

Abstract
Tailed bacteriophages and herpesviruses load their capsids with DNA through a tunnel formed by the portal protein assembly. Here we describe the X-ray structure of the bacteriophage SPP1 portal protein in its isolated 13-subunit form and the pseudoatomic structure of a 12-subunit assembly. The first defines the DNA-interacting segments (tunnel loops) that pack tightly against each other forming the most constricted part of the tunnel; the second shows that the functional dodecameric state must induce variability in the loop positions. Structural observations together with geometrical constraints dictate that in the portal–DNA complex, the loops form an undulating belt that fits and tightly embraces the helical DNA, suggesting that DNA translocation is accompanied by a ‘mexican wave' of positional and conformational changes propagating sequentially along this belt.
Keywords: bacteriophage SPP1, DNA translocation, molecular motor, viral portal protein, X-ray crystallography
Introduction
During viral particle assembly, tailed bacteriophages and herpesviruses package their DNA into a preformed viral shell using a molecular motor. The central component of this motor is the portal protein, a circular oligomer with a central tunnel through which the DNA is translocated into the viral procapsid during the assembly process, or ejected out of the mature capsid during the host cell infection (Hendrix, 1978). Viral particle assembly starts by the formation of a procapsid with a portal protein embedded in one of the twelve five-fold icosahedral symmetric vertices of the shell (Figure 1). At a later stage, a complex composed of the viral ATPase and DNA binds to the portal vertex to form a DNA translocating molecular motor. In bacteriophage φ29, the motor can generate a force of up to 57 pN, making it one of the most powerful molecular motors discovered so far (Smith et al, 2001). Such a force is needed to pump DNA against the high internal pressure that increases as the viral DNA is encapsidated.
Figure 1.
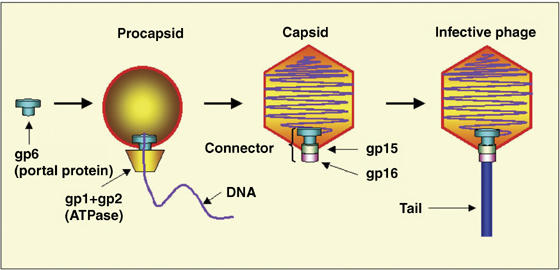
Bacteriophage SPP1 assembly. Double-stranded DNA is translocated into the procapsid through the portal protein, which together with the viral ATPase, forms a molecular motor. After termination of packaging, head completion proteins (gp15 and gp16) bind to the portal protein forming a head-to-tail connector. Tail attachment to the connector yields the infective phage particle.
Portal proteins of different phages and herpesviruses show no detectable similarity in the amino-acid sequence and exhibit large variations in their subunit molecular masses, for example, 36 kDa for phage φ29 and 57 kDa for phage SPP1 portals (Valpuesta and Carrascosa, 1994). Nevertheless, available electron microscopy (EM) data show that they all share a common turbine-like shape (Valpuesta and Carrascosa, 1994; Orlova et al, 1999; Trus et al, 2004). Although in all species, the portal protein is a central and essential component of the DNA-translocating machine, the organization of the molecular motor varies. In bacteriophage φ29, the motor consists of three coaxial macromolecular rings: the portal protein, the procapsid RNA (pRNA) and the ATPase (Simpson et al, 2000). There is no evidence for the presence of pRNA in other bacteriophages where an additional protein is normally required for DNA packaging, gp1 in the case of the Bacillus subtilis bacteriophage SPP1 (Chai et al, 1995). In contrast, a circular oligomeric ATPase (Simpson et al, 2000) appears to be a shared feature of the DNA translocating complexes, and such assemblies have been proposed for T3, T4 and lambda phages (Catalano, 2005). In bacteriophage SPP1, the molecular motor, consisting of three proteins (Figure 1)—gp1, gp2 (ATPase) and gp6 (portal protein)—powers translocation of the 45.9 kbp chromosome (Camacho et al, 2003; Oliveira et al, 2005). Different components of the DNA-translocating motor possess distinct rotational symmetries, for example, the capsid's vertex has five-fold symmetry and the B-form DNA has a 101 screw axis. In common with the herpesvirus portal protein (Trus et al, 2004), the portal protein of bacteriophage SPP1 can exist as a circular assembly with varying number of subunits: it is found as a 13-subunit assembly in its isolated form and as a 12-subunit assembly when integrated into the functional viral capsid (Orlova et al, 2003).
How exactly the molecular motor works has been the subject of much debate. The low-energy barriers to rotation of symmetry mismatching protein rings relative to each other led Hendrix (1978) to propose that DNA translocation is accompanied by rotation of the portal protein inside the capsid vertex. Different models of DNA translocation, all involving the rotation of the portal protein, were put forward following the EM image analysis of the SPP1 portal protein (Dube et al, 1993) and the determination of the X-ray structure of the φ29 portal protein (Simpson et al, 2000; Guasch et al, 2002). However, until the work presented here, the structural data did not characterize the segments defining the most constricted part of the internal tunnel (tunnel loops) that would be in close contact with the DNA during translocation. This issue is addressed by the X-ray structure of the SPP1 portal protein 13-mer and reconstruction of its 12-mer active form. The combination of structural data with geometrical and symmetry constraints imply that DNA translocation is accompanied by sequential conformational changes propagating along the belt of tunnel loops with the portal protein remaining engaged with the DNA.
Results and discussion
X-ray structure of the SPP1 portal protein 13-mer
The structure was solved by single isomorphous replacement with a mercury atom attached to C55, the only cysteine residue in the sequence. All 13 heavy atoms were located by a molecular replacement search against the isomorphous difference data using a 13-atom ring of variable radius as the search model. Analysis of omit-maps calculated after initial refinement of the structure did not reveal any significant deviations of individual subunits and their domains from the 13-fold symmetry. The final model was refined at 3.4 Å (Figure 2A and Table I). The 13 subunits of the 745 kDa portal protein are arranged around the central tunnel in a circular assembly with an overall diameter of ∼165 Å and a height of ∼110 Å (Figure 2B and C). Each subunit can be subdivided into regions that we have named clip, stem, wing and crown (Figure 2D). The clip forms the base of the portal protein and is expected to be exposed to the outside of the procapsid during viral particle assembly. This region has an α/β fold and is connected by a two-residue segment to helix α3 and by a five-residue segment to helix α5, both part of the stem. Helices α3 and α5 are tilted relative to the tunnel axis by ∼50° and ∼30°, respectively, and span the length of the stem. The stem connects the clip to the wing, which makes up the outer part of the molecule. This wing region is largely α-helical except for a distal β-sheet. Its 40-residue long helix α6 spans the subunit from the central tunnel to the peripheral rim of the wing. An extended 21-residue segment links this long helix to the crown, which consists of three α-helices connected by short turns and 40 additional C-terminal residues that are disordered in this structure.
Figure 2.
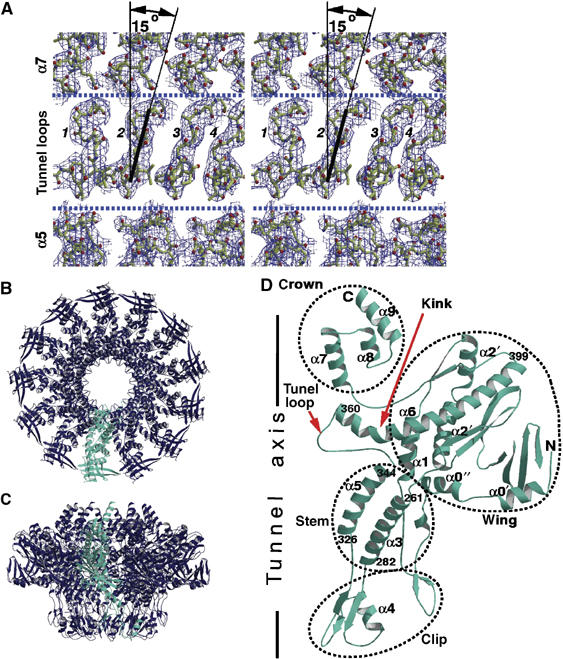
X-ray structure of the SPP1 portal protein 13-subunit assembly. (A) Averaged, weighted 2∣Fo∣−∣Fc∣ electron density maps corresponding to the tunnel loops and viewed from the tunnel axis (vertical) toward the protein surface. The area with four tunnel loops (numbered 1–4) is outlined by dashed lines to show that there is room for mobility. One of the loops is fitted with a line to show its tilt with respect to the vertical axis. (B, C) Ribbon diagrams of the portal protein along and perpendicular to the 13-fold axis. (D) Single subunit with helices numbered to match the secondary structure of the φ29 portal protein (Simpson et al, 2000). The first and last residues of helices α3, α5 and α6 are indicated.
Table 1.
Data collection and refinement statistics
| Data collection | ||
| Data set | HgCl derivative | Native |
| Wavelength (Å) | 1.0040 | 0.9326 |
| ESRF station | ID 14-4 | ID 14-2 |
| Resolution range (Å) | 40.0–3.4 | 100–4.1 |
| Number of unique reflections | 111 804 | 64 126 |
| Redundancya | 4.0 | 5.9 |
| Completeness (%) | 99.8 | 99.9 |
| Reflections with I>3σI (%) | 73.0 | 70.5 |
| Rmergeb | 0.104 | 0.110 |
| Refinement and model correlation | ||
| Number of atoms | 39 260 | |
| Number of reflections used in refinement | 110 287 | |
| R-factorc | 0.288 | |
| Number of reflections used for Rfree | 1118 | |
| Rfreec | 0.319 | |
| Average atomic B-factor (Å2) | 140.1 | |
| Deviations from ideal geometryd | ||
| Bond distance (Å) | 0.010 (0.020) | |
| Angles (deg) | 1.1 (1.9) | |
| Non-crystallographic symmetry (Å) | 0.15 (Cα) | |
| 0.25 (overall) | ||
| The average number of observations of the same reflection. | ||
| The value of the merging R-factor between equivalent measurements of the same reflection, RI=∑∣I−〈I〉∣/∑I. | ||
| Crystallographic R-factor, R(free)=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣. For calculation of Rfree, the sum was taken over randomly chosen 1% of reflections, which were excluded from the refinement. All other reflections in the range 40.0–3.4 Å were used in the refinement. | ||
| R.m.s. deviations from the standard values are given with target values within parentheses. | ||
Helix α5 is connected to α6 by a 15-residue loop (345–359) that we refer to as the tunnel loop as it protrudes into the tunnel with a seven-residue segment lining its internal surface. The body of this seven-residue segment is tilted at ∼15° to the tunnel axis as seen in Figure 2A. The loops from adjacent subunits do not make any direct hydrogen-bonding interactions with each other but make extensive van der Waals contacts that stabilize their conformation and position in the tunnel. Their tilt ensures conjugation between their positions. The belt formed by these loops defines the most constricted area of the tunnel with a diameter of 27 Å in the 13-mer (Figure 2B). Another well-defined narrowing of the tunnel, with a somewhat larger diameter, is located at the base of the portal protein in the clip. It is formed by short loops, residues 312–315, connecting two β-strands.
Three-dimensional mapping of critical mutations
The crystal structure makes it possible to rationalize the results of previous mutational studies on the SPP1 portal protein. A number of point mutations in the gene encoding the portal protein impair DNA packaging, without disrupting the assembly of the portal protein oligomer or its incorporation into the procapsid (Isidro et al, 2004a, 2004b). The corresponding residues are shown on Figure 3. Many of these line the central tunnel or are part of the tunnel loop (Figure 3). Impairment of DNA packaging caused by single amino-acid substitutions V347A, V347 M, S350P and G360V in the tunnel loop (Isidro et al, 2004a) suggests that the structural organization of this loop is essential for DNA translocation. Mutations of residues D318, E334 and E352 suggest that the negative charge of the tunnel is important for function. Interestingly, the E352G mutation in the tunnel loop reduces the efficiency of DNA translocation into capsids (Isidro et al, 2004b). At the same time, this substitution reduces ATP hydrolysis by the packaging motor (Oliveira et al, 2006), indicating a crosstalk between the tunnel loop and the viral ATPase. Further support for the portal-ATPase crosstalk hypothesis is provided by the single amino-acid substitution T319A. This mutation results in a poor stimulation of the ATPase activity and in diminished DNA packaging (Oliveira et al, 2006), suggesting that T319 is located within a path of signal/force transmission between the tunnel loop/helix-5 and the ATPase. A number of other mutations in the same area as T319 indicate residues (yellow in Figure 3) that may play similar roles. A cluster of mutations at the base of the portal protein, in the clip (pink in Figure 3), that also impair DNA packaging are likely to disrupt interactions of the portal protein with the ATPase during DNA translocation and with gp15 after termination of packaging (Lurz et al, 2001; Orlova et al, 2003). Finally, several mutations in the crown (green in Figure 3) indicate importance of a proper structural organization of this area for DNA translocation.
Figure 3.
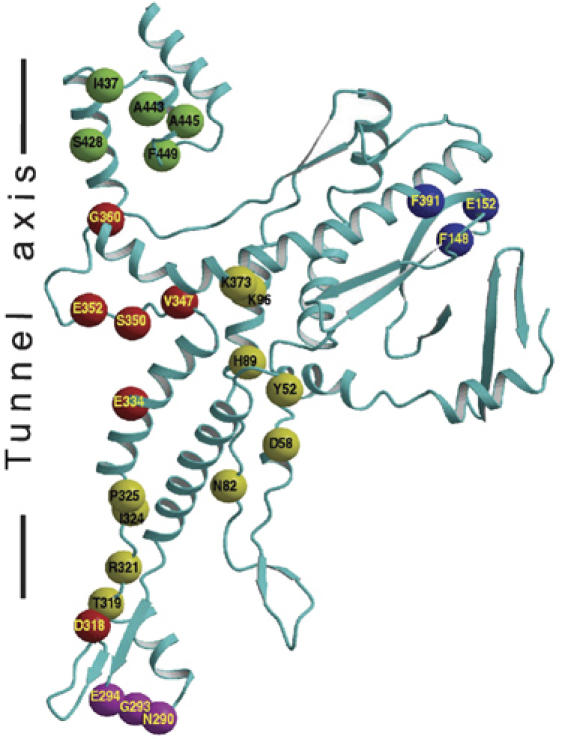
Location of mutations that affected specifically DNA packaging (Isidro et al, 2004a, 2004b). The SPP1 portal protein monomer is shown as a ribbon. The residues, shown as spheres centered at their Cα atoms, are subdivided into five groups: (red) residues in the tunnel loop and residues contributing to the negative charge of the tunnel's surface; (magenta) residues at the base of the portal protein that are likely to interact with the ATPase and gp15; (yellow) residues that could be involved in signal-force transmission between the portal protein and the ATPase and/or carry a structural role; (green) residues involved in stabilization of the crown; and (blue) residues with unassigned function.
Comparison with the φ29 portal protein structure: evolutionary implications
Although there is no detectable amino-acid sequence similarity between the portal proteins of SPP1 and φ29 phages (Supplementary Figure S1), the three-dimensional structures of the two proteins contain cores with a strikingly similar fold. In particular, the fold of both proteins in the region spanning helix α3 to helix α6 is identical (Figure 4A). This segment makes up 46% of the φ29 portal protein sequence. Furthermore, the β-sheet in the wing domain of each protein appears to have the same topology, although a detailed comparison is not possible because part of the SPP1 portal protein wing domain is disordered. On the other hand, two protein segments disordered in the φ29 portal protein structure (Simpson et al, 2000; Guasch et al, 2002) are observed in the structure of the SPP1 portal protein. These are the tunnel loop defining the narrowest region of the SPP1 portal tunnel, and the 24 C-terminal residues, that by analogy with the SPP1 portal protein could form a crown in the φ29 portal protein. A longer C-terminal region and a considerably larger wing (Figure 4A) account for the 21 kDa larger molecular mass of the SPP1 portal protein.
Figure 4.
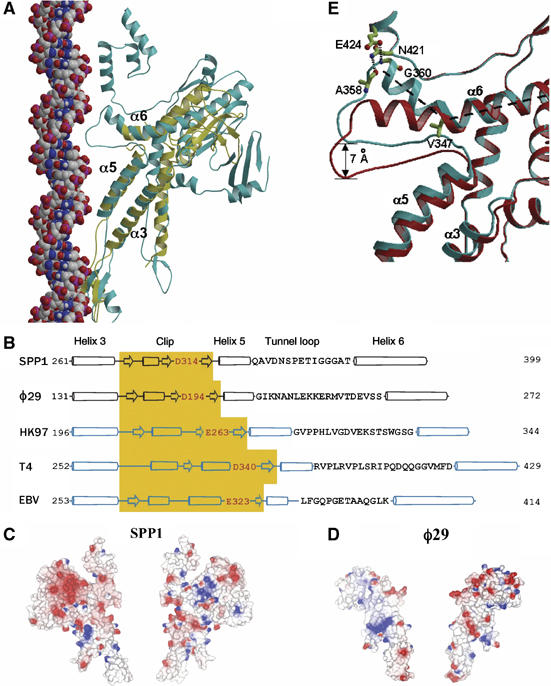
Structural conservation. (A) Single subunits of SPP1 (cyan) and φ29 (yellow) portal proteins are superimposed. B-form DNA (van der Waals model) is positioned along the tunnel to show the relative size and match between the tunnel loop and the major groove of the DNA. (B) Secondary structure alignment of the central part of the polypeptide chain, α3–α6. For HK97, T4 and Epstein–Barr (EBV) portal proteins predicted secondary structures are shown. The positions of α3–α6 segments were validated by a number of criteria described in Materials and methods; the negatively charged residue at the tunnel entrance is highlighted in red. Tunnel loop sequences are shown as single letter code; the length of the cylinders (α-helices) and arrows (β-sheets) are proportional to the predicted length. (C, D) Molecular surfaces of two opposing subunits of SPP1 and φ29 portal proteins colored according to electrostatic potential. (E) Two extreme states of the SPP1 tunnel loop: the cyan from the crystal structure with residues stabilizing the kinked conformation of helix α6 shown in ball and stick, and the red obtained by modeling a straightened conformation of this helix.
The α3–α6 segment conserved in φ29 and SPP1 phages is likely to be found in portal proteins from other phages and herpesviruses, where secondary structure predictions indicate a very similar pattern of helices and strands with comparable length and spacing (Figure 4B). This conserved core segment is likely to be an ancient structure, retained during evolution to serve a common function in genome packaging.
It has already been shown that the major capsid proteins of bacteriophages HK97, P22, φ29, T4, epsilon 15 and herpes simplex virus have an identical fold (Wikoff et al, 2000; Baker et al, 2005; Morais et al, 2005; Jiang et al, 2006). The α-helical structure of the scaffolding protein that assists the association of the major capsid protein was also shown to be conserved (Sun et al, 2000; Morais et al, 2003). The similarity between the three-dimensional structures of φ29 and SPP1 portal proteins provides additional evidence for the proposal that the dsDNA tailed bacteriophages diverged from a common ancestor, which was the root of the lineage formed by tailed phages and herpesviruses (Bamford et al, 2005). Equally this conservation suggests that the mechanism of DNA translocation is similar in all these systems.
Subunit–subunit interactions
The 13-subunit assembly of gp6 is mostly stabilized within the clip, which contains a tightly packed ring of three-stranded β-sheets each made up of two strands from one subunit and one strand from an adjacent subunit. This is the only area with direct main-chain/main-chain inter-subunit hydrogen bonds. Within the rest of the structure, only 12 side-chain hydrogen-bonding interactions are observed between adjacent subunits. As the contact area between adjacent subunits is 2800 Å2, this is equivalent to 4.3 hydrogen bonds per 1000 Å2. This contrasts with the average value of seven hydrogen bonds per 1000 Å2 of contact area calculated for 36 dimeric proteins (Jones and Thornton, 1996). For these proteins, 35% of interface atoms are polar compared to 39% in the portal protein. Thus, in spite of having a more polar interface, the portal protein (excluding the clip domain) has only about half the expected number of direct inter-subunit hydrogen bonds. The 2.1 Å resolution X-ray structure of the φ29 portal protein (Guasch et al, 2002) shows a very similar situation, with most inter-subunit hydrogen-bonding contacts in the clip area (residues 162–197). Again, the rest of the structure appears to be loosely bound, with only 4.4 hydrogen bonds per 1000 Å2 (nine per ∼2050 Å2) of contact area. The low number of direct hydrogen bonds between the polar contacting surfaces suggests flexibility at the subunit–subunit interface. This hypothesis is further supported by the ability of the SPP1 portal protein to assemble into rings containing 12 (Dube et al, 1993; Lurz et al, 2001), 13 (Dube et al, 1993) and 14 (unpublished) subunits and by the observed deviation of individual subunits in the bacteriophage φ29 portal protein from the 12-fold symmetry (Simpson et al, 2001; Xiang et al, 2006). In both SPP1 and φ29 portal proteins (Simpson et al, 2001), the lack of good contacts between neighboring subunits is compensated by charge complementarity (Figure 4C and D) between the opposing surfaces of neighboring subunits.
Molecular lever
The most distinctive feature of the portal protein is that the long helix α6 contains a 135° kink (Figure 2D). This unusual conformation is stabilized by interactions with the C-terminus of helix α5, which is approximately perpendicular to α6. Helix α5 itself has hydrophobic interactions with the roughly parallel helix α3. Two direct hydrogen bonds (A358–N421 and G360–E424) linking the tunnel loop and the N-terminus of α6 to the crown further stabilize this kinked conformation (Figure 4E).
As the tunnel loop connects helices α5 and α6, the mutual orientation of these helices is critical for its position in the tunnel. The conservation of the three-helical substructure (α3, α5 and α6) in the portal proteins of bacteriophages SPP1 and φ29 (Figure 4A) and its apparent conservation in other viruses (Figure 4B) strongly suggest that this substructure is important for DNA translocation. Mutagenesis and biochemical data (Isidro et al, 2004b; Oliveira et al, 2006) support this hypothesis with a group of five different mutant proteins (Figure 3) leaving little doubt about the involvement of the tunnel loops in the process. One of the mutated residues, V347, is in the tunnel loop with its side chain underpinning the kink in the helix α6 (Figure 4E). Its substitutions to alanine (smaller) or methionine (larger) abolish DNA packaging. These substitutions would alter the kink in helix α6 and/or the conformation of the loop.
To investigate possible conformational changes in the portal protein and in particular the potential movements of the tunnel loop, we performed normal mode analysis calculations. These showed low-energy modes that correspond to the up and down movement of the loop along the direction of the tunnel (Supplementary Movie S1). Inspection of the structure shows that the structural motif comprising α5 and α6 could function as a molecular lever acting on the tunnel loop, in which a slight vertical shift of helix α5 is associated with a much larger shift of the N-terminal end of helix α6 and the tunnel loop, in the direction of tunnel axis (Figure 4E). The straightened helix α6 gains five additional α-helical hydrogen bonds, which are otherwise disrupted by the kink of α6. Between the two positions shown in Figure 4E, the end of the loop moves by about 7 Å along the tunnel. Reconstruction of the pseudoatomic structure of the 12-mer, described in the following section, provides experimental evidence for the variability in the positions of tunnel loops within the functional dodecameric assembly.
Pseudoatomic structure of the SPP1 portal protein 12-subunit assembly
The X-ray analysis was carried out for the isolated SPP1 portal protein, which forms a 13-subunit assembly. However, inside phage particles, the protein forms a 12-subunit assembly (Lurz et al, 2001). EM maps of the SPP1 portal protein are available both for the isolated 13-subunit oligomer (9 Å resolution) and for the connector (10 Å resolution; Figure 5A and B)—an assembly purified from viral capsids consisting of the 12-mer portal protein in a coaxial complex with two other viral components (Orlova et al, 2003). We generated a pseudoatomic structure of the 12-subunit state by fitting subunits from the X-ray structure of the 13-mer into the EM map of the connector, using the spherically averaged phased translation function in MOLREP (Vagin and Isupov, 2001). To assess the accuracy of the procedure, we first generated a pseudoatomic model for the isolated 13-subunit assembly for which both crystal and EM structures are available. The reconstructed structure was obtained by fitting subunits as rigid bodies into the EM maps, and proved to be in excellent agreement with the X-ray structure, with the r.m.s. deviation calculated over Cα atoms of the whole oligomer of 0.44 Å. All the regions in the protomer had clear density in the EM maps (data not shown).
Figure 5.
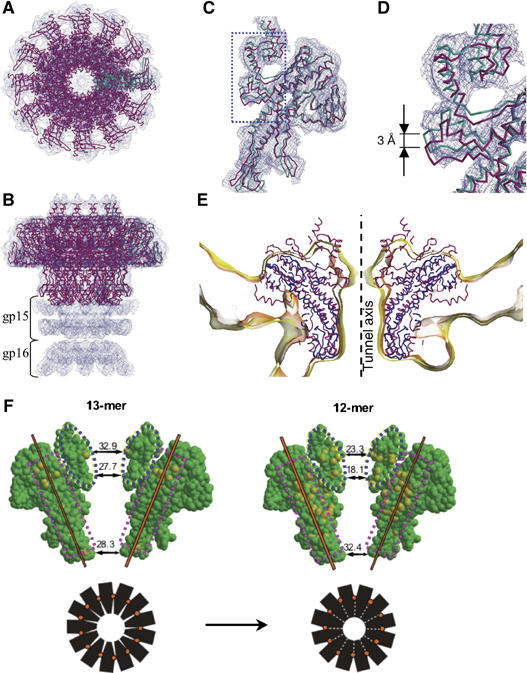
Pseudoatomic structure of the 12-subunit assembly. (A–D) EM maps of the SPP1 connector (Orlova et al, 2003) with the fitted model of the portal protein. (A, B) Two orthogonal views of the connector. (C, D) Single subunit of the 12-mer (magenta), superimposed with a single subunit from the 13-mer (cyan). (E) Cα models of SPP1 (magenta) and φ29 (blue) portal proteins are fitted into five-fold averaged EM maps of φ29 proheads (Morais et al, 2005). The EM density is contoured at two different levels with the yellow map at a 25% lower contouring level than the pink map. For clarity, only a 16 Å slice of the maps and models is shown. (F) Changes in subunit–subunit contacts during 13- to 12-mer transition. Two diametrically opposite subunits of the 13-mer (left) and 12-mer (right) structures with van der Waals size of the tunnel are shown for the clip, tunnel loop and crown areas. Atoms forming short inter-subunit contacts (<3 Å) to the next subunit are highlighted in yellow. The two contact areas (dashed pink boxes) are different; the subunits in the 12-mer ‘roll' around the inter-subunit rotation axis (orange) to pack much more snugly. This forces substantial conformational changes in the crown and the tunnel loop (dashed blue ovals). The simple packing model schematized in black shows how the 12-mer tunnel diameter is considerably reduced by this rocking motion.
A similar fitting into the EM map of the SPP1 connector gave a model for the 12-subunit assembly of the portal protein. This model had acceptable subunit contacts except for the crown and the tunnel loop regions. Here the EM maps indicated positional rearrangements; so the initial fitting was refined by dissecting each subunit into segments and docking them as rigid bodies while maintaining 12-fold symmetry constraints. The resulting model is shown in Figure 5A–D. The crown and a segment comprising the tunnel loop and 10 N-terminal residues of α6 undergo positional changes. Of particular interest is the ∼3 Å movement of the N-terminal end of α6 towards helix α5. As predicted for the molecular lever formed by these helices, this shift correlates with a smaller shift of α5. In addition, there is a rearrangement in the clip region in an outer loop that interacts with gp15, a connector protein that binds to gp6 (Orlova et al, 2003). Weak electron density corresponding to the exposed tunnel loop (residues 353–355; Figure 5C and D) suggests variability in conformation and/or position. Similarly, in the crystal structures of the 12-subunit assembly of the φ29 portal protein (Simpson et al, 2000; Guasch et al, 2002), 17 amino acids (229–245) that could form a tunnel loop are disordered. Inspection of the EM maps of the φ29 portal protein embedded in the procapsid (Morais et al, 2005) reveals narrowing of the tunnel in the same area as observed in the SPP1 portal protein (Figure 5E). The density in this region, as well as for the putative crown, is weaker than average, suggesting conformational variability in these parts of the φ29 portal. Taken together, the evidence from the φ29 and SPP1 portal protein X-ray structures and the EM reconstructions suggests that the tunnel loops' conformations are variable in the 12-mer assembly.
The diameter of the tunnel loop belt is substantially decreased on the transition of gp6 from a 13-mer to a 12-mer. Assuming that all 12 tunnel loops have the same conformation, the diameter decreases by ∼10 Å (Figure 5F), a substantially greater contraction than a simple scaling down by a factor of 12/13. This is a result of the reorientation of the subunits and the different interactions within the 12-mer and the 13-mer assemblies. The changing pattern of inter-subunit contacts (Figure 5F, pink boxes) in the 13-mer → 12-mer switch suggests that (i) the greater part of adjacent subunits move as a rigid body around a rotation axis tilted with respect to the tunnel axis and (ii) the crown and the tunnel loop (blue ovals) undergo additional conformational changes to avoid clashes. The data suggest that in the 13-mer assembly, there is sufficient room to allow all 13 tunnel loops to obey the circular symmetry, whereas this is not so for the 12-mer assembly.
Portal protein rotation
The transient nature of the portal protein–DNA complex and the predominantly glycine/proline-rich character of tunnel loops (Figure 4B) favor shape-matching interactions between the moving components of the molecular motor. Together with tight contacts caused by the small diameter of the tunnel, such interactions with DNA imply the presence of a pronounced energy minimum. This favorable state will repeat at the specific positions of the protein and the DNA, that is, when the portal protein is rotated by every 30° around its 12-fold symmetry axis and/or when the DNA is rotated by 36° and translated by 1 bp (101 symmetry). The energy minima are shown on Figure 6; DNA translocation will be associated with a certain path through these minima.
Figure 6.
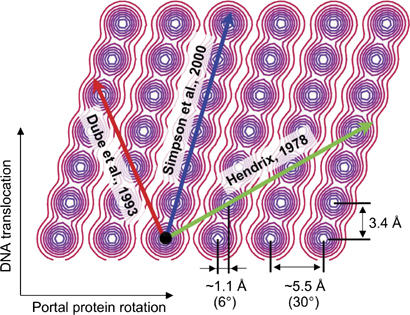
Energy landscape generated by the mismatching symmetries of portal protein and DNA. The X- and Y-axis correspond to the relative rotation and translation of the portal protein and the DNA, respectively. The relative scale on X and Y is such that distances on the drawing are proportional to those at the interface between the DNA and the tunnel loops. The energy minima form a periodic pattern. The horizontal separation between the minima corresponds to the observed 5.5 Å distance between the tips of the neighboring tunnel loops and the vertical separation corresponds to the 3.4 Å (1 bp) translation of the DNA. The combination of the two rotations (36–30°) effectively results in 6° horizontal angular separation between the minima, which corresponds to 1.1 Å horizontal displacement between the tips of the tunnel loops.
Figure 6 shows three pathways connecting adjacent minima. The first corresponds to the model of DNA translocation considered by Hendrix (1978), which involves 36°/bp rotation of the portal protein with respect to the DNA (‘nut and bolt' mechanism). The second (Dube et al, 1993) was proposed for the 13-mer assembly and, when adapted for the 12-mer assembly, involves 9°/bp rotation of the portal protein in the opposite direction. The third is a feature of the model of DNA translocation proposed by Simpson et al (2000) and involves 6°/bp rotation. It should be emphasized that the relative rotation of the portal protein and the DNA occurs for all rational pathways.
The scheme (Figure 6) allows classifying possible translocation pathways in terms of total displacements occurring at the interaction interface. The pathway suggested by Simpson et al (2000) corresponds to the shortest route between the adjacent energy minima. Transitions between the same energy minima could be achieved via different structural events. Simpson et al (2000) suggested a ‘peristaltic pump' mechanism of DNA translocation, controlled by the inclination of tunnel helices, where movements within all 12 subunits are synchronous. Another mechanism following the same pathway, proposed later by Guasch et al (2002), is based on the assumptions that the portal protein remains rigid and the DNA translocation is achieved by its electrostatic interactions with lysine side-chain nitrogens, which form two rings in the portal protein tunnel (with a diameter of ∼31 Å). In both mechanisms, proposed in the absence of structural information about the tunnel loops, the rotational symmetry of the portal protein assembly remains preserved. In our mechanistic model, described below, DNA translocation follows the same pathway (Simpson et al, 2000). However, based on the structural data reported above, we argue that the tunnel loops are arranged asymmetrically around the DNA and that the DNA translocation is accompanied by sequential conformational changes in loop positions.
Model of the portal protein–DNA complex
Structural observations on the SPP1 portal protein impose constraints on possible models of portal protein complex with DNA. We note that the diameter of the 12-mer tunnel varies along its length and at its most constricted part, the belt formed by the tunnel loops, the van der Waals diameter would be only ∼18 Å if all the loops were in the same conformation (Figure 5F). As the van der Waals diameter of B-form DNA is at least 5 Å larger, several sugar-phosphate groups would clash with the tunnel loops, if DNA were positioned symmetrically into the 12-subunit assembly. Our mechanistic model avoids such clashes while providing a basis for the process of translocation through two rearrangements away from a simplistic symmetrical organisation. Firstly, the DNA is shifted so that it is no longer coaxial with the portal protein, allowing a number of adjacent tunnel loops to penetrate the major groove (Figure 7A and B). Secondly, sequential sliding of tunnel loops relative to each other in the form of a ‘mexican wave' (Figure 7C) will eliminate remaining clashes with the DNA while maintaining the observed coordination between neighboring loops. Such a rearrangement is consistent with their tight packing next to each other at van der Waals distances and the 15° tilt observed in the 13-mer assembly (Figure 2A), with their apparent mobility (Supplementary Movie S1 and Figure 5D and E) and even tighter packing (Figure 5F) in the 12-mer and with their predominantly glycine/proline-rich character (Figure 4B).
Figure 7.
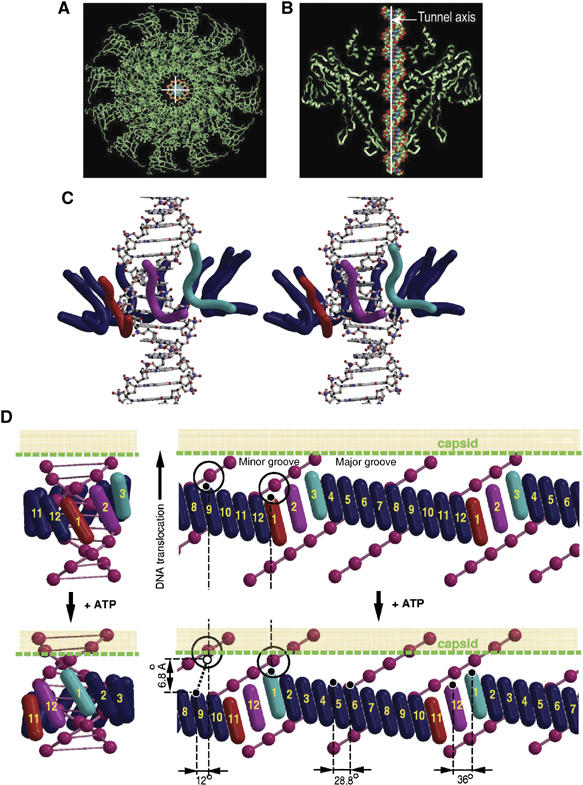
DNA translocation. (A, B) Top and side views of the pseudoatomic structure of the portal protein dodecamer with B-form DNA fitted into the tunnel. The double helix is shifted relative to the rotational axis of the portal protein (white cross/line) to avoid clashes with tunnel loops. (C) Stereo view of the proposed arrangement of tunnel loops (ribbons drawn along the main-chain atoms of residues 350–360) around the DNA (ball and stick). Loops occupying the three states inside the major groove are colored red, magenta and cyan, whereas the remaining nine loops are in dark blue. (D) Mechanistic model of DNA translocation. Two sequential states of the portal protein/DNA complex before (top) and after (bottom) consumption of one ATP molecule by the ATPase. A three-dimensional model (left) is sliced open (right) to give a representation with the horizontal axis corresponding to the angular position inside the tunnel; two periods of DNA are shown with two phosphates circled to provide reference points. The tilted tunnel loops (cylinders) and DNA (pink spheres centered at phosphates) are shown to scale. Numbers designate specific loops and colors designate specific conformational states. The three states—red, magenta and cyan—propagate along the circle of loops. There will be larger angular separation for the three loops occupying these three states, compared to the rest, allowing the three loops to dip into the major groove. An idealized mechanistic model of the transition between the top and bottom states requires 12° rotation of the bulk of the portal protein relative to the DNA. During this transition, loops 2–12 move with respect to the DNA, whereas loop 1 does not change its position with respect to the DNA, and together with the DNA moves by 6.8 Å relative to the capsid.
The size and shape of tunnel loops observed in the X-ray structure of the 13-subunit assembly matches the size and shape of the major groove of B-form DNA (Figure 7B). The geometry suggests that three tunnel loops could be fitted inside the major groove at a time. Because of tight contacts, loops would have to slide into and out of the groove between adjacent phosphates. Thus, the three loops occupying the red, magenta and cyan states inside the major groove (Figure 7C and D) must follow the 101 symmetry of the DNA. The 6.8 Å amplitude span along the DNA axis between the two extreme states (red and cyan) is consistent with the potential straightening of helix α6 (Figure 4E). The remaining nine loops (dark blue in Figure 7C and D) occupy intermediate vertical positions, with all loops forming a continuous undulating belt tightly embracing the double-helical DNA. The exact position and conformation of each loop and the nature of their interactions with the DNA are not the issue here, only that each loop will be in a unique position in relation to the DNA. The proposed crude model is based on the assumption that the DNA in the portal protein–DNA complex contains 10 base pairs per period. However, B-form DNA contains ∼10.5 base pairs per period and DNA conformation as well as conformations of tunnel loops within the portal–DNA complex may be affected by their local interactions. Nevertheless, the proposed structural organization of the complex could be easily adapted for such cases by small adjustments in angular spacing between adjacent loops.
DNA translocation
Based on the model of the portal protein complex with the DNA, we can now discuss a possible scenario of events during DNA translocation. Loops occupying the three structural states that fit the 101 symmetry of the DNA (red, magenta and cyan in Figure 7C and D) ensure engagement of the portal protein with the DNA at all stages of DNA translocation. Movement of the DNA through the tunnel would cause these three structural states to propagate sequentially along the belt of 12 tunnel loops (Supplementary Movie S2) resulting in local symmetry adjustments and generating a torque that facilitates the 12° rotation of the bulk of the portal protein with respect to the DNA helix per each step of the molecular motor. The restructuring of the tunnel loops occupying the three states inside the major groove is accompanied by positional rearrangement of remaining loops so that the net changes per cycle of the molecular motor are as follows (Figure 7D): (i) cyclic permutation of loop positions (1–3, 2–4, …, 11–1, 12–2); (ii) DNA translocation by 2 base pairs; (iii) rotation of the portal protein by 12° degrees relative to DNA; (iv) hydrolysis of one ATP molecule by the viral ATPase. Such a scenario of events is consistent with structural observations, mutagenesis data (Isidro et al, 2004b), crosstalk between the portal protein tunnel loops and the ATPase (Oliveira et al, 2006), consumption of one ATP molecule per two translocated base pairs (∼6.8 Å) (Guo et al, 1987; Morita et al, 1993) and coordinated action of the motor subunits (Chemla et al, 2005). The overall negatively charged internal surface of the tunnel, a feature that is conserved in the φ29 portal protein (Simpson et al, 2001), will facilitate movement of the DNA and ensure that interactions controlling this movement take place only at the sections of the tunnel narrower than the diameter of the DNA.
Loop ‘1', which rests with respect to the DNA during the whole cycle of the molecular motor (Figure 7D), allows one to consider the mechanism within a general framework of mechano-chemical reactions, where a chemical event (ATP hydrolysis) forces one structural element (loop ‘1') to occupy a certain position (to sink into the major groove of DNA) where it remains on duty in its individual local energy minima during the most of the motor cycle and rectifies the Brownian motion of the remaining elements towards the next global minima of the whole system.
The rotation of the portal protein with respect to procapsid has been recently questioned for bacteriophage T4 (Baumann et al, 2006), but there is no experimental evidence supporting or opposing the relative rotation of the portal protein and DNA. In principle, it is possible to adopt the proposed conformational changes for the case involving no portal–DNA rotation. However, the energy and structural considerations described earlier support mutual rotation between the portal protein and DNA. Indeed, all pathways through the energy minima of the DNA–portal protein complex involve rotation (Figure 6). Moreover, the expected sliding of loops between adjacent phosphates of DNA inside the tight portal–DNA complex suggests a structural basis for the proposed rotational adjustment.
Structural and biochemical data (Oliveira et al, 2006) suggest that the tunnel loops and ATPase communicate via the molecular lever (Figure 4E), where movements of the tunnel loop are associated with alterations in the position of helix α5. Concomitant adjustments between helices α5 of adjacent subunits would be facilitated by the flexible nature of the subunit–subunit interface. The portal–DNA interactions described above are applicable independent of whether the force is applied to the DNA by the portal protein in response to conformational changes generated by the ATPase or whether the viral ATPase itself performs the power stroke, pushing the DNA into the capsid. In the latter case, the sequential nature of interactions between the tunnel loops and DNA could serve to coordinate ATP hydrolysis events with the position and symmetry of the DNA.
Materials and methods
Crystallization and X-ray data collection
Diffracting crystals were obtained for the SPP1 portal protein gp6SizA with the amino-acid substitution N365K. This mutation reduces the length of encapsidated DNA but does not affect the DNA packaging process (Tavares et al, 1992). Purification and crystallization have been described previously (Jekow et al, 1998). The best crystals were obtained using the hanging-drop vapor diffusion. Solution with 8 mg/ml of protein was mixed in a 1:1 ratio with the reservoir solution containing 20% PEG 400, 100 mM CaCl2, 50 mM HEPES pH 7.6 and 10% glycerol, which acted also as a cryoprotectant. The diffraction quality of the crystals was improved from ∼4.5 to ∼3.5 Å by the addition of 0.5–2.5 mM HgCl2 directly to the protein solution. Crystals belong to the C2221 space group with a=174.3, b=221.4 and c=421.9 Å. X-ray data from native protein crystals and from the mercury chloride derivative were collected at 100K using synchrotron radiation at the ESRF storage ring (Table I) to 4.1 and 3.4 Å, respectively. The data were processed using DENZO and SCALEPACK (Otwinowski and Minor, 1997).
X-ray structure determination
Structure was determined using the CCP4 suite of crystallographic programs (Collaborative Computational Project N4, 1994). The crystal asymmetric unit contained one 13-mer of molecular mass 745 kDa (Jekow et al, 1998) and the self-rotation function showed a clear 13-fold symmetry. The isomorphous difference data (but not the anomalous) for the Hg derivative showed a similar pattern. As each subunit contains only one cysteine residue (C55), it was expected that only 13 Hg atoms would bind to the complex. Neither the isomorphous nor the anomalous difference Patterson syntheses showed a clear vector pattern, and automated Patterson search and direct method procedures failed to find a solution. The structure was solved by imposing 13-fold symmetry constraints during the heavy atom search, using the procedure implemented for structure determination of TRAP protein 11-mer (Antson et al, 1995). A series of models for a molecular replacement search against the isomorphous differences were constructed as planar rings of 13 equally positioned atoms with radii varying from 10 to 100 Å and a step size of 1 Å. The correlation coefficient (CC) of the rotation function calculated by AMORE (Navaza, 2001) peaked at 21.8% for the 41.0 Å radius model (next CC=13.5%), and the translation function gave a clear solution with a CC of 15.3% (next CC=12.7%). Initial phasing was performed at 4.1 Å using native data and both isomorphous and anomalous signals. For the refinement of heavy atoms, we used our own program (AA Lebedev, unpublished), which takes into account external phases during the refinement (in this case, phases from averaged map). The phases were extended to 3.4 Å using an iterative procedure of averaging alternated by heavy atom refinement and phase combination. The model was built using QUANTA (Accelrys) and refined using REFMAC (Murshudov, 1997). Initially, only the α-helical region around the tunnel was visible in the electron density map. This was built as polyalanine segments. The first model contained 44.6% of the final structure although the direction of some segments was incorrect. Several rounds of refinement imposing non-crystallographic symmetry restraints, followed by rebuilding into the 13-fold averaged map, allowed the correction and expansion of the model. Owing to the limited resolution, TLS parameters but not individual atomic B factors were refined. The final model contains all residues except for 26 N-terminal residues and 37 C-terminal residues; a segment of 69 residues (170–238) in the wing can only be partially modeled by a 30-residue polyalanine segment.
Fitting the X-ray structure into the EM map of the connector
The connector contains a 12-mer of gp6 in association with 12-mers of gp15 and gp16 (Orlova et al, 2003). Molecular replacement techniques implemented in MOLREP (Vagin and Teplyakov, 1997) were used to fit the X-ray atomic model into the EM map. The spherically averaged phased translation function (Vagin and Isupov, 2001) was first used to position the center of mass of the search model, and then a phased rotation function was used to find the orientation (this is the other way round compared to the conventional molecular replacement approach, which does not use phase information). Two adjacent subunits from the X-ray structure of the 13-mer were used as a search model. The protocol was repeated for both original and mirror EM maps with map scale factors ranging from 0.9 to 1.2. The best CC was obtained for a set of six dimers making up a 12-mer, for a map scaled by 1.08. CC was 15% better for the map with a correct hand. The 12 subunits were initially refined as rigid bodies with strict 12-fold symmetry constraints; during this procedure the CC increased from 0.19 to 0.25. The fitting revealed differences in conformation between the gp6 molecules in the X-ray and the EM structures. To best interpret this and exclude the effect of gp15 and gp16 on the packing, each monomer was dissected into 13 domains for further symmetry-constrained rigid-body refinement. The division into domains was made following manual inspection to allow compact parts of each subunit, such as the crown, the β-sheet of the wing and the tunnel loop together with the N-terminal portion of helix α6 (up to the kink), to move independently. The symmetry-constrained rigid-body fitting did not result in unfavorable contacts, except for few areas shown in Figure 5F. The domain movements mentioned in the main text refer to a mean coordinate shift between the two positions—before and after the rigid-body fitting, averaged over all atoms of a particular domain. Improvement in the fitting was monitored by further increase in the CC (to 0.36).
Secondary structure prediction
For HK97, T4 and Epstein–Barr portal protein secondary structures were predicted using PSIPRED (Jones, 1999; McGuffin et al, 2000). Similar predictions for the SPP1 and the φ29 portal proteins resulted in secondary structure elements whose length and position were in good agreement with those observed in the X-ray structures. In all cases, the segment lining the walls of the central tunnel is 30–31 residues long and contains a β-strand followed by an α-helix. This segment is rich in negatively charged residues (usually 4–6 Asp/Glu amino acids). In all shown sequences, there is a negatively charged residue at the entrance of the tunnel (highlighted in red in Figure 4B). Alignment of homologous sequences shows that this is a conserved feature although in a few cases, there is Asn, Ser or Gly in this position.
Normal mode analysis
Normal mode analysis calculations were performed with the X-ray structure of the 13-mer using the elNemo server (Suhre and Sanejouand, 2004). To imitate stabilizing effects of the capsid embracing the portal protein, the exterior segments of the portal protein were represented by all atoms, whereas the segments lining the tunnel including the tunnel loop were represented by their Cα atoms. The tunnel loop makes only two hydrogen-bonding interactions with the crown and should, therefore, be able to move independently. The crown was therefore excluded during calculations of normal modes but repulsion interactions with it were accounted for by restricting the span of the motion. With this approach, 11 out of 25 lowest frequency modes involved significant motions of the tunnel loops. Supplementary Movie S1 corresponds to one of these modes.
Figures and movies were produced by BOBSCRIPT (Esnouf, 1997).
Supplementary Material
Supplementary Information
Supplementary Figure S1
{kind=link}
Supplementary Movie S1
Supplementary Movie S2
Acknowledgments
We are grateful to Thomas A Trautner for his interest in the work and useful discussions. We thank Garib Murshudov for useful suggestions during structure refinement, and Petra Jekow and Asita Stiege for their contribution during crystallization. We also thank Guy Dodson, Félix Rey, David Waterman and Tony Wilkinson for critically reading the manuscript and for useful suggestions. This work was supported by Wellcome Trust fellowship to AAA, Wellcome Trust project grant to AAA and EJD, FCT fellowship to AI, grants from FCT, FRM and ATIPE/CNRS to PT, NIH grant to AAL and BBSRC grants to AAV and EVO. We thank ESRF for provision of synchrotron facilities through the block allocation to York and Sean McSweeney for help during the data collection at beamline ID14-4.
Crystallographic coordinates and structure factors have been deposited with the Protein Data Bank under accession code 2jes.
References
- Antson AA, Otridge J, Brzozowski AM, Dodson EJ, Dodson GG, Wilson KS, Smith TM, Yang M, Kurecki T, Gollnick P (1995) The structure of trp RNA-binding attenuation protein. Nature 374: 693–700 [DOI] [PubMed] [Google Scholar]
- Baker ML, Jiang W, Rixon FJ, Chiu W (2005) Common ancestry of herpesviruses and tailed DNA bacteriophages. J Virol 79: 14967–14970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford DH, Grimes JM, Stuart DI (2005) What does structure tell us about virus evolution? Curr Opin Struct Biol 15: 655–663 [DOI] [PubMed] [Google Scholar]
- Baumann RG, Mullaney J, Black LW (2006) Portal fusion protein constraints on function in DNA packaging of bacteriophage T4. Mol Microbiol 61: 16–32 [DOI] [PubMed] [Google Scholar]
- Camacho AG, Gual A, Lurz R, Tavares P, Alonso JC (2003) Bacillus subtilis bacteriophage SPP1 DNA packaging motor requires terminase and portal proteins. J Biol Chem 278: 23251–23259 [DOI] [PubMed] [Google Scholar]
- Catalano CE (2005) Viral Genome Packaging: Genetics, Structure, and Mechanism. New York: Springer-Verlag [Google Scholar]
- Chai S, Lurz R, Alonso JC (1995) The small subunit of the terminase enzyme of Bacillus subtilis bacteriophage SPP1 forms a specialized nucleoprotein complex with the packaging initiation region. J Mol Biol 252: 386–398 [DOI] [PubMed] [Google Scholar]
- Chemla YR, Aathavan K, Michaelis J, Grimes S, Jardine PJ, Anderson DL, Bustamante C (2005) Mechanism of force generation of a viral DNA packaging motor. Cell 122: 683–692 [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project N4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Dube P, Tavares P, Lurz R, van Heel M (1993) The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO J 12: 1303–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf RM (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graph Model 15: 132–134, 112–133 [DOI] [PubMed] [Google Scholar]
- Guasch A, Pous J, Ibarra B, Gomis-Ruth FX, Valpuesta JM, Sousa N, Carrascosa JL, Coll M (2002) Detailed architecture of a DNA translocating machine: the high-resolution structure of the bacteriophage phi29 connector particle. J Mol Biol 315: 663–676 [DOI] [PubMed] [Google Scholar]
- Guo P, Peterson C, Anderson D (1987) Prohead and DNA-gp3-dependent ATPase activity of the DNA packaging protein gp16 of bacteriophage phi 29. J Mol Biol 197: 229–236 [DOI] [PubMed] [Google Scholar]
- Hendrix RW (1978) Symmetry mismatch and DNA packaging in large bacteriophages. Proc Natl Acad Sci USA 75: 4779–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidro A, Henriques AO, Tavares P (2004a) The portal protein plays essential roles at different steps of the SPP1 DNA packaging process. Virology 322: 253–263 [DOI] [PubMed] [Google Scholar]
- Isidro A, Santos MA, Henriques AO, Tavares P (2004b) The high-resolution functional map of bacteriophage SPP1 portal protein. Mol Microbiol 51: 949–962 [DOI] [PubMed] [Google Scholar]
- Jekow P, Schaper S, Gunther D, Tavares P, Hinrichs W (1998) Crystallization and preliminary X-ray crystallographic studies of the 13-fold symmetric portal protein of bacteriophage SPP1. Acta Crystallogr D 54 (Part 5): 1008–1011 [DOI] [PubMed] [Google Scholar]
- Jiang W, Chang J, Jakana J, Weigele P, King J, Chiu W (2006) Structure of epsilon15 bacteriophage reveals genome organization and DNA packaging/injection apparatus. Nature 439: 612–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT (1999) Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292: 195–202 [DOI] [PubMed] [Google Scholar]
- Jones S, Thornton JM (1996) Principles of protein–protein interactions. Proc Natl Acad Sci USA 93: 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurz R, Orlova EV, Gunther D, Dube P, Droge A, Weise F, van Heel M, Tavares P (2001) Structural organisation of the head-to-tail interface of a bacterial virus. J Mol Biol 310: 1027–1037 [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16: 404–405 [DOI] [PubMed] [Google Scholar]
- Morais MC, Choi KH, Koti JS, Chipman PR, Anderson DL, Rossmann MG (2005) Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of phi29. Mol Cell 18: 149–159 [DOI] [PubMed] [Google Scholar]
- Morais MC, Kanamaru S, Badasso MO, Koti JS, Owen BA, McMurray CT, Anderson DL, Rossmann MG (2003) Bacteriophage phi29 scaffolding protein gp7 before and after prohead assembly. Nat Struct Biol 10: 572–576 [DOI] [PubMed] [Google Scholar]
- Morita M, Tasaka M, Fujisawa H (1993) DNA packaging ATPase of bacteriophage T3. Virology 193: 748–752 [DOI] [PubMed] [Google Scholar]
- Murshudov GN (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J (2001) Implementation of molecular replacement in AMoRe. Acta Crystallogr D 57: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Oliveira L, Alonso JC, Tavares P (2005) A defined in vitro system for DNA packaging by the bacteriophage SPP1: insights into the headful packaging mechanism. J Mol Biol 353: 529–539 [DOI] [PubMed] [Google Scholar]
- Oliveira L, Henriques AO, Tavares P (2006) Modulation of the viral ATPase activity by the portal protein correlates with DNA packaging efficiency. J Biol Chem 281: 21914–21923 [DOI] [PubMed] [Google Scholar]
- Orlova EV, Dube P, Beckmann E, Zemlin F, Lurz R, Trautner TA, Tavares P, van Heel M (1999) Structure of the 13-fold symmetric portal protein of bacteriophage SPP1. Nat Struct Biol 6: 842–846 [DOI] [PubMed] [Google Scholar]
- Orlova EV, Gowen B, Droge A, Stiege A, Weise F, Lurz R, van Heel M, Tavares P (2003) Structure of a viral DNA gatekeeper at 10 Å resolution by cryo-electron microscopy. EMBO J 22: 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Simpson AA, Leiman PG, Tao Y, He Y, Badasso MO, Jardine PJ, Anderson DL, Rossmann MG (2001) Structure determination of the head-tail connector of bacteriophage phi29. Acta Crystallogr D 57: 1260–1269 [DOI] [PubMed] [Google Scholar]
- Simpson AA, Tao Y, Leiman PG, Badasso MO, He Y, Jardine PJ, Olson NH, Morais MC, Grimes S, Anderson DL, Baker TS, Rossmann MG (2000) Structure of the bacteriophage phi29 DNA packaging motor. Nature 408: 745–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DE, Tans SJ, Smith SB, Grimes S, Anderson DL, Bustamante C (2001) The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature 413: 748–752 [DOI] [PubMed] [Google Scholar]
- Suhre K, Sanejouand YH (2004) ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res 32: W610–W614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Parker MH, Weigele P, Casjens S, Prevelige PE Jr, Krishna NR (2000) Structure of the coat protein-binding domain of the scaffolding protein from a double-stranded DNA virus. J Mol Biol 297: 1195–1202 [DOI] [PubMed] [Google Scholar]
- Tavares P, Santos MA, Lurz R, Morelli G, de Lencastre H, Trautner TA (1992) Identification of a gene in Bacillus subtilis bacteriophage SPP1 determining the amount of packaged DNA. J Mol Biol 225: 81–92 [DOI] [PubMed] [Google Scholar]
- Trus BL, Cheng N, Newcomb WW, Homa FL, Brown JC, Steven AC (2004) Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J Virol 78: 12668–12671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30: 1022–1025 [Google Scholar]
- Vagin AA, Isupov MN (2001) Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr D 57: 1451–1456 [DOI] [PubMed] [Google Scholar]
- Valpuesta JM, Carrascosa JL (1994) Structure of viral connectors and their function in bacteriophage assembly and DNA packaging. Q Rev Biophys 27: 107–155 [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE (2000) Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289: 2129–2133 [DOI] [PubMed] [Google Scholar]
- Xiang Y, Morais MC, Battisti AJ, Grimes S, Jardine PJ, Anderson DL, Rossmann MG (2006) Structural changes of bacteriophage phi29 upon DNA packaging and release. EMBO J 25: 5229–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Figure S1
Supplementary Movie S1
Supplementary Movie S2