, Adelman JP (2004) Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci 24: 5301–5306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Steinberg JP, Huganir RL, Linden DJ (2003) Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 300: 1751–1755 [DOI] [PubMed] [Google Scholar]
- Edgerton JR, Reinhart PH (2003) Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol-London 548: 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M (1985) Calcium release from sarcoplasmic-reticulum. Curr Topics Memb Trans 25: 181–230 [Google Scholar]
- Finlayson K, McLuckie J, Hern J, Aramori I, Olverman HJ, Kelly JS (2001) Characterisation of I-125-apamin binding sites in rat brain membranes and HEK293 cells transfected with SK channel subtypes. Neuropharmacology 41: 341–350 [DOI] [PubMed] [Google Scholar]
- Flucher BE (1992) Structural-analysis of muscle development—transverse tubules, sarcoplasmic-reticulum, and the triad. Dev Biol 154: 245–260 [DOI] [PubMed] [Google Scholar]
- FranziniArmstrong C, Protasi F (1997) Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev 77: 699–729 [DOI] [PubMed] [Google Scholar]
- Furutani K, Okubo Y, Kakizawa S, Iino M (2006) Postsynaptic inositol 1,4,5-trisphosphate signaling maintains presynaptic function of parallel fiber-Purkinje cell synapses via BDNF. Proc Natl Acad Sci USA 103: 8528–8533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JH, Tamas G, Aronov D, Yuste R (2003) Calcium microdomains in aspiny dendrites. Neuron 40: 807–821 [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Ichikawa R, Takechi H, Inoue Y, Aiba A, Sakimura K, Mishina M, Hashikawa T, Konnerth A, Watanabe M, Kano M (2001) Roles of glutamate receptor delta 2 subunit (GluR delta 2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J Neurosci 21: 9701–9712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K, Takeshima H (2001) Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol 154: 1059–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M (2002) The molecular organization of cerebellar long-term depression. Nat Rev Neurosci 3: 896–902 [DOI] [PubMed] [Google Scholar]
- Kakizawa S, Miyazaki T, Yanagihara D, Iino M, Watanabe M, Kano M (2005) Maintenance of presynaptic function by AMPA receptor-mediated excitatory postsynaptic activity in adult brain. Proc Natl Acad Sci USA 102: 19180–19185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa S, Yamasaki M, Watanabe M, Kano M (2000) Critical period for activity-dependent synapse elimination in developing cerebellum. J Neurosci 20: 4954–4961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S (1995) Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell 83: 1223–1231 [DOI] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, Tonegawa S (1997) Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron 18: 71–79 [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Fujimichi R, Araishi K, Kawahara S, Kano M, Aiba A, Kirino Y (2002) mGluR1 in cerebellar Purkinje cells is required for normal association of temporally contiguous stimuli in classical conditioning. Eur J Neurosci 16: 2416–2424 [DOI] [PubMed] [Google Scholar]
- Kohda K, Inoue T, Mikoshiba K (1995) Ca2+ release from Ca2+ stores, particularly from ryanodine-sensitive Ca2+ stores, is required for the induction of ltd in cultured cerebellar Purkinje-cells. J Neurophysiol 74: 2184–2188 [DOI] [PubMed] [Google Scholar]
- Leitges M, Kovac J, Plomann M, Linden DJ (2004) A unique PDZ ligand in PKC alpha confers induction of cerebellar long-term synaptic depression. Neuron 44: 585–594 [DOI] [PubMed] [Google Scholar]
- Levitan IB (2006) Signaling protein complexes associated with neuronal ion channels. Nat Neurosci 9: 305–310 [DOI] [PubMed] [Google Scholar]
- Linden DJ, Connor JA (1995) Long-term synaptic depression. Annu Rev Neurosci 18: 319–357 [DOI] [PubMed] [Google Scholar]
- McCormick DA, Thompson RF (1984) Cerebellum—essential involvement in the classically-conditioned eyelid response. Science 223: 296–299 [DOI] [PubMed] [Google Scholar]
- Meissner G (1994) Ryanodine receptor Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol 56: 485–508 [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Fukaya M, Shimizu H, Watanabe M (2003) Subtype switching of vesicular glutamate transporters at parallel fibre–Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosc 17: 2563–2572 [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Hashimoto K, Shin HS, Kano M, Watanabe M (2004) P/Q-type Ca2+ channel alpha 1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci 24: 1734–1743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F, Fukaya M, Abe H, Wakabayashi K, Watanabe M (2000) Developmental changes in expression of the three ryanodine receptor mRNAs in the mouse brain. Neurosci Lett 285: 57–60 [DOI] [PubMed] [Google Scholar]
- Moriguchi S, Nishi M, Komazaki S, Sakagami H, Miyazaki T, Masumiya H, Saito SY, Watanabe M, Kondo H, Yawo H, Fukunaga K, Takeshima H (2006) Functional uncoupling between Ca2+ release and afterhyperpolarization in mutant hippocampal neurons lacking junctophilins. Proc Natl Acad Sci USA 103: 10811–10816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Watanabe M, Isobe T, Kondo H, Inoue Y (1998) Cytological compartmentalization in the staggerer cerebellum, as revealed by calbindin immunohistochemistry for Purkinje cells. J Comp Neurol 395: 112–120 [DOI] [PubMed] [Google Scholar]
- Namiki S, Kakizawa S, Hirose K, Iino M (2005) NO signalling decodes frequency of neuronal activity and generates synapse-specific plasticity in mouse cerebellum. J Physiol-London 566: 849–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M, Hashimoto K, Kuriyama K, Komazaki S, Kano M, Shibata S, Takeshima H (2002) Motor discoordination in mutant mice lacking junctophilin type 3. Biochem Biophys Res Commun 292: 318–324 [DOI] [PubMed] [Google Scholar]
- Nishi M, Sakagami H, Komazaki S, Kondo H, Takeshima H (2003) Coexpression of junctophilin type 3 and type 4 in brain. Mol Brain Res 118: 102–110 [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Mosbacher J, Rivard A, Cingolani LA, Oliver D, Stocker M, Adelman JP, Fakler B (2001) Control of electrical activity in central neurons by modulating the gating of small conductance Ca2+-activated K+ channels. J Biol Chem 276: 9762–9769 [DOI] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P (2004) Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci USA 101: 9474–9478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M (2004) Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci 5: 758–770 [DOI] [PubMed] [Google Scholar]
- Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K (2000) Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell 6: 11–22 [DOI] [PubMed] [Google Scholar]
- Tohgo A, Eiraku M, Miyazaki T, Miura E, Kawaguchi S, Nishi M, Watanabe M, Hirano T, Kengaku M, Takeshima H (2006) Impaired cerebellar functions in mutant mice lacking DNER. Mol Cell Neurosci 31: 326–333 [DOI] [PubMed] [Google Scholar]
- Verkhratsky A (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85: 201–279 [DOI] [PubMed] [Google Scholar]
- Womack MD, Chevez C, Khodakhah K (2004) Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci 24: 8818–8822 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Table I
Supplementary Information