

Abstract
The hematopoietic system provides an attractive model for studying growth factor-controlled expansion and differentiation of cells in relation to receptor routing and its consequences for signal transduction. Suppressor of cytokine signaling (SOCS) proteins regulate receptor signaling partly via their ubiquitin ligase (E3)-recruiting SOCS box domain. Whether SOCS proteins affect signaling through modulating intracellular trafficking of receptors is unknown. Here, we show that a juxtamembrane lysine residue (K632) of the granulocyte colony-stimulating factor receptor (G-CSFR) plays a key role in receptor routing and demonstrate that the effects of SOCS3 on G-CSF signaling to a major extent depend on this lysine. Mutation of K632 causes accumulation of G-CSFR in early endosomes and leads to sustained activation of signal transducer and activator of transcription 5 and ERK, but not protein kinase B. Myeloid progenitors expressing G-CSFR mutants lacking K632 show a perturbed proliferation/differentiation balance in response to G-CSF. This is the first demonstration of SOCS-mediated ubiquitination and routing of a cytokine receptor and its impact on maintaining an appropriate signaling output.
Keywords: G-CSF receptor, lysosomal routing, receptor ubiquitination, signal attenuation, SOCS3
Introduction
In the past 5 years, a major role has been demonstrated for protein ubiquitination in the control of endocytosis and intracellular routing of receptor tyrosine kinases, particularly of the epidermal growth factor (EGF) receptor (reviewed in Haglund et al, 2003; Marmor and Yarden, 2004; Miaczynska et al, 2004; Polo and Di Fiore, 2006). For instance, upon ubiquitination by the E3 ligase Cbl, EGF receptor associates with multiple proteins containing an ubiquitin interaction motif (UIM), which guides EGF receptors into the different compartments of the endocytotic pathway. One such UIM-containing protein is hepatocyte growth factor receptor substrate (Hrs), implicated in the sorting of ubiquitinated cargo from early endosomes towards late endosomes, the lumen of multivesicular bodies and finally to lysosomes (Haglund et al, 2003). Receptor endocytosis is no longer considered as a mechanism solely involved in termination of signaling. Rather, it became clear that signaling processes proceed in endosomes, and that temporal and spatial dissociation as well as integration of signals may occur during endocytosis (Miaczynska et al, 2004; Polo and Di Fiore, 2006). This form of cell signaling orchestration is believed to contribute significantly to cellular processes such as cell fate determination, migration and differentiation (Polo and Di Fiore, 2006). However, owing to a paucity of appropriate model systems, insights into the impact of receptor routing on fine-tuning of cellular responses in response to growth factors are still scarce.
Granulocyte colony-stimulating factor (G-CSF) is the main cytokine involved in the production of neutrophilic granulocytes (Avalos et al, 1990). Activation of the G-CSF receptor (G-CSFR), a member of the class I cytokine receptor superfamily, supports the proliferation, survival and differentiation of neutrophilic progenitor cells (van de Geijn et al, 2003). Similar to most cytokine receptors, G-CSFR activates multiple signaling pathways fuelled by activation of JAK kinases. For instance, JAK-phosphorylated tyrosine-based motifs become docking sites for SH2-containing signaling molecules, such as signal transducer and activator of transcription (STAT) 3 (Ward et al, 1999a; Shao et al, 2006), components of the p21Ras/Raf/MAPK pathway (Rausch and Marshall, 1997; de Koning et al, 1998) and suppressor of cytokine signaling 3 (SOCS3) (Hortner et al, 2002; Hermans et al, 2003). In addition, the membrane-proximal cytoplasmic region of G-CSFR is involved in the activation of protein kinase B (PKB), a serine/threonine kinase implicated in cell survival and in the activation of STAT5 (Dong and Larner, 2000).
G-CSF stimulates proliferation and survival of myeloid progenitor cells, followed by a cell cycle arrest and differentiation towards neutrophils. Probably, this complex response requires attenuation of each of the signaling functions of G-CSFR during receptor endocytosis and intracellular routing. In support of this notion, perturbed signaling by G-CSFR truncation mutants, found in patients with acute myeloid leukemia preceded by severe chronic neutropenia, is to a large extent due to their inability to undergo endocytosis (Hunter and Avalos, 1999; Ward et al, 1999c).
Routing of class I cytokine receptors has been studied only to a limited extent and appears markedly heterogeneous. Most members studied thus far, for example, the growth hormone receptor (GHR) (van Kerkhof and Strous, 2001), leptin receptor (LR) (Uotani et al, 1999), prolactin receptor (PRLR) (Kelly et al, 1991), interleukin (IL)-9 receptor (Yen et al, 2000), gp130 (Dittrich et al, 1996; Blanchard et al, 2000) and the IL-2 receptor beta chain (Yen et al, 2000), follow the endosomal/lysosomal pathway. Endocytosis of LR depends on ubiquitination of two lysine residues in its cytoplasmic tail (Belouzard and Rouille, 2006). The ubiquitin-conjugating system is also required for endocytosis of GHR, but this does not depend on the cytoplasmic lysines of GHR (Govers et al, 1999; Alves dos Santos et al, 2001). In contrast, the Ub-conjugating system is not essential for internalization of PRLR (Lu et al, 2005). The thrombopoietin receptor (TPOR) forms a noteworthy exception, as it does not route to late endosomes and lysosomes but is rapidly recycled to the plasma membrane (Dahlen et al, 2003; Royer et al, 2005). This diversity in control of endocytosis and lysosomal routing emphasizes the importance of intracellular trafficking for the specific regulatory functions of the different receptors of this superfamily.
Human G-CSFR contains five cytoplasmic lysines (K632, K672, K681, K682 and K762) that are candidates for ubiquitination and all are conserved between humans and mice. We recently reported that cytoplasmic lysines in the G-CSFR play a key role in its steady-state membrane expression and protein stability, and provided evidence that WD40 and SOCS box-containing protein (Wsb) negatively control forward routing of G-CSFR depending on these lysines (Erkeland et al, 2006). However, whether ubiquitination of one or more of these lysine residues is crucial for ligand-induced G-CSFR endocytosis, trafficking to early and late endosomes, which Ub ligases might be involved and how this influences G-CSF-induced proliferation and differentiation signaling remained unknown.
Studies in conditional knockout mice have established that SOCS3 is a major negative regulator of G-CSF-induced neutrophil production in vivo (Croker et al, 2004; Kimura et al, 2004). SOCS proteins can inhibit cytokine signaling via multiple mechanisms: that is, by competition for SH2-containing signaling substrates in receptor binding, by inhibition of JAK kinase activity through their kinase inhibitory region or by recruitment of elongins B and C to the SOCS box to form an E3 Ub-ligase complex for degradation of signaling substrates (Kile et al, 2002). In an earlier report, we have shown that the SOCS box has a major role in SOCS3-mediated inhibition of G-CSF signaling (van de Geijn et al, 2004b), but how this is achieved remained unclear.
In this study, we show that a G-CSFR mutant lacking all five conserved cytoplasmic lysine residues (K5R) internalizes normally but fails to route to lysosomes. K5R is retained in early endosome antigen 1 (EEA1) and Hrs-positive endocytotic vesicles, where it continues to activate STAT5 but not PKB. Subsequent mutational analysis demonstrated that membrane-proximal lysine K632 is most crucial for lysosomal routing of the G-CSFR and for attenuation of G-CSF-induced STAT activity, cell proliferation and for the induction of terminal neutrophilic differentiation of myeloid 32D cells. We then show that ubiquitination of G-CSFR-K632 and lysosomal routing of the G-CSFR both depend on the integrity of the SOCS3 recruitment site (Y729) of G-CSFR. Finally, we demonstrate that inhibition of STAT5 activation by SOCS3 fully depends on the cytoplasmic lysines of G-CSF-R, in particular K632. These results reveal a major role of SOCS3 in G-CSFR routing from early (Hrs-positive) to late (Rab7-positive) endosomes and lysosomes and highlight the importance of receptor routing for an appropriate balance of proliferation and differentiation of myeloid progenitors in response to G-CSF.
Results
Cytoplasmic lysines are not essential for ligand-induced endocytosis of G-CSFR
Previously, we reported that internalization of the G-CSFR is guided by two adjacent internalization motifs, one of them containing a serine-type dileucine motif (Ward et al, 1999c; Aarts et al, 2004). To determine a possible role of cytoplasmic lysine residues in endocytosis and routing, we first compared internalization kinetics of wild-type (wt) G-CSFR and mutant K5R, in which cytoplasmic lysines had been replaced by arginines, in 32D cells. As shown in Figure 1A, the rates of G-CSF-induced internalization of wt and K5R G-CSFR were similar. In addition, the amount of cell surface receptors did not substantially increase within 2 h after G-CSF withdrawal (Figure 1B). Moreover, no colocalization was found between internalized anti-G-CSFR antibodies and a marker for recycling endosomes (TRITC-conjugated transferrin) from 30 min post-internalization onwards (data not shown). These results establish that cytoplasmic lysines does not contribute significantly to ligand-induced G-CSFR endocytosis and that internalized wt or K5R G-CSFR do not massively recycle to the plasma membrane.
Figure 1.
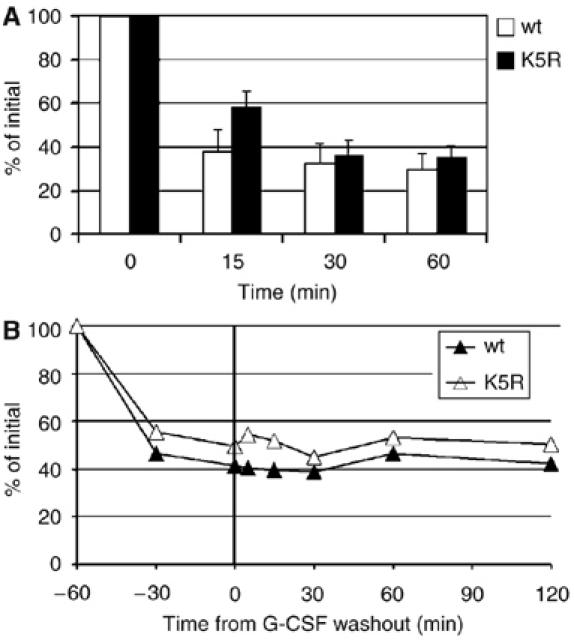
Cytoplasmic lysines of G-CSFR are not involved in ligand-induced receptor internalization. (A) Flow cytometric analysis of internalization kinetics of G-CSFR in 32D clones expressing wt G-CSFR (open bars) or mutant K5R (filled bars). G-CSFR cell surface expression was assessed at various time points after addition of G-CSF. Data represent the mean percentages of fluorescence compared to the fluorescence at t=0 min from three different clones. (B) G-CSFR wt and K5R are not rapidly recycled to the plasma membrane. G-CSF was added at t=−60 min and washed out at t=0. Cells were then resuspended in the presence of cycloheximide and analyzed for cell surface expression of G-CSFR as outlined in (A). The mean fluorescence at t=−60 min was set at 100%. This result is representative of three experiments with three independent 32D clones.
Lysosomal routing of G-CSFR depends on cytoplasmic lysines
To examine the post-endocytic fate of the internalized G-CSFR, nonactivating antibodies directed against an extracellular epitope of G-CSFR were used to track receptor localization by confocal laser scanning microscopy (CLSM) (Aarts et al, 2004). HeLa cells transfected with wt G-CSFR were allowed to bind G-CSFR antibodies and G-CSF for 20 min at 16°C, washed and then transferred to 37°C to permit internalization. Fifteen minutes after transfer, the internalized G-CSFR resided predominantly in EEA1-positive early endosomes (Figure 2A, upper panel). From 30 min onwards, the G-CSFR accumulated in Rab7-positive late endosome/lysosomes, which was complete after 2 h (Figure 2A, lower panel). These results show that the G-CSFR follows the endosomal–lysosomal degradation route.
Figure 2.
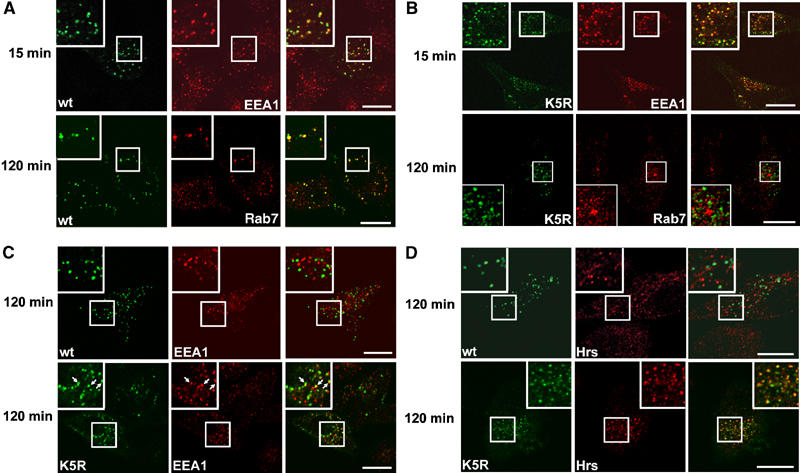
Role of G-CSFR lysines in lysosomal routing. HeLa cells ectopically expressing wt or K5R G-CSFR were incubated with G-CSFR antibodies and G-CSF for 20 min at 16°C, washed and transferred to 37°C for 15 or 120 min as indicated. Subsequently, cells were fixed and stained for internalized G-CSFR (green, left panels) and organelle markers EEA1, Rab7 or Hrs (red, middle panels) and analyzed by CLSM. Merged pictures are shown in the right panels, indicating colocalization in yellow. Insets show enlargements of the boxed areas. (A) Presence of wt G-CSFR in EEA1-positive early endosomes at 15 min and in Rab7-positive late endosomes/lysosomes at 120 min after internalization. (B) K5R is present in EEA1-positive endosomes at 15 min, but is not detectable in Rab7-positive endosomes/lysosomes at 120 min after internalization. (C) Presence of K5R (arrows) but not wt G-CSFR in EEA1-positive endosomes at 120 min after internalization. (D) Presence of K5R but not wt G-CSFR in Hrs-positive endosomes 120 min after internalization. Scale bar, 10 μm.
We then asked whether replacement of the cytoplasmic lysines affected intracellular trafficking of the G-CSFR. Similar to the wt G-CSFR, mutant K5R accumulated in EEA1-positive early endosomes 15 min after induction of internalization (Figure 2B, upper panel). However, contrary to the wt G-CSFR, no transfer of K5R to Rab7-positive late endosomes had occurred after 2 h (Figure 2B, lower panel). Instead, a fraction of K5R was retained in EEA1-positive early endosomes (Figure 2C, lower panel, arrows), which was not the case for wt G-CSFR (Figure 2C, upper panel). Even 4 h after internalization, mutant K5R was still found in early endosomes, indicating a block or severe delay in lysosomal routing (Supplementary Figure 1). A large fraction of K5R colocalized with Hrs (Figure 2D, lower panel), a marker for prelysosomal endosomes. In contrast, wt G-CSFR colocalized with Hrs at 30 min (Supplementary Figure 2), but not at 2 h post-internalization (Figure 2D, upper panel). Together, these data show that internalized G-CSFR route to an Hrs-positive endosomal compartment via a lysine independent mechanism. Subsequently, one or more lysines in the cytoplasmic tail are required for routing to lysosomes. Colocalization of the wt G-CSFR or mutant K5R with caveolin 1 was not observed (data not shown), indicating that G-CSFR internalization via the raft/caveolar endocytotic pathway does not occur under these conditions.
As cell context may potentially influence routing mechanisms, we validated the results obtained in HeLa cells in myeloid 32D cells. Because Rab7 immunoreactivity is low in these cells, antibodies against lysosome-associated membrane protein-1 (LAMP-1) were used instead to mark the lysosomal compartment (Granger et al, 1990). Kinetics of lysosomal routing of G-CSFR in 32D cells were similar to those in HeLa cells: 1 h after receptor activation, most wt G-CSFR accumulated in lysosomes (Figure 3, upper panel), whereas K5R did not (Figure 3, lower panel).
Figure 3.
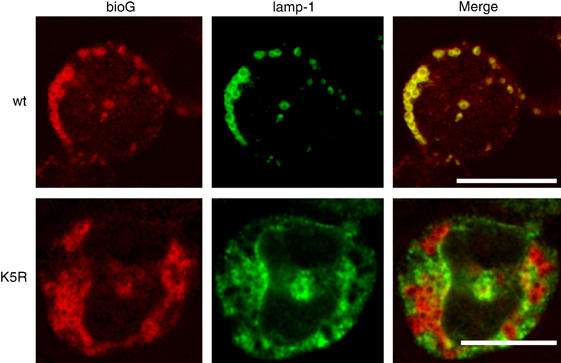
Intracellular localization of K5R G-CSFR in myeloid 32D cells. 32D clones expressing wt or K5R G-CSFR were allowed to bind biotinylated G-CSF (bio-G) for 60 min at 4°C, washed and transferred to 37°C for an additional 60 min. Subsequently, cells were spun on glass slides, fixed, immunostained for bio-G and LAMP-1 and analyzed by CLSM. Scale bar, 10 μm.
Differential involvement of internalization motifs and cytoplasmic lysines in attenuation of G-CSF signaling
To compare the effects of receptor internalization and lysosomal routing on G-CSF signaling, we studied the kinetics of signaling off-switch from wt G-CSFR, mutant K5R and a mutant lacking the internalization domain (d749–769). 32D and Ba/F3 clones with comparable expression levels of wt, d749–769 or K5R G-CSFR were used (Figure 4A and data not shown). First, we assessed the kinetics of downregulation of G-CSF-induced STAT5 activity in 32D cells by electrophoretic mobility shift assay (EMSA) following G-CSF washout after 10 min of stimulation (Figure 4B). Compared with wt G-CSFR, both d749–769 and K5R G-CSFR showed a significant delay in the attenuation of STAT5 activity (Figure 4B). Western blot analysis with phosphospecific antibodies against signaling substrates showed that phosphorylation of STAT3, STAT5 and ERK induced by mutants K5R and d749–769 is prolonged compared with wt G-CSFR after G-CSF washout (Figure 4C). In contrast, PKB activitation was prolonged after activation of d749–769, but normally attenuated after activation of mutant K5R (Figure 4C). These results indicate that PKB activation is abrogated immediately upon receptor internalization, whereas activation of STAT3, STAT5 and Erk continues upon routing of the G-CSFR to early endosomes. The latter experiments were performed in Ba/F3 cells instead of 32D cells, because 32D cells were less optimal for Western blot analysis, probably due to higher levels of protease activity. A quantification of these data is presented in Supplementary Table 1.
Figure 4.
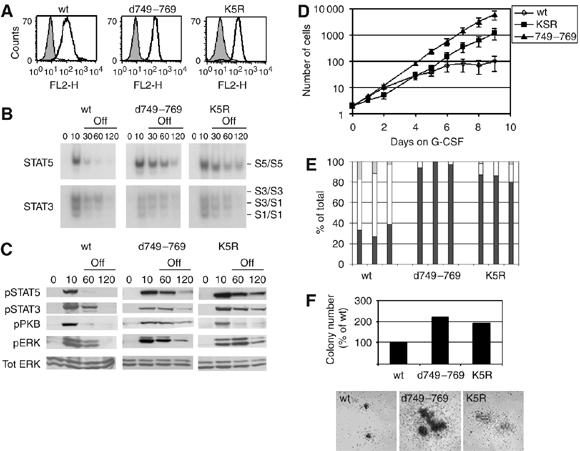
Role of internalization motif and cytoplasmic lysines in G-CSF signal attenuation and myeloid differentiation. (A) Flow cytometric analysis of membrane expression of wt, d749–769 or K5R G-CSFR in 32D cells. Bold histograms: cells stained with anti-G-CSFR and GAM-PE; shaded histograms: cells stained with GAM-PE only. (B) 32D cells expressing wt or mutant G-CSFR were deprived of IL-3 and serum for 4 h, incubated with G-CSF for 10 min at 37°C, washed and kept at 37°C in medium without factors. Nuclear extracts were assayed at the indicated time points by EMSA for activated STAT5 using β-casein oligonucleotides (C) Ba/F3 cells expressing wt or mutant G-CSFR were treated as described in (B) and lysates immunoblotted and stained with anti-pSTAT5, anti-pSTAT3, anti-pPKB, anti-pERK and restained with anti-ERK to verify equal loading. Results are representative of three independent experiments in different Ba/F3 clones. (D) Proliferation of 32D.cl8.6 cells expressing wt or mutant G-CSFR during 9 days of culture in the presence of G-CSF. Numbers of viable cells were determined at the indicated times. Data represent mean values±s.e.m. of three clones per construct. (E) Differential counts of cells taken from cultures shown in panel (D). On day 8, three clones per construct were analyzed and scored for percentages of blast cells (black bars), band forms (gray bars) or mature neutrophils (white bars). (F) G-CSF-induced colony formation by Csfr3−/− primary bone marrow cells after retroviral transduction of wt or mutant G-CSFR constructs. Upper panel: mean colony numbers per 25 000 infected bone marrow cells from triplicate colony assays. Lower panel: photomicrograph of representative colonies showing differences in colony size. Data are representative of two independent experiments. No G-CSF-induced colonies were observed with Csfr3−/− bone marrow cells transduced with empty vector.
Subsequently, we assessed the consequences of removal of the internalization domain and substitution of the lysine residues for G-CSF-induced proliferation and differentiation. As demonstrated previously, 32D cells expressing wt G-CSFR proliferated 6–8 days and differentiated into neutrophilic granulocytes from day 6 onwards (Figure 4D and E) (Ward et al, 1999a). In contrast, 32D cells expressing d749–769 or K5R continued with proliferation in response to G-CSF at the expense of terminal granulocytic differentiation (Figure 4D and E). Colony assays with G-CSFR-deficient (Csf3r−/−) bone marrow cells, retrovirally transduced with wt, d749–769 or K5R G-CSFR and cultured in G-CSF-supplemented semi-solid medium, were then performed. Both the numbers and size of G-CSF-induced colonies formed by d749–769- or K5R G-CSFR-expressing bone marrow cells exceeded those of wt G-CSFR transduced cells (Figure 4F), indicating that both defective internalization and lysosomal routing result in a hyperproliferative response of primary hematopoietic progenitors to G-CSF. Notably, the internalization defect had a more pronounced impact on proliferation signaling than the lysosomal routing defect, which might relate to the fact that G-CSF-induced PKB activation by the internalization defective mutant is sustained, whereas it is downregulated with normal kinetics after activation of mutant K5R (Figure 4C and Supplementary Table 1) (Dong and Larner, 2000).
G-CSFR-K632 is the most critical lysine for lysosomal routing and attenuation of G-CSF signaling
To determine which of the five cytoplasmic lysines of G-CSFR are involved in lysosomal routing and termination of signaling, we constructed single lysine substitution mutants. Mutant K632R failed to accumulate in Rab7-positive lysosomes 2 h after internalization, whereas a significant proportion of mutants K672R, K681R, K682R and K762R colocalized with Rab7 at this time point (Figure 5A and Supplementary Figure 3). EMSAs on stable 32D clones expressing either one of these mutants showed that only mutation of G-CSFR-K632 resulted in prolonged STAT activation following ligand washout (Figure 6A, upper panel). To further substantiate this result, we made G-CSFR constructs with single lysine residues added back into K5R (mKA to mKE). Only the add-back of K632 (mKA) restored the attenuation of STAT activation at a rate comparable to wt G-CSFR, whereas adding back each of the other lysines had little or no effect (Figure 6A, lower panel). In G-CSF-containing medium, 32D/K632R cells showed sustained and enhanced proliferation in response to G-CSF, whereas clones expressing the other lysine substitution mutants were arrested in proliferation and differentiated comparably to 32D/wt cells (Figure 6B). Together, these results establish that G-CSFR-K632 plays a key role in lysosomal receptor degradation, termination of G-CSF-induced STAT activation and inhibition of proliferation signaling.
Figure 5.
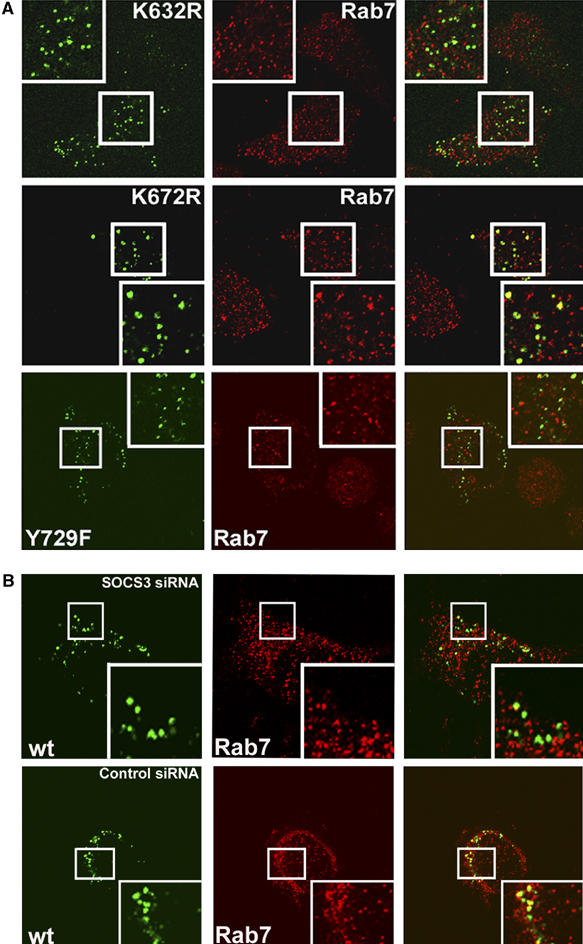
Intracellular localization of G-CSFR mutants. (A) HeLa cells were transfected with G-CSFR mutants K632R, K672R and Y729F and examined for localization in Rab7-positive endosomes or lysosomes after 120 min as detailed in Figure 2. (B) HeLa cells were cotransfected with wt G-CSFR and 30 nM SOCS3 siRNA (upper panel) or 30 nM control siRNA (lower panel), and analyzed as outlined above.
Figure 6.
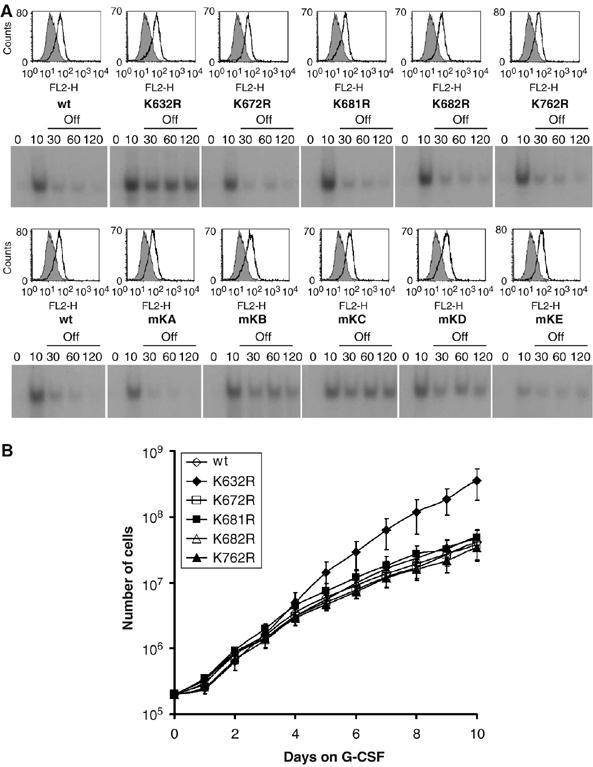
Involvement of G-CSFR K632 in attenuation of G-CSF-induced STAT5 activation and cell proliferation. (A) STAT5 EMSA on 32D cells expressing wt G-CSFR or single K to R substitution mutants (upper panel) or single add-back mutants (lower panel). Cells were deprived of IL-3 and serum, incubated for 10 min at 37°C with 100 ng/ml G-CSF, washed to remove G-CSF and subjected to EMSA. FACS histograms indicate expression levels of different mutants (see Figure 4B for explanation). (B) Proliferation of 32D cells expressing wt or single K to R substitution mutants cultured in the presence of G-CSF. Numbers of viable cells were determined at the indicated time points. Data represent mean values±s.e.m. of three independent clones.
G-CSFR is ubiquitinated on K632
To investigate whether G-CSFR lysines are acceptors for ubiquitination, we transfected cells with hemagglutinin (HA)-tagged G-CSFR constructs and FLAG-tagged Ub and subjected these to HA immunoprecipitation and FLAG Western blot analysis. G-CSFR immunoprecipitates showed FLAG-immunoreactive bands with estimated molecular sizes between 140 and 170 kDa (Figure 7A). In contrast, no significant FLAG immunoreactivity was detected in immunoprecipitates from cells transfected with K5R, indicating that ubiquitination is receptor lysine specific. Mutant K5R was significantly more abundant than G-CSFR wt in cell lysates, as was expected based on its reduced lysosomal routing. However, because only a minor fraction (approximately 5%) of the total G-CSFR protein is present on the plasma membrane (Aarts et al, 2004; Harada et al, 2005), these results may reflect predominantly intracellular G-CSFR. To examine specifically ubiquitination of G-CSFR expressed on the cell membrane, cells were incubated with biotinylated G-CSF (bio-G) and ligand-bound G-CSFR was precipitated using streptavidin-coated magnetic beads. FLAG immunoreactivity was observed with wt G-CSFR but not with K5R (Figure 7B). In addition, substantial FLAG immunoreactivity of mKA was seen, indicating that G-CSFR-K632 is subject to ubiquitination. Notably, substitution mutant K632R is still robustly ubiquitinated, indicating that other lysines in G-CSFR also serve as substrates for E3 ligases (Supplementary Figure 4). Additional experiments with single add-back mutants indicated that K672, K681, K682 and to a lesser extent K762 are ubiquitinated (data not shown). Having established that these lysines are not critical for lysosomal routing in the endocytotic pathway, they may possibly be involved in other functions, such as forward routing and protein stability, as recently proposed (Erkeland et al, 2006). The E3 ligase involved in ubiquitination of these lysines is not SOCS3, as is evident from the observation that G-CSFR mutant Y729F, lacking the SOCS3 docking site (Hortner et al, 2002; van de Geijn et al, 2004a) is ubiquitinated (Supplementary Figure 4).
Figure 7.
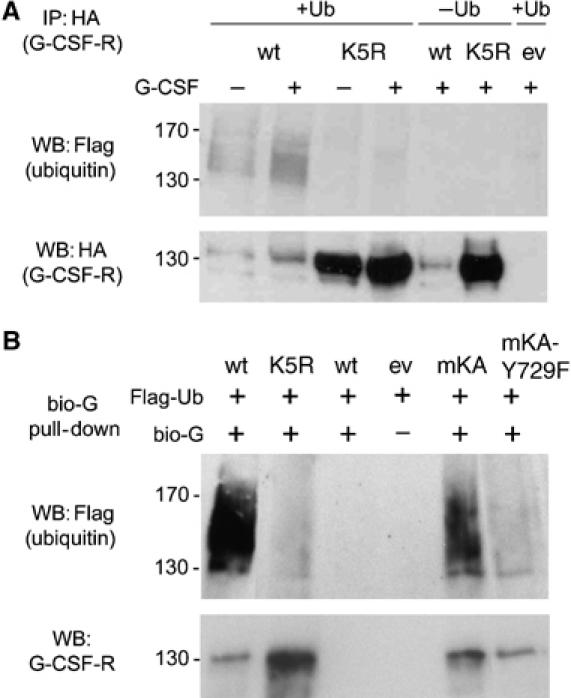
Ubiquitination of G-CSFR. (A) Phoenix E cells cotransfected with HA-tagged wt or K5R G-CSFR and FLAG-tagged Ub were incubated with (+) or without (−) G-CSF for 30 min, and HA immunoprecipitates were analyzed by Western blotting using anti FLAG (upper panel) or anti-HA antibodies (lower panel). (B) A similar setup as in (A), but instead of HA immunoprecipitation, cells were incubated with bio-G for 20 min at 16°C and for 30 min at 37°C and subsequently G-CSFR was pulled down with streptavidin-coated magnetic beads.
Involvement of SOCS3 in ubiquitination and lysosomal routing of G-CSFR
Previously, it was shown that the inhibitory action of SOCS3 in G-CSF-induced STAT5 activation to a major extent depends on the SOCS-box (van de Geijn et al, 2004b). This raises the question whether SOCS3 may be directly involved in the ubiquitination of G-CSFR, specifically on K632. We addressed this possibility using a G-CSFR mutant that lacks the SOCS3 recruitment site Y729. Substitution of Y729 by phenylalanine resulted in a significantly reduced ubiquitination of G-CSFR-K632 (compare mKA to mKA/Y729F; Figure 7B). This led us to examine the possible involvement of SOCS3 in the lysosomal routing of G-CSFR. Two hours after internalization, no substantial colocalization of G-CSFR mutant Y729F with Rab7 was seen (Figure 5A), indicating that loss of SOCS3 recruitment to Y729 prevents lysosomal routing of G-CSFR. To further substantiate the role of SOCS3 in the lysosomal routing of G-CSFR, we inhibited its expression using siRNA in wt G-CSFR-expressing cells. In these experiments, the efficacy of transfection, as detected by siGlo Cyclophilin B siRNA, exceeded 90% (data not shown). As expected, SOCS3-siRNA but not control siRNA significantly reduced colocalization of G-CSFR with Rab7 2 h after G-CSF-induced internalization (Figure 5B). Protein stability assays in which lysosomal activity is inhibited by NH4CL-mediated deacidification confirmed that lysosomes play a major role in G-CSFR degradation (Supplementary Figure 5).
G-CSFR-K632 is the most critical lysine for the inhibitory effects of SOCS3 on G-CSF-induced STAT5 activation
SOCS3 causes a dose-dependent reduction of G-CSF-induced STAT5 activity from the wt G-CSFR in luciferase reporter assays (van de Geijn et al, 2004b). Using this assay, we asked whether SOCS3-mediated inhibition of G-CSF signaling directly depends on the cytoplasmic lysines of the G-CSFR. As demonstrated previously, inhibition of STAT5 activation was lost completely after mutation of Y729 of the G-CSFR (mutant Y729F) (Figure 8A). Strikingly, mutant K5R was also hardly inhibited by SOCS3, indicating that lysines of the G-CSFR are crucial for SOCS3-controlled attenuation of STAT5 activation (Figure 8A). In contrast, adding back K632 (mKA) fully restored sensitivity to SOCS3 (Figure 8A). These inhibitory effects of SOCS3 on wt and mKA G-CSFR were lost upon removal of the SOCS box, consistent with the role of the SOCS box domain in recruiting ubiquitin ligase activity (Figure 8B). Individual add-backs of the lysines K672 (mKB), K681 (mKC) or K682 (mKD) restored SOCS3 responses only at the highest concentrations, whereas adding back K762 (mKE) had even less effect (Figure 8C). These results identify the juxtamembrane lysine K632 as the most critical target within the G-CSFR for SOCS3-mediated termination of STAT5 activation. In contrast to SOCS3, the inhibitory effect of SOCS1 on STAT5 activation was not abrogated by mutation of Y729 of G-CSFR, supporting the notion that recruitment to G-CSFR Y729 is required for SOCS3, but not SOCS1 to inhibit STAT5 activation (Figure 8D). This result complies with a study showing that phosphorylated Y729-containing peptides have high affinity for the SH2 domain of SOCS3, but not SOCS1 (Hortner et al, 2002), but is in apparent conflict with a study showing some Y729-dependent inhibitory effects of SOCS1 on STAT5 activation (Zhuang et al, 2005). However, also in this latter study, the authors failed to demonstrate a physical interaction between G-CSFR-Y729 and SOCS1. Notably, the inhibitory effects of SOCS1 were not affected by mutation of K632 or other lysines in the G-CSFR cytoplasmic domain (Figure 8D), further arguing against a direct effect of SOCS1 on G-CSFR routing. Two recent studies have shown that SOCS2 interacts with and degrades SOCS3, thereby playing a unique role in cytokine signaling (Tannahill et al, 2005; Piessevaux et al, 2006). We therefore investigated the effects of SOCS2 on STAT5 activation by wt G-CSFR and mutants, but found that SOCS2 had no effect on G-CSF-induced STAT5 activation (Figure 8E) and did not neutralize the inhibitory effects of ectopically expressed SOCS3 (data not shown).
Figure 8.
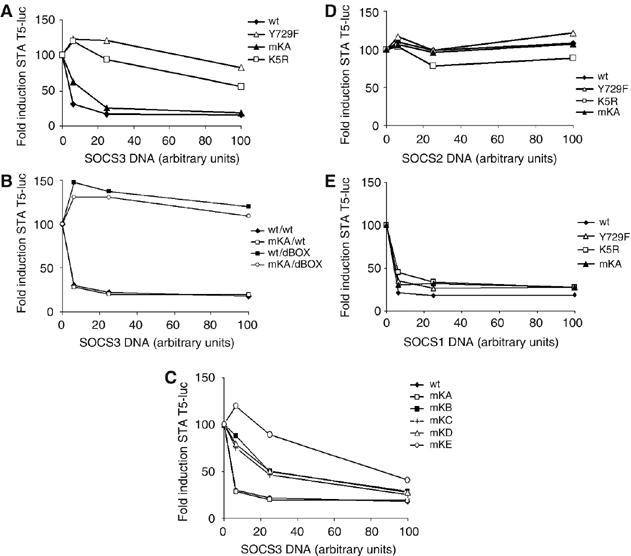
Role of lysines of G-CSFR in SOCS3-mediated inhibition of STAT5 activation. HEK293 cells were transfected with the indicated G-CSFR constructs, increasing amounts of SOCS3 (A, C), SOCS3 and SOCS3dBox (B), SOCS 2 (D) and SOCS1 (E) and a STAT5-responsive luciferase construct. SOCS protein levels were determined by Western blotting, of which a representative analysis is shown in Supplementary Figure 6. For each G-CSFR construct, G-CSF-induced luciferase reporter activity in the absence of SOCS was set at 100%. Data are representative of three independent experiments each performed in triplicate.
Discussion
Role of receptor lysine K632 in G-CSFR routing and control of G-CSF signaling
In this study, we have shown that G-CSFR routing from early endosomes to lysosomes depends on the integrity of a single lysine, G-CSFR-K632, in the juxtamembrane cytoplasmic region and that maintaining an appropriate balance of G-CSF-induced signals largely depends on this mechanism. In contrast, G-CSFR internalization was not affected by substitution of any of the five conserved cytoplasmic lysines for arginines. This result differs from a recent study on the related LR, which showed that two lysines in the cytoplasmic domain are critical for internalization (Belouzard and Rouille, 2006). The reason for this discrepancy might relate to the fact that the LR lacks tyrosine- or dileucine-based endocytosis motifs found in other class I cytokine receptor chains such as G-CSFR and gp130, and instead carries a Ub-based novel internalization motif (Belouzard and Rouille, 2006). However, it remains possible that the Ub conjugation system controls G-CSFR endocytosis via an indirect mechanism, as has been demonstrated for GHR (Govers et al, 1999).
An important question is which effector protein(s) linked to K632 is responsible for lysosomal routing of G-CSFR. We considered the UIM-containing protein Hrs as the most plausible candidate, as it has been shown to form a multivalent complex, including other UIM-containing proteins such as STAM1/STAM2 and Eps15, that guides endocytosed membrane proteins to lysosomes (Bache et al, 2003). However, we were unable to demonstrate association of Hrs to G-CSFR in immunoprecipitation assays. Furthermore, siRNA-mediated knockdown of Hrs did not alter off-rate kinetics of STAT5 activation as seen with G-CSFR mutants lacking K632 (data not shown). It thus remains to be determined which proteins or protein complexes are crucial for ligand-induced lysosomal routing of G-CSFR and attenuation of STAT5 activation.
Consequences of G-CSFR routing for G-CSF signaling
Another major objective was to determine how intracellular trafficking impacts on G-CSF signaling. From the off-rate experiments with G-CSFR mutant K5R, which internalizes normally but fails to route to late endosomes and lysosomes, it can be deduced that activation of STAT5 and ERK continues in early endosomes. In contrast, PKB activation by K5R was terminated following G-CSF washout with kinetics similar to wt G-CSFR, whereas it was sustained with internalization-defective G-CSFR mutant d749–769 (Figure 4C). These findings suggest that distinct mechanisms of signal attenuation are involved in the termination of PKB versus STAT5 and ERK activation, depending on the intracellular localization of G-CSFR. Although this needs further study, several potential mechanisms can be envisaged. For instance, as demonstrated for the insulin receptor and the tyrosine phosphatase PTP1B (Romsicki et al, 2004), an endosome-associated phosphatase activity might be involved that affects PKB but not STAT5 and ERK. Alternatively, divergence in intracellular routing of signaling intermediates, for example, through caveolin-mediated internalization, may cause dissociation of the PI-3K/PKB pathway from the G-CSFR complex in early clathrin-coated endosome, as was recently shown for the abrogation of PI-3K/PKB signaling after internalization of integrin receptors (del Pozo et al, 2005).
SOCS3 as a key regulator of G-CSFR routing and STAT5 activation
Together with previous reports, our present data support a model in which SOCS3, recruited to the phosphorylated Y729 of G-CSFR following receptor activation, inhibits G-CSF signaling by two mechanisms (Figure 9). The first mechanism engages the kinase inhibitory region (KIR) of SOCS3 to inhibit JAK kinase activity and has been documented for a number of receptors (Kile and Alexander, 2001). The second, new mechanism supported by our data involves ubiquitination of the G-CSFR protein on a membrane proximal lysine, which triggers routing of the G-CSFR from an Hrs-positive compartment to lysosomes. Although functionally distinct, these mechanisms do not necessarily have to act independently. For instance, the unidentified protein(s) recruited to Ub-K632 and involved in lysosomal routing may be negatively controlled by JAK activity, which would imply that the KIR of SOCS3 indirectly also controls lysosomal routing of G-CSFR. Conversely, the SOCS box of SOCS3 may also be involved in degradation of JAK proteins (Kile and Alexander, 2001), which in turn could have an effect on routing of G-CSFR.
Figure 9.
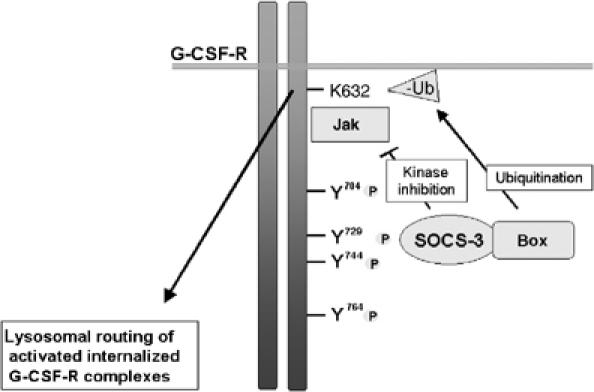
Model of SOCS3-controlled termination of G-CSF signaling. Following activation by G-CSF, G-CSFR recruits SOCS3 via phosphorylated tyrosine residue Y729. SOCS3 inhibits JAK activity via its kinase inhibitory region to inactivation of G-CSFR. This may also be achieved in part by direct recruitment of SOCS3 to JAK itself (Kile and Alexander, 2001). A second and novel mechanism involves recruitment of E3 ligase activity, leading to ubiquitination of K632 of G-CSFR. This triggers G-CSFR transition from Hrs-positive endosomes to lysosomes, attenuating activation of STAT5.
The K632 of G-CSFR resides at position –5 relative to the box 1 domain (PGIPSP). Intriguingly, this lysine is conserved among several other class I cytokine receptors, for example, leptin receptor, gp130, IL-4R and IL-7R. This might be suggestive of a general role of this conserved juxtamembrane lysine and merits investigations aimed at determining whether control of receptor routing by members of the SOCS protein family may be a more common theme in cytokine receptor signaling.
Materials and methods
Antibodies
Mouse-anti-human CD114 (G-CSF receptor) and MoAb anti-EEA-1 were purchased from Becton-Dickinson/PharMingen (San Diego, CA). Rat anti-Lamp-1 (clone 1D4B) was obtained from Research Diagnostics (Flanders, NJ). Mouse anti-HA MoAb, goat anti-EEA-1, rabbit anti-Rab7, and rabbit anti-STAT5a and anti-STAT5b were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). MoAb anti-pSTAT5 (STAT5A-pY694, STAT5B-pY699) was from Upstate Biotechnology Inc. (Charlottesville, VA) and anti-pSTAT3 (pY705) from Cell Signaling Technology Inc. (Danvers, MA). Texas Red-conjugated streptavidin (Caltag Laboratories, Burlingame, CA) was used for detection of biotinylated G-CSF (bio-G). MoAbs against pPKB, pERK and total ERK were from Santa Cruz. FLAG M2 MoAb was purchased from Sigma (St Louis, MO). Rabbit anti-Hrs was a gift from Dr H Stenmark (Department of Biochemistry, Institute for Cancer Research, The Norwegian Radium Hospital and The University of Oslo, Oslo, Norway). Dye-coupled immunoglobulin (Ig) antibodies used in CLSM were purchased from Molecular Probes (Invitrogen, Breda, The Netherlands): donkey anti-mouse Alexa Fluor 488 (for G-CSFR), goat anti-rabbit Alexa Fluor 546 (for Rab7 and Hrs), rabbit anti-goat Alexa Fluor 546 (for EEA1) and goat anti-rat Alexa-Fluor 488 (Lamp-1). Goat anti-mouse Ig coupled to phycoerythrin (GAM-PE) used in flow cytometry was from Dako (Dako BV, Heverlee, Belgium).
Expression constructs
The pBabe expression constructs of wt G-CSFR, d749–769 and Y729F have been described previously (Ward et al, 1999b; Aarts et al, 2004). The K632R, K672R, K681R, K682R, K762R and K5R G-CSFR constructs were made in pLNCX containing wt G-CSFR using the Stratagene QuickChange site directed mutagenesis kit (Stratagene, La Jolla, CA). The single lysine add-back mutants were made in a similar way using K5R as template. C-terminal double HA-tagged versions of wt and mutant G-CSFR were created by PCR using a 5′ oligonucleotide spanning a region in the cDNA corresponding to amino acids 590–595 (GRFR7) and a 3′ oligonucleotide containing a segment spanning the amino acids immediately upstream of the stop codon of the G-CSFR, a double HA tag and a restriction site. All mutant constructs were verified by sequencing. Expression vectors for SOCS1, SOCS2, SOCS3 and SOCS3Δbox and reporter constructs have been described previously (van de Geijn et al, 2004b). FLAG-tagged Ub in pCDNA3 was a gift from Dr B van der Reijden (Nijmegen, The Netherlands).
Cell culture, transfection, retroviral transduction and introduction of siRNA
Ba/F3 cells and 32D.cl8.6 cells, a subline of the IL-3-dependent murine myeloid 32Dcl3 cell line that lacks endogenous G-CSFR (de Koning et al, 1998), were maintained in RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), 100 IU/ml penicillin, 100 μg/ml streptomycin and murine IL-3 (10 ng/ml) at 37°C and 5% CO2. To obtain Ba/F3 cells expressing wt or mutant G-CSFR, cells were electroporated as described (de Koning et al, 1996). Subsequently, cells were diluted and expanded in 96-well plates in IL-3-containing medium supplemented with puromycin (1 μg/ml) to obtain individual clones. 32D.cl8.6 clones expressing different G-CSFR constructs were obtained by retroviral transduction as described (Aarts et al, 2004). HeLa and Phoenix E cells were cultured in Dulbecco's modified Eagle's medium (DMEM, glucose 4.5 g/l) supplemented with 10% FCS, 100 IU/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere at 37°C and 7% CO2. HeLa cells were plated on glass coverslips and transfected with G-CSFR constructs using lipofectamin (Invitrogen). Phoenix E cells were transfected using the calcium phosphate precipitation method. To reduce SOCS3 expression, HeLa cells (2 × 105) were transfected using an siRNA transfection kit (Mirus Bio Corporation, Madison WI, USA) according to the manufacturer's instructions. The siRNAs used were SOCS3 human ON-TARGET plus siRNA SMARTpool (Dunn et al, 2005), siCONTROL non-targeting siRNA #1 and siGLO cyclophylin B siRNA, all obtained from Dharmacon, Chicago, IL, USA.
Immunoprecipitation and Western blotting
Phoenix E cells (G Nolan, Stanford, CA) were transfected with HA-tagged wt or mutant-G-CSFR and FLAG-tagged Ub using the calcium phosphate precipitation method. After 48 h, cells were stimulated with G-CSF (100 ng/ml) for 30 min or left untreated, washed with cold PBS and resuspended in lysis buffer containing 20 mM Tris–HCl pH 8.0, 137 mM NaCl, 10 mM EDTA, 100 mM NaF, 1% NP-40, 10% glycerol, 2 mM Na3VO4 and 1 mM Pefablock SC, 50 μg/ml aprotinin, 50 μg/ml leupeptin, 50 μg/ml bacitracin and 50 μg/ml iodoacetamide. Immunoprecipitation was performed using anti-HA antibodies and protein G–Sepharose beads. Alternatively, G-CSFR pull-down was performed with biotinylated G-CSF (bio-G), prepared using a biotin labeling kit according to the manufacturer's instructions (Roche Molecular Biochemicals, Mannheim, Germany). Cells were incubated with bio-G (10 μg/ml) for 15 min at 16°C and for another 30 min at 37°C. Subsequently, cells were washed with cold PBS, resuspended in lysis buffer and incubated with streptavidin-conjugated magnetic beads (Dynabeads, Invitrogen) for 1 h at 4°C. Beads were washed and resuspended in 1 × Laemlli buffer (pH 11) and supernatants subjected to Western blotting as described (Ward et al, 1999b).
Immunocytochemistry and confocal microscopy
To visualize postendocytic receptor trafficking in situ, cells were incubated with G-CSFR antibodies (2.5 μg/ml) and G-CSF (100 ng/ml) for 20 min at 16°C, washed (PBS) and transferred to 37°C. Subsequently, cells were permeabilized by treatment for 5 min with 0.05% saponin, fixed with 3% paraformaldehyde and stained for fluorescence microscopy (Raiborg et al, 2002). To visualize receptor internalization in 32D cells, cells were incubated with bio-G for 1 h at 4°C, washed (PBS) and transferred to 37°C to induce internalization. After washing (PBS), cells were spun onto glass slides, fixed (4% paraformaldehyde and 0.05% gluteraldehyde) and stained for CLSM (Aarts et al, 2004). Coverslips were examined in the multitrack detection mode of a Zeiss LSM 510 instrument equipped with argon/HeNe lasers and a × 63 Planapochromat oil immersion objective.
Cell proliferation assay and cytology
To determine the proliferation and differentiation characteristics of the 32D.cl8.6 cells transduced with various G-CSFR constructs in response to G-CSF, cells were washed three times in Hank's balanced salt solution (HBSS) and seeded at a density of 2 × 105 cells/ml in RPMI 1640 medium, supplemented with 10% FCS and 100 ng/ml human G-CSF. Viable cells were counted daily using a CASY1 electronic cell counter (Schärfe-System, Reutlingen, Germany) and readjusted to 2 × 105 cells/ml. To analyze granulocytic differentiation, cells were spun onto glass slides and examined by light microscopy after May–Grünwald–Giemsa staining.
Bone marrow cells
Hematopoietic progenitor cells were collected from the femurs and tibiae of 8- to 12-week-old Csf3r−/− mice as described (Hermans et al, 2003). Cells were washed twice in HBSS/5% FCS/0.5% bovine serum albumin (BSA) and prestimulated for 2 days at a final concentration of 5 × 105 cells/ml in Cell Gro (SCGM BE SP047; Boehringer Ingelheim Bioproducts Partnership, Heidelberg, Germany), supplemented with a cytokine cocktail composed of murine IL-3 (10 ng/ml), human Flt3-ligand, human thrombopoietin, murine stem cell factor (100 ng/ml) and granulocyte macrophage-colony-stimulating factor (2 U/ml). Retroviral infection was performed with viral supernatant of Phoenix E cells as described (Erkeland et al, 2003).
Colony assay
Bone marrow cells were plated in triplicate at a density of 5 × 104 cells in 1 ml of methylcellulose containing medium supplemented with 30% fetal bovine serum (FBS), 1% BSA, 0.1 mM 2-mercaptoethanol, 2 mM L-glutamine and G-CSF (20 U/ml) as described (Hermans et al, 2003). Colonies consisting of more than 50 cells were counted on day 7 of culture.
Flow cytometric analysis of G-CSFR
G-CSFR-expressing 32D.cl8.6 transfectants (0.5 × 106 cells) were incubated at 4°C for 1 h with G-CSFR antibody in PSA consisting of PBS supplemented with 1% FCS and 0.02% NaN3 to block endocytosis. After washing, cells were incubated with GAM-PE for 1 h at 4°C, washed and analyzed on a FACS Calibur flow cytometer (Becton Dickinson). For internalization experiments, 32D cells (0.5 × 106 cells per time point) were incubated with anti-G-CSFR antibodies in the presence or absence of 100 ng/ml G-CSF for 1 h at 4°C and subsequently incubated for 0, 15, 30 or 60 min at 37°C. Subsequently, cells were washed in PSA and incubated for 1 h at 4°C with GAM-PE. To measure G-CSFR rerouting, cells incubated with G-CSF for 60 min were washed with PSA, incubated without G-CSF at 37°C for indicated times in the presence of cycloheximide (10 μg/ml), incubated with GAM-PE for 1 h at 4°C and analyzed after a final wash in PSA. Peak channel values of FITC fluorescence histograms were taken as a measure of average G-CSFR densities.
Electrophoretic mobility shift assay
32D cells were washed in HBSS and deprived of serum and factors for 4 h at 37°C in RPMI at a density of 1 × 106 cells/ml. Subsequently, cells were stimulated with G-CSF (100 ng/ml) for 10 min, washed twice with HBSS and incubated for 30, 60 or 120 min in RPMI in the absence of G-CSF. Preparation of nuclear extracts and STAT5 EMSA were carried out as described (de Koning et al, 2000).
Luciferase assay
Luciferase assays to assess the effects of increasing levels of SOCS proteins on STAT5 activity induced by wt and mutant G-CSFR were performed as described (van de Geijn et al, 2004a, 2004b). In brief, HEK293 cells were transfected with the indicated G-CSFR constructs, increasing amounts of SOCS constructs and a STAT5-responsive luciferase construct. After 2 days, cells were stimulated with G-CSF for 6 h and assayed for luciferase activity. G-CSF-induced luciferase reporter activity without transfected SOCS constructs was set at 100%. All experiments were performed in triplicate and data presented are representative of three independent experiments.
Supplementary Material
Supplementary Material
Acknowledgments
This work was supported by a grant from the Dutch Cancer Society 'KWF kankerbestrijding'. We thank Dr Marieke von Lindern for advice on the manuscript and Marijke Valkhof for technical assistance.
References
- Aarts LH, Roovers O, Ward AC, Touw IP (2004) Receptor activation and 2 distinct COOH-terminal motifs control G-CSF receptor distribution and internalization kinetics. Blood 103: 571–579 [DOI] [PubMed] [Google Scholar]
- Alves dos Santos CM, ten Broeke T, Strous GJ (2001) Growth hormone receptor ubiquitination, endocytosis, and degradation are independent of signal transduction via Janus kinase 2. J Biol Chem 276: 32635–32641 [DOI] [PubMed] [Google Scholar]
- Avalos BR, Gasson JC, Hedvat C, Quan SG, Baldwin GC, Weisbart RH, Williams RE, Golde DW, DiPersio JF (1990) Human granulocyte colony-stimulating factor: biologic activities and receptor characterization on hematopoietic cells and small cell lung cancer cell lines. Blood 75: 851–857 [PubMed] [Google Scholar]
- Bache KG, Raiborg C, Mehlum A, Stenmark H (2003) STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J Biol Chem 278: 12513–12521 [DOI] [PubMed] [Google Scholar]
- Belouzard S, Rouille Y (2006) Ubiquitylation of leptin receptor OB-Ra regulates its clathrin-mediated endocytosis. EMBO J 25: 932–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard F, Duplomb L, Wang Y, Robledo O, Kinzie E, Pitard V, Godard A, Jacques Y, Baumann H (2000) Stimulation of leukemia inhibitory factor receptor degradation by extracellular signal-regulated kinase. J Biol Chem 275: 28793–28801 [DOI] [PubMed] [Google Scholar]
- Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, Cluse LA, Sutherland KD, Hartley L, Williams E, Zhang JG, Hilton DJ, Nicola NA, Alexander WS, Roberts AW (2004) SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity 20: 153–165 [DOI] [PubMed] [Google Scholar]
- Dahlen DD, Broudy VC, Drachman JG (2003) Internalization of the thrombopoietin receptor is regulated by 2 cytoplasmic motifs. Blood 102: 102–108 [DOI] [PubMed] [Google Scholar]
- de Koning JP, Schelen AM, Dong F, van Buitenen C, Burgering BM, Bos JL, Lowenberg B, Touw IP (1996) Specific involvement of tyrosine 764 of human granulocyte colony-stimulating factor receptor in signal transduction mediated by p145/Shc/GRB2 or p90/GRB2 complexes. Blood 87: 132–140 [PubMed] [Google Scholar]
- de Koning JP, Soede-Bobok AA, Schelen AM, Smith L, van Leeuwen D, Santini V, Burgering BM, Bos JL, Lowenberg B, Touw IP (1998) Proliferation signaling and activation of Shc, p21Ras, and Myc via tyrosine 764 of human granulocyte colony-stimulating factor receptor. Blood 91: 1924–1933 [PubMed] [Google Scholar]
- de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D, Lowenberg B, Touw IP (2000) STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1). Oncogene 19: 3290–3298 [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Balasubramanian N, Alderson NB, Kiosses WB, Grande-Garcia A, Anderson RG, Schwartz MA (2005) Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat Cell Biol 7: 901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich E, Haft CR, Muys L, Heinrich PC, Graeve L (1996) A di-leucine motif and an upstream serine in the interleukin-6 (IL-6) signal transducer gp130 mediate ligand-induced endocytosis and down-regulation of the IL-6 receptor. J Biol Chem 271: 5487–5494 [DOI] [PubMed] [Google Scholar]
- Dong F, Larner AC (2000) Activation of Akt kinase by granulocyte colony-stimulating factor (G-CSF): evidence for the role of a tyrosine kinase activity distinct from the Janus kinases. Blood 95: 1656–1662 [PubMed] [Google Scholar]
- Dunn SL, Bjornholm M, Bates SH, Chen Z, Seifert M, Myers MG Jr (2005) Feedback inhibition of leptin receptor/Jak2 signaling via Tyr1138 of the leptin receptor and suppressor of cytokine signaling 3. Mol Endocrinol 19: 925–938 [DOI] [PubMed] [Google Scholar]
- Erkeland SJ, Aarts LH, Irandoust M, Roovers O, Klomp A, Valkhof M, Gits J, Eyckerman S, Tavernier J, Touw IP (2006) Novel role of WD40 and SOCS box protein-2 in steady-state distribution of granulocyte colony-stimulating factor receptor and G-CSF-controlled proliferation and differentiation signaling. Oncogene 2006, Sept. 25, PMID: 17001306 [E-pub ahead of print] [DOI] [PubMed] [Google Scholar]
- Erkeland SJ, Valkhof M, Heijmans-Antonissen C, Delwel R, Valk PJ, Hermans MH, Touw IP (2003) The gene encoding the transcriptional regulator Yin Yang 1 (YY1) is a myeloid transforming gene interfering with neutrophilic differentiation. Blood 101: 1111–1117 [DOI] [PubMed] [Google Scholar]
- Govers R, ten Broeke T, van Kerkhof P, Schwartz AL, Strous GJ (1999) Identification of a novel ubiquitin conjugation motif, required for ligand-induced internalization of the growth hormone receptor. EMBO J 18: 28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger BL, Green SA, Gabel CA, Howe CL, Mellman I, Helenius A (1990) Characterization and cloning of lgp110, a lysosomal membrane glycoprotein from mouse and rat cells. J Biol Chem 265: 12036–12043 [PubMed] [Google Scholar]
- Haglund K, Di Fiore PP, Dikic I (2003) Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci 28: 598–603 [DOI] [PubMed] [Google Scholar]
- Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, Ohtsuka M, Matsuura K, Sano M, Nishi J, Iwanaga K, Akazawa H, Kunieda T, Zhu W, Hasegawa H, Kunisada K, Nagai T, Nakaya H, Yamauchi-Takihara K, Komuro I (2005) G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak–Stat pathway in cardiomyocytes. Nat Med 11: 305–311 [DOI] [PubMed] [Google Scholar]
- Hermans MH, van de Geijn GJ, Antonissen C, Gits J, van Leeuwen D, Ward AC, Touw IP (2003) Signaling mechanisms coupled to tyrosines in the granulocyte colony-stimulating factor receptor orchestrate G-CSF-induced expansion of myeloid progenitor cells. Blood 101: 2584–2590 [DOI] [PubMed] [Google Scholar]
- Hortner M, Nielsch U, Mayr LM, Johnston JA, Heinrich PC, Haan S (2002) Suppressor of cytokine signaling-3 is recruited to the activated granulocyte-colony stimulating factor receptor and modulates its signal transduction. J Immunol 169: 1219–1227 [DOI] [PubMed] [Google Scholar]
- Hunter MG, Avalos BR (1999) Deletion of a critical internalization domain in the G-CSFR in acute myelogenous leukemia preceded by severe congenital neutropenia. Blood 93: 440–446 [PubMed] [Google Scholar]
- Kelly PA, Djiane J, Postel-Vinay MC, Edery M (1991) The prolactin/growth hormone receptor family. Endocr Rev 12: 235–251 [DOI] [PubMed] [Google Scholar]
- Kile BT, Alexander WS (2001) The suppressors of cytokine signalling (SOCS). Cell Mol Life Sci 58: 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile BT, Schulman BA, Alexander WS, Nicola NA, Martin HM, Hilton DJ (2002) The SOCS box: a tale of destruction and degradation. Trends Biochem Sci 27: 235–241 [DOI] [PubMed] [Google Scholar]
- Kimura A, Kinjyo I, Matsumura Y, Mori H, Mashima R, Harada M, Chien KR, Yasukawa H, Yoshimura A (2004) SOCS3 is a physiological negative regulator for granulopoiesis and granulocyte colony-stimulating factor receptor signaling. J Biol Chem 279: 6905–6910 [DOI] [PubMed] [Google Scholar]
- Lu JC, Piazza TM, Schuler LA (2005) Proteasomes mediate prolactin-induced receptor down-regulation and fragment generation in breast cancer cells. J Biol Chem 280: 33909–33916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MD, Yarden Y (2004) Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene 23: 2057–2070 [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Pelkmans L, Zerial M (2004) Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol 16: 400–406 [DOI] [PubMed] [Google Scholar]
- Piessevaux J, Lavens D, Montoye T, Wauman J, Catteeuw D, Vandekerckhove J, Belsham D, Peelman F, Tavernier J (2006) Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J Biol Chem 281: 32953–32966 [DOI] [PubMed] [Google Scholar]
- Polo S, Di Fiore PP (2006) Endocytosis conducts the cell signaling orchestra. Cell 124: 897–900 [DOI] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H (2002) Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol 4: 394–398 [DOI] [PubMed] [Google Scholar]
- Rausch O, Marshall CJ (1997) Tyrosine 763 of the murine granulocyte colony-stimulating factor receptor mediates Ras-dependent activation of the JNK/SAPK mitogen-activated protein kinase pathway. Mol Cell Biol 17: 1170–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romsicki Y, Reece M, Gauthier JY, Asante-Appiah E, Kennedy BP (2004) Protein tyrosine phosphatase-1B dephosphorylation of the insulin receptor occurs in a perinuclear endosome compartment in human embryonic kidney 293 cells. J Biol Chem 279: 12868–12875 [DOI] [PubMed] [Google Scholar]
- Royer Y, Staerk J, Costuleanu M, Courtoy PJ, Constantinescu SN (2005) Janus kinases affect thrombopoietin receptor cell surface localization and stability. J Biol Chem 280: 27251–27261 [DOI] [PubMed] [Google Scholar]
- Shao H, Xu X, Jing N, Tweardy DJ (2006) Unique structural determinants for Stat3 recruitment and activation by the granulocyte colony-stimulating factor receptor at phosphotyrosine ligands 704 and 744. J Immunol 176: 2933–2941 [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Elliott J, Barry AC, Hibbert L, Cacalano NA, Johnston JA (2005) SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol Cell Biol 25: 9115–9126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uotani S, Bjorbaek C, Tornoe J, Flier JS (1999) Functional properties of leptin receptor isoforms: internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes 48: 279–286 [DOI] [PubMed] [Google Scholar]
- van de Geijn GJ, Aarts LH, Erkeland SJ, Prasher JM, Touw IP (2003) Granulocyte colony-stimulating factor and its receptor in normal hematopoietic cell development and myeloid disease. Rev Physiol Biochem Pharmacol 149: 53–71 [DOI] [PubMed] [Google Scholar]
- van de Geijn GJ, Gits J, Touw IP (2004b) Distinct activities of suppressor of cytokine signaling (SOCS) proteins and involvement of the SOCS box in controlling G-CSF signaling. J Leukoc Biol 76: 237–244 [DOI] [PubMed] [Google Scholar]
- van de Geijn GJ, Gits J, Aarts LH, Heijmans-Antonissen C, Touw IP (2004a) G-CSF receptor truncations found in SCN/AML relieve SOCS3-controlled inhibition of STAT5 but leave suppression of STAT3 intact. Blood 104: 667–674 [DOI] [PubMed] [Google Scholar]
- van Kerkhof P, Strous GJ (2001) The ubiquitin–proteasome pathway regulates lysosomal degradation of the growth hormone receptor and its ligand. Biochem Soc Trans 29: 488–493 [DOI] [PubMed] [Google Scholar]
- Ward AC, Hermans MH, Smith L, van Aesch YM, Schelen AM, Antonissen C, Touw IP (1999a) Tyrosine-dependent and -independent mechanisms of STAT3 activation by the human granulocyte colony-stimulating factor (G-CSF) receptor are differentially utilized depending on G-CSF concentration. Blood 93: 113–124 [PubMed] [Google Scholar]
- Ward AC, Smith L, de Koning JP, van Aesch Y, Touw IP (1999b) Multiple signals mediate proliferation, differentiation, and survival from the granulocyte colony-stimulating factor receptor in myeloid 32D cells. J Biol Chem 274: 14956–14962 [DOI] [PubMed] [Google Scholar]
- Ward AC, van Aesch YM, Schelen AM, Touw IP (1999c) Defective internalization and sustained activation of truncated granulocyte colony-stimulating factor receptor found in severe congenital neutropenia/acute myeloid leukemia. Blood 93: 447–458 [PubMed] [Google Scholar]
- Yen CH, Yang YC, Ruscetti SK, Kirken RA, Dai RM, Li CC (2000) Involvement of the ubiquitin-proteasome pathway in the degradation of nontyrosine kinase-type cytokine receptors of IL-9, IL-2, and erythropoietin. J Immunol 165: 6372–6380 [DOI] [PubMed] [Google Scholar]
- Zhuang D, Qiu Y, Haque SJ, Dong F (2005) Tyrosine 729 of the G-CSF receptor controls the duration of receptor signaling: involvement of SOCS3 and SOCS1. J Leukoc Biol 78: 1008–1015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material