

Abstract
Tubules are the building blocks of epithelial organs and form in response to cues derived from morphogens such as hepatocyte growth factor (HGF). Relatively little is known about signaling pathways that orchestrate the cellular behaviors that constitute tubule development. Here, using three-dimensional cell cultures of Madin–Darby canine kidney cells, we show that the ARF6 GTPase is a critical determinant of tubule initiation in response to HGF. ARF6 is transiently activated during tubulogenesis and perturbing the ARF6 GTP/GDP cycle by inducible expression of ARF6 mutants defective in GTP binding or hydrolysis, inhibits the development of mature tubules. Further, we show that activation of ARF6 is necessary and sufficient to initiate tubule extension. The effect of ARF6 on tubule initiation is two-fold. First, ARF6 regulates the subcellular distribution of the GTPase, Rac1, to tubule extensions. Second, ARF6-induced ERK activation regulates Rac1 activation during tubule initiation through the expression of the receptor for urokinase type plasminogen activator. Thus, we have identified a cellular apparatus downstream of ARF6 activation, which regulates membrane and cytoskeleton remodeling necessary for the early stages of tubule development.
Keywords: ARF6, epithelia, tubulogenesis, Rac1
Introduction
Epithelial organs are typically made up of two types of building blocks, cysts and tubules. Connecting cysts and tubules can generate complex structures; for example, in the vertebrate lung, branching tubules terminate in alveolar cysts. Here, we investigate signaling pathways and cellular mechanisms that underlie the process of tubule development, also referred to as tubulogenesis. Tube networks are found in many organs including the lung, kidney, trachea, the gastrointestinal and urinary tracts and the vasculature. Tubes usually comprise of a cell monolayer that encloses a hollow lumen and is often surrounded by additional cell layers that form a barrier between the lumen and other cellular compartments. It is still unclear how epithelial tubes elongate and become functionally polarized with a fluid-filled hollow lumen. Further investigation into the mechanisms and regulation of tubulogenesis is important not only to define fundamental aspects of development but also to understand the cellular basis of a variety of disease states.
A number of approaches have been used to study tubulogenesis, including in vivo studies, genetically tractable systems, and cell culture, each with a distinct set of attributes and limitations (Lubarsky and Krasnow, 2003). The ability of epithelial cells to form cysts and tubules and adopt tissue-like conformations when grown in a three-dimensional (3D) extracellular matrix has made 3D cell culture, a powerful approach to investigate the molecular and cellular basis of epithelial morphogenesis (O'Brien et al, 2002; Debnath and Brugge, 2005). Cysts and tubules formed in 3D culture comprise of spherical and cylindrical lumen enclosing monolayers respectively. MDCK (Madin–Darby canine kidney) cells self-organize to form cysts when grown in collagen or matrigel, and these cysts develop tubules when exposed to mesenchymal growth factors such as hepatocyte growth factor (HGF) or scatter factor (O'Brien et al, 2002).
As elegantly described by Mostov and colleagues, HGF-induced tubulogenesis involves an initial HGF-dependent phase of tubule initiation followed by an HGF-independent phase involving tubule maturation (O'Brien et al, 2002). Tubule initiation is characterized by a partial EMT, which would allow ‘pioneer' cells from a cyst to become invasive and project long basal membrane extensions into the extracellular matrix. This is followed by cell proliferation to form single cell layers that extend radially from the cyst. Therefore, cytosketelal and membrane remodeling events are likely vital to initiate tubule extension. The second phase is characterized by a morphogenetic switch wherein HGF signaling is downregulated, cells redifferentiate and form multilayered chords. At this step, lateral membrane contacts and cell polarity are restored and new lumen formation is initiated to promote the development of chains into cords and ultimately, mature tubules. Thus to generate a tubule, a wide range of cellular processes including migration, proliferation and polarization must be precisely coordinated, both spatially and temporally.
Much remains to be understood about the cellular processes that govern the establishment of tubule architecture. Although signaling pathways involving the activation of many growth factor receptors can induce the formation of tubes, the process is probably best characterized with respect to the activation of the met receptor by HGF (O'Brien et al, 2002; Rosario and Birchmeier, 2003). Distinct HGF downstream effectors have been shown to regulate the different stages of tubulogenesis. In this regard, two downstream HGF targets, ERK (extracellular signal regulated kinase) and MMPs (matrix metalloproteases), have been shown to play a regulatory role during tubule initiation and maturation respectively (O'Brien et al, 2004). Other potential targets of HGF that regulate tubule outgrowth are regulatory molecules that allow the expansion of the basal plasma membrane. In this regard, the exocyst complex, which facilitates the docking of vesicles at the basolateral membrane, has been shown to promote membrane extension during tubulogenesis (Lipschutz et al, 2000).
Here, we have investigated the role of the ARF6 GTPase in HGF-induced tubulogenesis. ARF6 is a member of ARF family of Ras-related GTPases and has been shown to control endocytic traffic and actin remodeling in a variety of cell types (Donaldson, 2003; D'Souza-Schorey and Chavrier, 2006). Pertinent to this study, ARF6 has been shown to function downstream of HGF signaling during adherens junction disassembly (Palacios et al, 2001, 2002; Palacios and D'Souza-Schorey, 2003), epithelial cell migration (Palacios et al, 2001; Santy and Casanova, 2001b; Powelka et al, 2004) and tumor cell invasion (Tague et al, 2004; Li et al, 2006). Understanding how ARF6 might regulate the development of glandular structures is particularly significant in light of a recent study examining the role of ARF6 in liver development, which showed that ARF6-null embryos are defective in HGF-induced chord formation (Suzuki et al, 2006). Here, we show that ARF6 is activated during tubule initiation and aberrant ARF6 cycling blocks tubulogenesis. We show that not only does ARF6 regulate the subcellular distribution of the Rac1 GTPase at tubule extensions, but that ARF6-induced ERK activation is required for Rac1 activation and consequently for cytoskeleton remodeling during tubule initiation. We also demonstrate that uPAR signaling downstream of the ARF6 → ERK pathway is critical for Rac1 activation during tubule outgrowth. In summary, we describe the key components of a signaling module downstream of HGF signaling, which is required for the early stages of tubule development.
Results
ARF6 is activated during the early stages of tubule development
As ARF6 GTPase regulates cellular processes such as actin remodeling and membrane recycling, both of which can directly impinge on tubule development, we hypothesized that ARF6 regulates one or more stages of tubulogenesis. To test this hypothesis, we first investigated whether endogenous ARF6 is activated during the different stages of tubule development. For the investigations described here, stable MDCK cysts grown in 3D culture were subject to continuous treatment with HGF for 4 days to generate mature tubules. As described earlier, tubulogenesis involves an initial HGF-dependent phase involving a partial EMT and is characterized by individual cells from a cyst becoming invasive and producing long basal membrane extensions into the extracellular matrix. This is followed by cell proliferation to form single file chains. After chain formation, cells redifferentiate, lateral membrane contacts and cell polarity is restored and new lumen formation is initiated to promote the development of chains into cords and mature tubules. The sequential stages involved in HGF-induced MDCK tubulogenesis is shown in Figure 1A.
Figure 1.
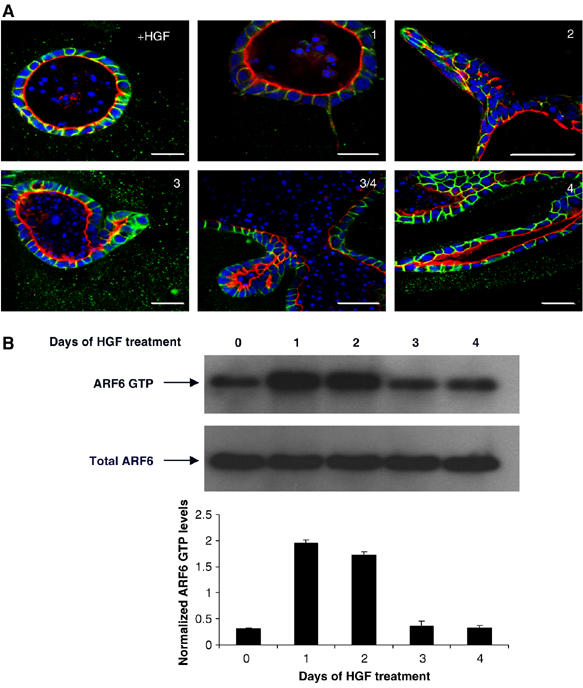
ARF6 is transiently activated during tubule development. (A) MDCK-I cells cysts in matrigel were treated with 60 ng/ml HGF for 4 days to develop tubules. HGF causes cysts to form tubules through a series of sequential events as described in the text. In cross-section, fully developed spherical MDCK cysts appear as a monolayer of cells surrounding a central lumen. The apical plasma membrane stained for actin (red) faces the lumen and basolateral membranes are labeled for E-cadherin (green). Cell nuclei are stained in blue. Intermediate structures formed at day 1 (extensions), day 2 (chains) and day 3 (cords) and matures tubules formed at day 4 are shown. Scale bar, 10 μm. (B) MDCK cysts exposed to HGF for varying times as indicated were lysed and subjected to the MT-2 pull-down assay to detect cellular levels of endogenous ARF6-GTP. Cell lysates were also examined for total ARF6 by Western blotting procedures. Band density was measured by densitometry and the ratios of ARF6-GTP to total ARF6 are shown. Values are the mean of three separate experiments ±s.d. (error bars). ARF6-GTP levels are transiently increased over basal levels at the early stages of tubule development.
To examine the activity of endogenous ARF6 during tubule development, MDCK cells in 3D culture were harvested at different time points post-HGF treatment as above and analyzed for endogenous ARF6-GTP levels using the MT2 pull-down assay, described previously (Schweitzer and D'Souza-Schorey, 2002). As seen in Figure 1B, there was a transient increase in the ARF6-GTP levels for 2 days after treatment with HGF, which returned to basal level at day 3. This transient activation of ARF6 suggests its requirement during tubule initiation.
Perturbing the ARF6-GTP/GDP cycle disrupts the normal process of epithelial tubule development
To explore the functional significance of ARF6 activation during tubule development, we assessed the effect of disrupting the ARF6 GTPase cycle on tubulogenesis by expression of ARF6 mutants locked in the active GTP- or inactive GDP-bound conformations. For these studies, we made use of MDCK Tet-Off cell lines, MDCKARF6-Q and MDCKARF6-T (Prigent et al, 2003), capable of stable and inducible expression of HA-tagged constitutively active ARF6 mutant [ARF6(Q67L)-HA] and dominant-negative ARF6 mutant, [ARF6(T27N)-HA], respectively. Cells were allowed to form fully developed cysts in the presence of doxycycline (Dox), after which Dox was removed from the culture medium to allow for mutant ARF6 expression. This was followed by addition of HGF to allow tubule formation. As seen in Figure 1A, approximately 60% of parental MDCK cells formed single extensions, although occasionally, two or three extensions were also observed. In contrast, more than 80% of MDCKARF6-Q cells formed multiple tubular extensions (Figure 2A and B) that did not develop into mature tubules but instead appeared to disintegrate by day 4 post-treatment with HGF (Supplementary Figure 1). In all MDCKARF6-T cysts, on the other hand, tubule outgrowth was completely inhibited (Figure 2A and B). Immunolabeling for mutant ARF6 proteins revealed that ARF6-GTP localized to the basolateral membrane and extending tubules of cyst cells (Figure 2A). In contrast, ARF6-GDP exhibited significant cytoplasmic distribution relative to ARF6-GTP, in addition to some labeling at the membrane (Figure 2A). Quantitation of ARF6 mutant protein at the surface membrane and internal pools in cysts formed by MDCKARF6-Q and MDCKARF6-T cells are provided as Supplementary data (see Supplementary Figure 2). These findings on the localization and effects of the GTP/GDP-bound ARF6 mutants on HGF-treated cysts point toward a regulatory role for the ARF6 GTPase cycle in tubulogenesis.
Figure 2.
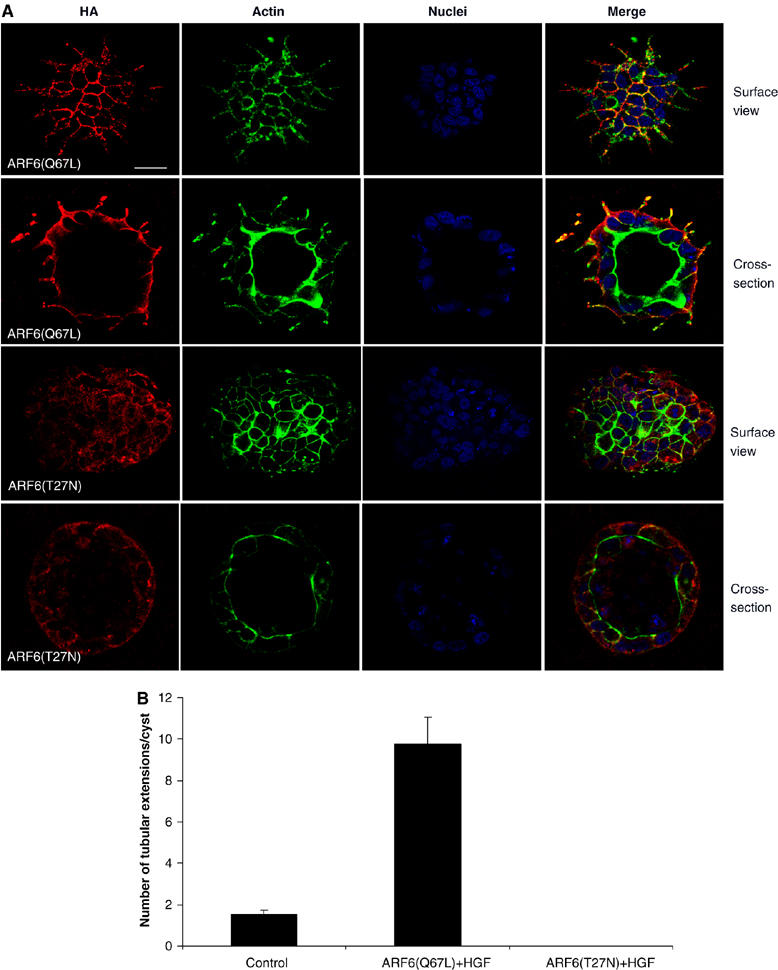
ARF6-GTP expression promotes the initiation of multiple tubules, whereas ARF6-GDP blocks tubulogenesis. (A) MDCKARF6-Q and MDCKARF6-T cells were grown in 3D to form fully developed cysts and then induced for HA-tagged mutant ARF6 expression by removal of Dox from the growth medium followed by exposure to HGF to promote tubulogenesis. Two days post-HGF treatment, cell cultures were fixed and labeled for HA (red) and actin (green). Cell nuclei are labeled blue. Images along the cyst surface and cross-sections through the cyst center are shown. Scale bar, 10 μm. (B) The number of tubule extensions emanating per cyst were scored. Greater than 50 cysts were analyzed for each experimental condition. Multiple tubules extensions were formed from MDCKARF6-Q cysts, whereas tubule initiation was blocked in MDCKARF6-T cells (image along the surface of the MDCKARF6-T cyst is shown). ARF6-GTP localized to tubule extensions.
ARF6 is required for ERK signaling during tubule development
We were interested in investigating the mechanism by which ARF6 regulates tubule formation. Recent studies have pointed toward an indispensable role for ERK signaling in the initial phase of tubulogenesis (O'Brien et al, 2004). Earlier studies from our laboratory and others' have shown that ARF6 is required for ERK activation in migratory tumor cells (Tague et al, 2004; Li et al, 2006). In addition, ARF6 is also activated downstream of HGF in epithelial cells (Santy and Casanova, 2001a; Palacios and D'Souza-Schorey, 2003). Thus, we investigated whether ARF6 regulates ERK activation downstream of HGF signaling during tubule formation. To this end, we examined the ERK activation profile in parental MDCK cells as well as in MDCKARF6-Q and MDCKARF6-T cell lines during tubule formation. For these studies, lysates of parental MDCK cells and MDCKARF6-Q and MDCKARF6-T cell lines were probed with antibodies that recognize the dually phosphorylated, activated form of ERK. Consistent with previous reports (O'Brien et al, 2004), ERK was transiently activated during the early stages of tubulogenesis (Figure 3A, upper panel). Notably, this spike in ERK activation occurred about the same time at which ARF6-GTP levels transiently increase during tubulogenesis. In MDCKARF6-Q cells, the downregulation of ERK activity, typically observed as tubules mature, was not observed. Instead, ERK activity was increased and persisted during tubulogenesis (Figure 3A, middle panel). This sustained ERK activation may explain the increased but immature tubular extensions observed in MDCKARF6-Q cell cultures. In support of this contention, we found that the formation of tubule extensions in MDCKARF6-Q cells was completely blocked in the presence of the MEK inhibitor, PD98059 (data not shown). Analyses of MDCKARF6-T lysates revealed a complete block in increased ERK activation following HGF treatment of cysts (Figure 3A, lower panel). Thus, just as ARF6 is required for ERK activation during tumor cell invasion, the above findings indicate that ARF6 activation is also required for the transient spike in ERK phosphorylation during epithelial tubule formation.
Figure 3.

(A) The ARF6 GTP/GDP cycle regulates ERK activation during tubulogenesis. Cysts formed by 3D culture of MDCKARF6-Q cells, MDCKARF6-T cells and parental MDCK cells were treated with HGF for the times indicated. At each time point, cultures were lysed, quantitated for protein content, followed by resolving equal amounts of lysates by SDS–PAGE. Gels were probed for ERK and phospho-ERK labeling using Western blotting procedures. Band density was measured by densitometry and the ratios of phospho-ERK to total ERK are shown. Values are the mean of at least three separate experiments ±s.d. (error bars). (B) ARF6-GTP expression is sufficient for tubule initiation. MDCKARF6-Q cells were grown in matrigel to form fully developed cysts and then induced to express HA-tagged mutant ARF6 by removal of Dox from the growth medium. Cysts were labeled for HA (red), actin (green) and nuclei (blue) and were visualized by laser scanning confocal microscopy. At 24–48 h post induction, tubular extensions from the basolateral surface were observed. Scale bar, 10 μm.
As ERK activation alone has been reported to initiate tubule extensions (O'Brien et al, 2004), we examined whether ARF6(Q67L) expression alone would be sufficient to induce tubule initiation in the absence of growth factor treatment. For these studies, fully developed cysts in 3D culture were induced for ARF6-GTP expression. Twenty-four to forty-eight hours later, more than 50% of cysts in culture developed structures that resembled those that are formed when parental MDCK cells are exposed to HGF for 24 h (Figure 3B), 24–48 h later. Further, only one or two extensions per cyst were observed. Thus the number of tubule extensions formed per cyst was about three-fold lower in the absence of growth factors. It is possible that the presence of HGF promotes a positive feedback loop that amplifies the signal required for the initiation of tubulogenesis. In addition, HGF might also turn on other ARF6-independent pathways that facilitate tubule outgrowth. We also found that ERK levels were constitutively upregulated post induction of ARF6-GTP expression (Supplementary Figure 3) and treatment with the MEK inhibitor, PD98059, abolished ARF6(Q67L)-induced tubule initiation (data not shown). Also, consistent with data described above, ARF6(Q67L)-induced structures did not develop into chains and chords, again suggesting that ERK downregulation is required for further tubule development. Taken together, the studies described above indicate that activation of ARF6 is necessary and sufficient to induce tubule initiation via a process that is dependent on ERK signaling.
ARF6 regulates Rac1 activity during tubulogenesis
Rac1, like ARF6, is member of Ras superfamily of small GTPases. Rac1 is an important regulator of actin remodeling in a variety of cell types including epithelia. Several laboratories have shown that ARF6-GTP can regulate Rac1 activity (Donaldson, 2003; D'Souza-Schorey and Chavrier, 2006). Given the requirement for cytoskeleton remodeling during the initial stages of tubule development, we examined the profile of Rac1 activation during tubule development and its regulation by ARF6. To this end, Rac1-GTP levels from cell lysates at different stages of tubule development were examined as described previously (Benard and Bokoch, 2002). As seen in Figure 4A, Rac1-GTP levels increase in response to HGF treatment and then revert to basal levels at the tubule maturation stage. However, in MDCKARF6-Q cultures, a relatively stronger and sustained activation of Rac1 was observed. In contrast, in MDCKARF6-T cell cysts, the increase in Rac1-GTP levels in response to HGF treatment was completely inhibited. These studies indicate that ARF6 regulates Rac1 activation, a step that appears to be critical for tubule initiation.
Figure 4a.
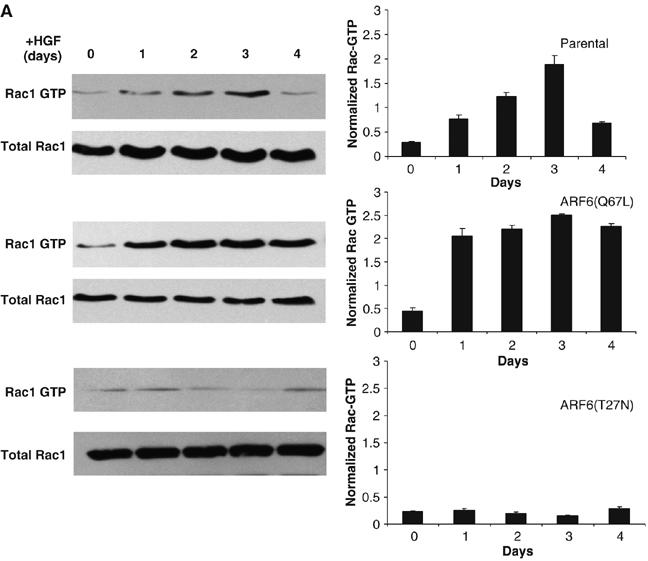
ARF6 regulates Rac1 activation and distribution. (A) Cysts formed by 3D culture of MDCKARF6-Q cells, MDCKARF6-T cells and parental MDCK cells were treated with HGF for the times indicated. At each time point, cultures were lysed, quantitated for protein content and Rac1-GTP was precipitated from equal amounts of cell lysates using the PAK pull-down assay. Rac1-GTP levels were detected by immunoblotting procedures. Cell lysates were also examined for total Rac1 expression by Western blotting procedures. Band intensities were quantified by densitometry and the ratio of Rac1-GTP to total Rac1 is shown. Values are the mean of at least three separate experiments ±s.d. (error bars).
Next, we examined the subcellular distribution of Rac1 in MDCKARF6-Q and MDCKARF6-T cells grown in 3D culture post-HGF treatment, relative to Rac1 distribution in HGF-induced tubules formed by parental MDCK cells. We found that Rac1 was localized largely to the basolateral membrane of cyst cells as well as to HGF-induced tubule extensions (Figure 4B). Little to no labeling was detected at the apical surface facing the cyst lumen. In MDCKARF6-Q cells, a significant amount of Rac1 along with ARF6-GTP was detected on the many tubules, extending radially from the cyst body (Figure 4B.) In marked contrast, however, in MDCKARF6-T cells the distribution of Rac1 at the basolateral surface was diminished. Instead, Rac1 was present in the cytoplasm, and some labeling toward the apical end of cells was also observed (Figure 4B). Quantitation of Rac1 at the surface membrane and internal pools in cysts formed by MDCKARF6-Q and MDCKARF6-T cells are provided as Supplementary data (see Supplementary Figure 4). Previous studies have suggested that ARF6 is involved in localizing Rac1 to the plasma membrane, a prerequisite to Rac1 functioning in cytoskeletal remodeling in response to some extracellular agonists (Radhakrishna et al, 1999; Zhang et al, 1999; Boshans et al, 2000). Here, we show that expression of dominant-negative ARF6 resulted in the mislocalization of Rac1 and its recruitment to the lateral membrane was perturbed. Taken together, these findings indicate the importance of Rac1 distribution and activation downstream of ARF6 signaling during tubule development.
Figure 4b.
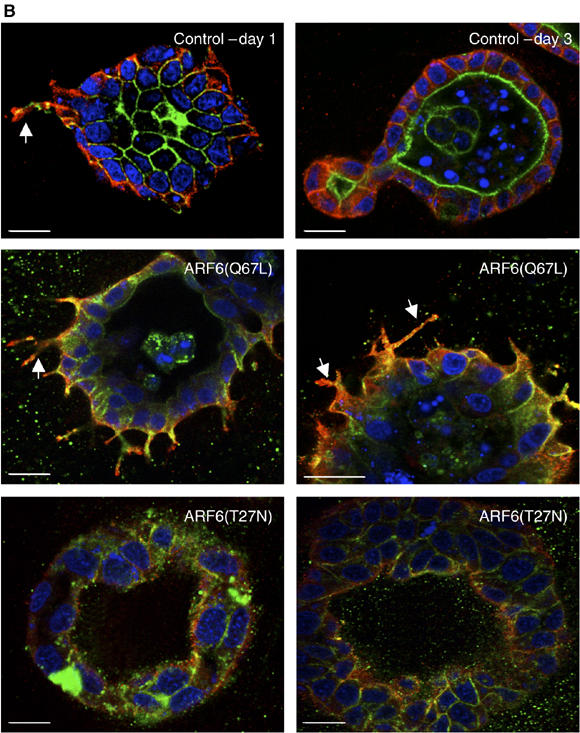
(B) Cysts formed from MDCKARF6-Q and MDCKARF6-T cells and parental MDCK cells were exposed to HGF. Parental cell cultures (upper panel) were labeled for Rac1 (red), actin (green) and cell nuclei (blue). 3D cultures of MDCKARF6-Q (middle panel) and MDCKARF6-T cells (lower panel) were labeled for Rac1 (red) and HA-ARF6 (green) and cell nuclei (blue). Only merged images are shown. Scale bar, 10 μm. Rac1 distribution on extending tubules is indicated by arrows. For clarity of Rac1 distribution, an image showing only Rac1 staining (red) and nuclei (blue) is shown in Supplementary Figure 7.
ARF6-dependent Rac1 activation requires ERK activity
The studies described above have shown that ARF6 is required for the activation of ERK as well as Rac1 during tubule development. Rac1 has been shown to activate ERK by recruitment of PAK1 (p21 adhesion kinase), which in turn phosphorylates upstream kinases, Raf-1 and MEK, of the Raf → MEK → ERK pathway (Frost et al, 1996; King et al, 2001). Thus, we questioned whether ARF6 via Rac1 might control the activation of MEK and/or ERK by promoting the activation of upstream kinases. To this end, we first determined whether Rac1 activation occurs upstream of ERK by monitoring Rac1-GTP levels in the presence of 20 μm PD98059, the MEK inhibitor. For these studies, MDCK cysts treated with HGF and PD98059 were subjected to PAK pull-down assays. Contrary to our expectations, we found that the HGF-induced increase in Rac1-GTP levels were abolished in the presence of PD98059 (Figure 5A), suggesting that Rac1 activation requires ERK activity, and likely occurs downstream of ERK during tubulogenesis. PD98059 also inhibited HGF-induced Rac1 activation in MDCKARF6-Q cultures (Figure 5A), supporting the hypothesis that ARF6-GTP-dependent ERK activation is required for and occurs upstream of Rac1 activation during epithelial tubule development.
Figure 5.
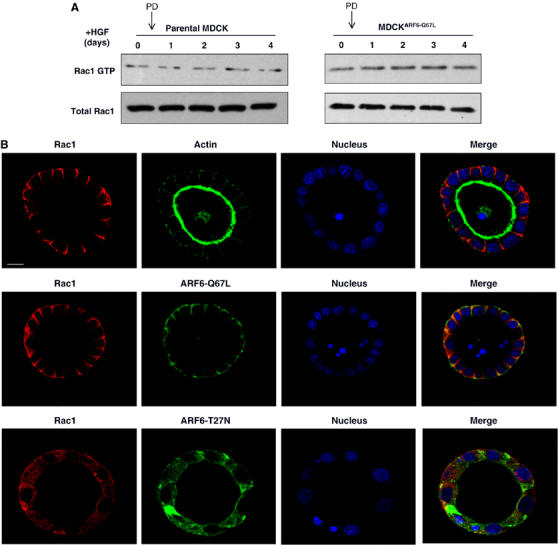
ARF6-dependent Rac1 activation but not Rac1 localization, requires ERK activity. (A) Cysts formed by 3D culture of MDCKARF6-Q and parental MDCK cells were treated with HGF for the times indicated, in the presence of 20 μM PD98059. Cultures were lysed, quantitated for protein and equal amounts of lysates were subjected to PAK pull-down assay to determine levels of Rac1-GTP, as described earlier. Cell lysates were also probed for total Rac1 levels. Treatment with PD98059 completely blocked the increase in Rac1-GTP activation in both parental as well as ARF6(Q67L)-expressing tubules (compare with Figure 4A). (B) Cysts formed from MDCKARF6-Q and MDCKARF6-T cells and parental MDCK cells were treated to HGF in the presence of 20 μM PD98059. Cysts were fixed, labeled for Rac1 (red), ARF6-HA or actin (green) and nuclei (blue) and processed for immunofluorescence microscopy. Images of cross-sections through the cyst center are shown. Scale bar, 10 μm.
We then examined whether the distribution of Rac1 was dependent on ERK activation. As shown in Figure 5B, treatment of cysts with PD98059 completely inhibited tubule formation, but had no effect on Rac1 distribution in parental MDCK cysts as well as MDCKARF6-Q and MDCKARF6-T cysts. These findings indicate that ARF6-regulated ERK activation is required to activate Rac1, but is not required for ARF6-mediated redistribution of Rac1 to the lateral membrane.
uPAR signaling is required for ARF6-ERK-dependent Rac1 activation during tubule development
The urokinse-type plasminogen activator receptor (uPAR) has been shown to be involved in the regulation of cell motility in variety of cell types (Blasi and Carmeliet, 2002; Kjoller, 2002). Furthermore, the DOCK180/Elmo/P130Cas complex, which mediates Rac1-GTP activation downstream of ARF6 (Santy et al, 2005), is also activated by uPAR signaling (Kjoller and Hall, 2001). In addition, activation of the Ras-ERK pathway has been shown to regulate uPAR expression via the AP1 transcription factor (Aguirre Ghiso et al, 1999; Dang et al, 1999; Muller et al, 2000; Vial et al, 2003). On the basis of these reports, we tested the hypothesis that ARF6/ERK-regulated Rac1 activation is regulated by uPAR expression during tubule development. Hence, we first examined the effects of expressing the ARF6 GTP-and GDP-bound mutants on uPAR mRNA and protein levels during tubule development. For these investigations, fully developed epithelial cysts formed from parental MDCK cells, MDCKARF6-Q cells, and MDCKARF6-T cells, were treated with HGF, followed by RNA isolation at various stages of tubule development. Quantitative RT–PCR was performed to examine uPAR mRNA levels as described in Materials and methods. As shown in Figure 6A, in 3D cultures of parental MDCK cells, there was an increase in uPAR transcription at day 1 and 2 post-HGF treatment, which correlated with when ERK activity is increased during tubulogenesis. In MDCKARF6-Q cultures, uPAR mRNA levels were higher than that seen in control cells and these levels persisted for days post HGF treatment. Contrary to these observations, no increase in uPAR transcription was detected in MDCKARF6-T cysts. It should be noted that uPAR protein levels correlated with its mRNA profile during tubule development both in the presence and absence of ARF6 mutants (Figure 6B).
Figure 6.
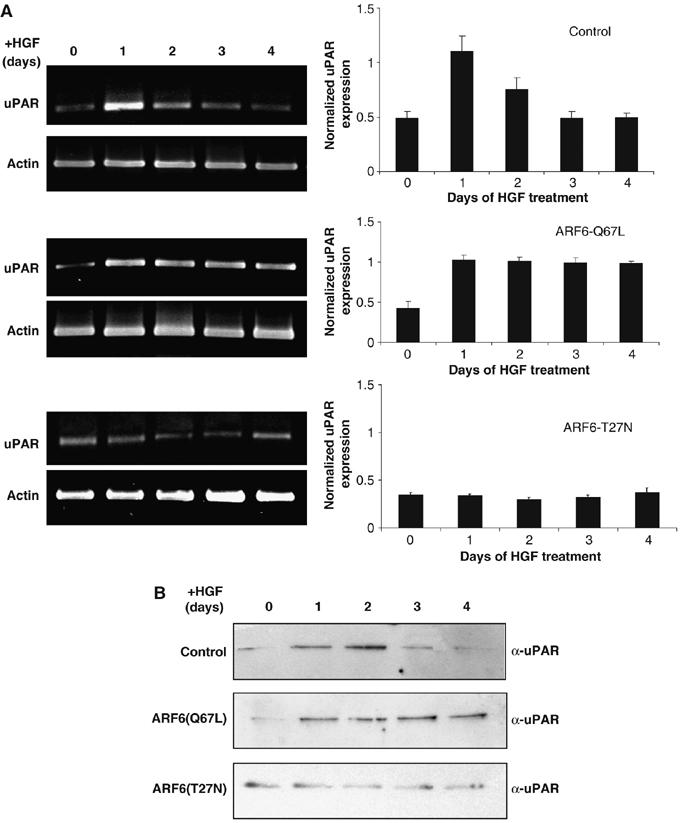
ARF6 GTP/GDP cycle regulates uPAR expression. (A) Cysts formed from MDCKARF6-Q cells, and MDCKARF6-T and parental MDCK cells were exposed to HGF for varying time periods as indicated and subjected to RT–PCR analysis to measure uPAR mRNA levels. Levels of uPAR mRNA were normalized to actin mRNA levels and the ratio of uPAR to actin mRNA is shown. Values are the mean of three separate experiments ±s.d. (error bars). (B) Lysates of cysts were also monitored for uPAR protein levels by Western blotting procedures. uPAR mRNA as well as protein levels are transiently increased at early stages (days 1 and 2) of tubulogenesis. In MDCKARF6-Q cells, there is a sustained increase in uPAR expression whereas in MDCKARF6-T cells, the increase in uPAR expression above basal levels is inhibited.
To determine that the increase in uPAR cellular levels was dependent on ERK activity, we examined the effect of PD98059 on uPAR transcription. Treatment with PD98059 blocked HGF-induced uPAR transcription in parental MDCK cells as well as MDCKARF6-Q cells (data not shown). These findings are consistent with a requirement for a HGF-induced signaling pathway involving ARF6 → ERK → uPAR, during the formation of epithelial tubules.
Next, we investigated whether inhibition of uPAR signaling would prevent Rac1 activation by examining the effect of (1) a ‘blocking' uPAR antibody, R3, and (2) the anti-tumor drug geldanamycin (GA), on tubule initiation. The monoclonal antibody, R3, which recognizes the D1 domain of uPAR, essential for binding of the receptor to uPA, has been shown to block Rac1 activation in cells transfected with uPAR (Kjoller and Hall, 2001). GA belongs to a group of compounds that has been thought to have an inhibitory effect on uPA activity as well as chapraones such as HSP90 (Xie et al, 2005). Although GA does not affect basal uPA activity, it is an extremely potent inhibitor of HGF-induced uPA activity, likely via its effect on a non-HSP90 molecular target, and effectively inhibits HGF/SF-meditated cell invasion (Webb et al, 2000; Xie et al, 2005). Analyses of Rac1-GTP levels in the presence of the R3 antibody as well as GA during the process of tubulogenesis revealed a marked decrease in Rac1 activation (Figure 7A). Further, whereas GA treatment resulted in greater than 80% decrease in the number of tubules formed per cyst in both parental as well as MDCKARF6-Q cells (Figure 7B), the presence of the R3 antibody completely abolished tubule initiation (Figure 7C). Of note is that cellular Rac1 was appropriately distributed to the basolateral surface of parental as well as MDCKARF6-Q cysts exposed to GA or R3. Thus although ARF6-regulated Rac1 targeting to the membrane is not perturbed, its activation at the membrane was significantly reduced. Taken together, these studies suggest that uPAR signaling downstream of HGF → ARF6 → ERK is required for Rac1-GTP activation during tubule development.
Figure 7.
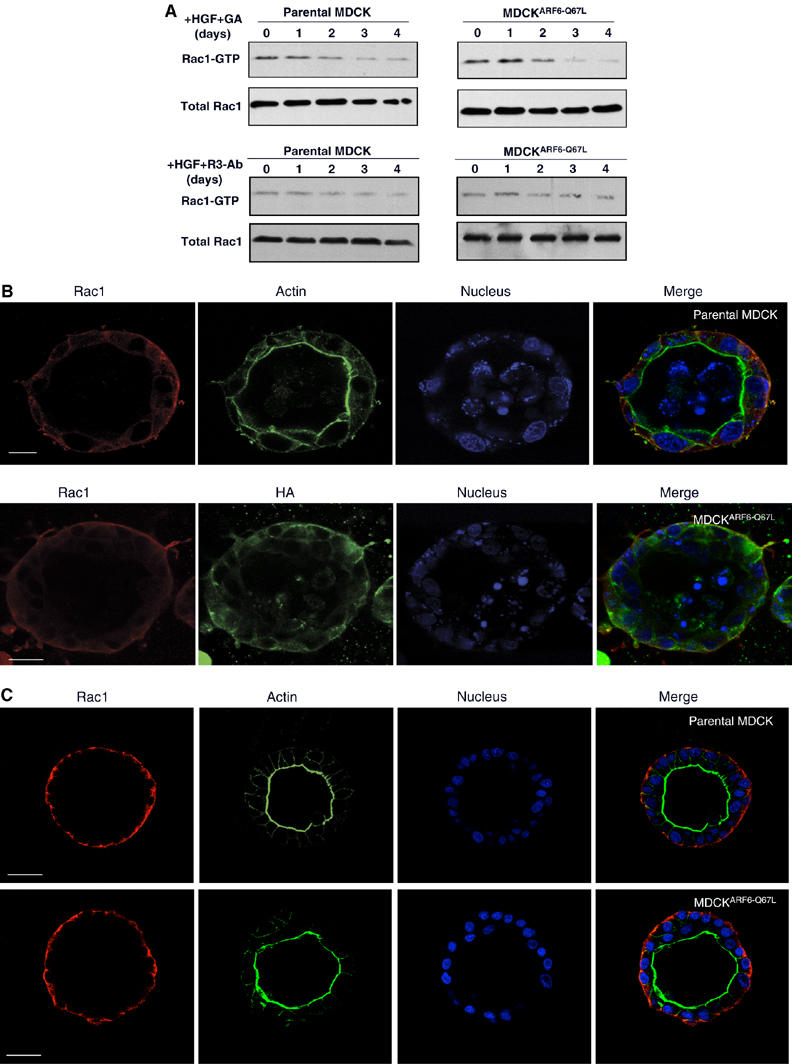
ERK-dependent Rac1 activation requires uPAR signaling. (A) Cysts formed by 3D culture of MDCKARF6-Q cells and parental MDCK cells were treated with HGF for the times indicated, in the presence of the R3 monoclonal antibody (50 μg/ml) or GA (1 μM/ml). Cultures were lysed, quantitated for protein content and Rac1-GTP was precipitated from cells using the PAK pull-down assay. Culture lysates were also probed for total Rac1 levels. Treatment with GA and the R3 antibody inhibits the increase in Rac1-GTP activation in both parental as well as ARF6(Q67L)-expressing tubules. (B) Cysts formed from MDCKARF6-Q and parental MDCK cells were exposed to HGF and GA for 4 days. Cultures were fixed and processed for immunofluorescence microscopy. Parental cell cultures (upper panel) were labeled for Rac1 (red), actin (green) and cell nuclei (blue). 3D cultures of MDCKARF6-Q cells were labeled for Rac1 (red) and HA or actin (green) and cell nuclei (blue). Scale bar, 10 μm. (C). Cysts formed from MDCKARF6-Q and parental MDCK cells were exposed to HGF and the anti-uPAR R3 antibody for 4 days. Cultures were fixed and labeled for Rac1 (red), actin (green) and cell nuclei (blue). Scale bar, 10 μm. Rac1 was present on the basolateral membrane. Tubule extensions formed from MDCKARF6-Q cells are dramatically diminished in the presence of GA or the R3 antibody (compare with Figures 2 and 4B).
Discussion
In the present study, we have described an important role for ARF6 in the regulation of cellular processes as well as specific signaling pathways, all of which affect tubule initiation. Perturbing the ARF6 GTP/GDP cycle by expression of ARF6-GTP- or ARF6-GDP-bound mutants inhibits the development of mature tubules. Whereas expression of a constitutively activated ARF6-GTP mutant increases the number of HGF-induced tubule extensions, albeit immature tubule extensions, expression of the dominant-negative ARF6-GDP mutant completely blocks tubule extension in response to HGF. Thus, nucleotide cycling on ARF6 is required to ensure normal progression of tubule development. We also show that sustained activation of ARF6 alone is sufficient to induce tubule initiation. A working model for the role of ARF6 in the early stages of tubule development is shown in Figure 8. ARF6-GTP facilitates tubule initiation via its effect on the subcellular distribution of Rac1, a well-known regulator of actin cytoskeleton remodeling. In addition, Rac1 activation during tubule initiation is dependent on ARF6-induced ERK activation. ERK signaling controls Rac1 activation through the expression of uPAR. According to this model, cells in which ERK is constitutively activated, endogenous ARF6 is still functional and Rac1 would be efficiently targeted to the cell surface, and thus constitutively activated ERK, would be expected to bypass the requirement for HGF, in full agreement with the study by O'Brien co-workers.
Figure 8.

Working model for the role of ARF6 in tubule initiation. The diagram shows the sequence for ARF6, ERK and Rac1 activation during epithelial tubulogenesis. HGF-induced ARF6 activation facilitates tubule initiation by promoting the recruitment of Rac1 to the cell surface. Rac1 activation at the plasma membrane is dependent on ARF6-induced ERK activation. ERK signaling controls Rac1 activation via the expression of uPAR. Rac1 activation downstream of uPAR activation may occur by recruitment of the DOCK180/Elmo complex, which has been shown to mediate Rac1 activation downstream of ARF6. According to this model, constitutively activated ERK would be expected to bypass the requirement for HGF, as endogenous ARF6 is still functional, and Rac1 would be efficiently targeted to the cell surface, in agreement with the study by O'Brien et al (2004).
The requirement for ARF6 in Rac1 distribution is manifested in the mislocalization of Rac1 to the lateral membrane of ARF6-T27N expressing cells. As a result cytoskeletal remodeling required for tubule initiation is inhibited. In contrast, the increased levels of Rac1 at the cell surface observed upon constitutive activation of ARF6 may explain the hypertubulation phenotype observed in MDCKARF6-Q cysts. These findings are consistent with previous studies demonstrating that ARF6-regulated membrane recruitment of Rac1 to the plasma membrane is required for the development of surface protrusions as in pseudopodia formation, membrane ruffling, cell migration, neurite extension and myoblast fusion (D'Souza-Schorey and Chavrier, 2006). ARF6 by itself can promote surface rearrangements via its effect on lipid metabolism (Donaldson, 2003; D'Souza-Schorey and Chavrier, 2006). In addition, ARF6 synergizes with Rac1 to facilitate cortical actin remodeling (Radhakrishna et al, 1999; Boshans et al, 2000).
ARF6-regulated ERK activation also appears to be critical for tubule extension. In fact, ARF6 activation coincides with ERK activation during tubule development. As described previously, we find that ERK activation is transiently upregulated during HGF-induced tubulogenesis. However, upon constitutive activation of ARF6, ERK activation persists, suggesting that GTP hydrolysis on ARF6 may facilitate ERK downregulation. It is also possible that ERK downregulation might be coupled to tubule maturation. As a result, immature tubules are formed in ARF6-Q67L-expressing cells. Consistent with this contention, previous reports have shown that prolonged ERK activation prevents redifferentiation during tubule development (O'Brien et al, 2004). ARF6-regulated ERK phosphorylation has previously been described and has been shown to be required for melanoma and glioma cell invasion (Tague et al, 2004; Li et al, 2006). Indeed, matrix invasion of tubules is highly reminiscent of the process of tumor cell invasion, so it is likely that these processes are governed by common regulatory mechanisms. A potential mechanism by which ARF6 might dictate ERK activation is by regulating the trafficking of the HGF receptor, c-Met. It should be noted that total levels of c-Met in parental MDCK, MDCKARF6-Q and MDCKARF6-T cysts are the same (see Supplementary Figure 5), so control of receptor downregulation is not a mechanism for modulation of ERK activation. However, altered distribution of the receptor as a result of ARF6-regulated receptor trafficking, cannot be ruled out at this time. ARF6-mediated recycling may also direct the trafficking of components of the ERK pathway to the cell surface to promote ERK signaling. In this regard, a study has shown that ERK, MEK and the scaffolding protein, kinase suppressor of Ras 1, localizes to the ARF6-regulated endosmal compartment in HeLa cells and ARF6 activation leads to increased ERK phosphorylation (Robertson et al, 2006). Of note, this study also demonstrated a complex epistatic relationship between ARF6 and ERK, as treatment of cells with an MEK inhibitor inhibited EFA6 (exchange factor for ARF6)-induced ARF6 activation, albeit to a small extent. However, the findings described here that (1) blocking ERK activation does not prevent the redistribution of ARF6 or Rac1 to the cell surface and (2) no buildup of phosphorylated ERK is observed in MDCKARF6-T cysts, suggests that ERK phosphorylation does not preceed ARF6 activation during tubule initiation.
Although the mechanism by which ARF6-GTP induces ERK activation has yet to be identified, we show here that it is critical for the activation of Rac1. Inhibition of the direct ERK activator, MEK, results in complete inhibition of Rac1 activation, suggesting that Rac1 activation is dependent on and occurs downstream of ERK activation during tubulogenesis. We also show that ERK-induced Rac1 activation is mediated by uPAR signaling. In support of these findings, we found that upon inhibition of uPAR signaling, Rac1 activation and formation of tubule extensions is abolished, although the distribution of Rac1 at the lateral surface is not perturbed. Furthermore, uPAR mRNA and protein levels transiently increase during tubulogenesis, an expression profile that appears to parallel ERK activation. The increase in uPAR expression persists in cells expressing the ARF6-GTP mutant and is blocked in the presence of MEK inhibitors. Rac1 activation downstream of uPAR activation may occur likely by the recruitment of the DOCK180/Elmo/P130Cas complex that has been shown to mediate Rac1 activation downstream of ARF6 (Santy et al, 2005), and is also activated by uPAR signaling (Kjoller and Hall, 2001). Rac1 activation has been reported to be essential for tubulogenesis, as the process is blocked by expression of dominant-negative Rac1 (Rogers et al, 2003). Thus, we have identified a signaling pathway involving HGF → ARF6 → ERK → uPAR → Rac1 that appears to be critical for cytoskeletal remodeling during tubule initiation.
In addition to the effect of ARF6 on Rac1 localization and activation, ARF6 may affect other cellular processes to promote tubule initiation. For instance, ARF6 activation may direct the recruitment of other cargo required for lateral membrane expansion and cytoskeletal remodeling during tubules extension and does so via the recruitment of the exocyst complex. ARF6-GTP was shown to interact with sec10, a subunit of the exocyst complex that localizes to the TGN as well to recycling endosomes, and redistributes to the cell surface upon ARF6 activation (Prigent et al, 2003). Indeed, vesicle delivery to the growing tip of the extending tubule is mediated by the exocyst complex (Lipschutz et al, 2000).
Tubule initiation is also thought to require a partial EMT, wherein cells lose their polarity and the ability to form cell–cell contacts. In this regard, ARF6 has been shown to regulate the stability of the adherens junctions via its effect on E-cadherin trafficking (Palacios et al, 2001, 2002). We find that E-cadherin exhibits a striking cytoplasmic distribution in ARF6(Q67L)-expressing tubules relative to normal MDCK tubules, whereas in cysts expressing the dominant-negative mutant, ARF6(T27N), E-cadherin labeling is largely at sites of cell–cell contact, even in the presence of HGF. Moreover, ERK inhibition completely blocks ARF6-GTP-regulated E-cadherin internalization (see Supplementary Figure 6). Whether this apparent requirement of ERK for cadherin internalization during tubulogenesis is direct or indirect is presently not known and is under investigation in our laboratory.
In summary, via its effect on cytoskeletal remodeling, membrane recycling and the disassembly of cell–cell contacts, the ARF6 GTPase likely orchestrates structural changes required for the initiation of tubule outgrowth. The process of tubulogenesis necessitates the coordinated regulation of cytoskeleton and membrane remodeling, cell adhesion and cell polarization. Further studies in vivo that build on the studies described here will provide further insight into the molecular basis of tube morphogenesis.
Materials and methods
Cell culture
Tet-Off MDCK I cells capable of regulated expression of HA-tagged ARF6-Q67L and HA-tagged ARF6-T27N, MDCKARF6-Q and MDCKARF6-T, respectively, were grown as described previously (Prigent et al, 2003). Cells were maintained in the presence of 1 μg/ml Dox, or switched to Dox-free medium to induce expression of ARF6 proteins. ARF6 mutant proteins are expressed within 24 h post induction. Parental MDCK cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin and streptomycin.
Antibodies and other reagents
Primary antibodies used in these studies were mouse anti-Rac1 (Transduction labs), mouse anti-ERK1/2 (Cell Signaling), Rabbit anti-phospho-ERK1/2 (Cell Signaling), mouse anti-E-cadherin (Iowa), mouse anti-uPAR (Zymed) and mouse anti-HA (Covance). The anti-uPAR mouse monoclonal antibody, R3, was a generous gift from Dr Gunilla Hoyer-Hansen (Finsen Laboratory, Rigshospitalet Copenhagen, Denmark). Secondary antibodies were goat anti-mouse FITC/goat anti-mouse Texas Red or goat anti rabbit FITC/goat anti-rabbit Texas Red (ICN and Molecular Probes). Nuclei were stained using Draq5 (Alexis Biochemicals) and actin filaments were stained with either Rhodamine phalloidin or FITC phalloidin (Molecular Probes). HGF and PD98059 were obtained from Calbiochem and GA was from AG Scientific Inc. (San Diego, CA). High-pure RNA isolation kit (FastStart High Fidelity PCR systems) was obtained from Roche Applied Sciences. iScript cDNA synthesis kit was from Bio-Rad. Matrigel was obtained from BD Biosciences.
Tubule formation in 3D cultures
Growth of cysts in Matrigel (an extract of the copious ECM produced by Englebreth-Holm-Swarm tumors) was performed as described previously (Debnath et al, 2003; Pollack et al, 1998). For MDCKARF6-Q and MDCKARF6-T cells, cyst development was allowed to proceed in presence of 1 μg/ml Dox. To allow the expression of ARF6-Q67L/ARF6-T27N, the medium was replaced with culture medium without Dox. 24 h post induction, fully formed cysts were treated with 60 ng/ml HGF to induce tubulogenesis and growth medium was replaced every 24 h with fresh media containing HGF for up to 4 days to form mature tubules. To downregulate p-ERK expression, 20 μm PD98059 was added to the medium. To block uPAR signaling, 1 μM/ml GA or 50 μg/ml of the R3 antibody was added to the culture medium.
To quantitate the number of tubule extensions formed per cyst, fluorescently labeled 3D cultures were visualized under a fluorescence microscope coupled to a Bio-Rad MRC 1024 scanning confocal three-channel system (see below). For each experimental condition, greater than 50 cysts were visualized and the number of extensions emanating per cyst was scored.
Immunofluorescence and biochemical analysis
3D cell cultures were processed for immunofluorescence and biochemical assays as previously described (Palacios et al, 2001; O'Brien et al, 2004). For generation of protein lysates for biochemical assays, cultures were first treated with 0.25% trypsin to separate the glandular structures from the matrigel. Cells were lysed in RIPA buffer followed by protein quantitation and then assayed for Rac1-GTP using the PAK pull-down assay (Benard and Bokoch, 2002), or for ARF6-GTP using the MT2 pull-down assay (Schweitzer and D'Souza-Schorey, 2002). For immunofluorescence microscopy, cells were fixed in 4% paraformaldehyde followed by staining with primary antibody for 4–5 h. This was followed by incubation with secondary antibody conjugated to fluorophore, as previously described (Palacios et al, 2001, 2002). Coverslips were mounted on glass slides using antifade mounting media, (Molecular Probes) and visualized by laser scanning confocal microscopy. Immunofluorescent imaging was accomplished using a Bio-Rad MRC 1024 scanning confocal three-channel system, which utilizes a krypton–argon laser with excitation filters for 488, 568 and 647 nm and Bio-Rad LaserSharp 2000 software version 4.0.
Quantitative RT–PCR
RNA was extracted from 3D cell culture lysates using high-pure RNA isolation kit (Roche Applied Sciences). RNA (2 μg) was used for cDNA synthesis, as described by the manufacturer (iScript cDNA synthesis kit, Roche Biochemicals). Serial dilutions of cDNA were used for quantitative PCR according to the manufacturer's instructions, along with the following cycles: 95°C for 30 s, 60°C for 30 s and 72°C for 1 m for 35 cycles. The following forward primer sequences were used for uPAR—AGA CAGGAGCTGCTTCCAGA, RV: TCCCTTGGACTGTCTTCCAG, and for actin—ACTGGGACGACATGGAGAAG, RV: CGTCGGGTAGTTCGTAGC TC. The PCR products were quantitated using a NanoDrop-ND 1000 spectrophotometer and resolved on agarose gels. Band densities were measured using an enhanced UltroScan XL densitometer (Pharmacia) and uPAR mRNA levels were normalized against actin mRNA.
Supplementary Material
Supplementary Information and Figures
Acknowledgments
We thank Dr Philippe Chavrier for the MDCKARF6-Q and MDCKARF6-T cell lines, Dr Gunilla Hoyer-Hansen for the anti-uPAR antibody, R3, and Drs Keith Mostov, Julie Donaldson and Jill Schweitzer for critical reading of the manuscript. This work was supported in part by grants from the American Cancer Society and the Leda J Sear's Trust to CD-S.
References
- Aguirre Ghiso JA, Alonso DF, Farias EF, Gomez DE, de Kier Joffe EB (1999) Deregulation of the signaling pathways controlling urokinase production. Its relationship with the invasive phenotype. Eur J Biochem 263: 295–304 [DOI] [PubMed] [Google Scholar]
- Benard V, Bokoch GM (2002) Assay of Cdc42, Rac, and Rho GTPase activation by affinity methods. Methods Enzymol 345: 349–359 [DOI] [PubMed] [Google Scholar]
- Blasi F, Carmeliet P (2002) uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol 3: 932–943 [DOI] [PubMed] [Google Scholar]
- Boshans RL, Szanto S, van Aelst L, D'Souza-Schorey C (2000) ADP-ribosylation factor 6 regulates actin cytoskeleton remodeling in coordination with Rac1 and RhoA. Mol Cell Biol 20: 3685–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza-Schorey C, Chavrier P (2006) ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 7: 347–358 [DOI] [PubMed] [Google Scholar]
- Dang J, Boyd D, Wang H, Allgayer H, Doe WF, Wang Y (1999) A region between -141 and -61 bp containing a proximal AP-1 is essential for constitutive expression of urokinase-type plasminogen activator receptor. Eur J Biochem 264: 92–99 [DOI] [PubMed] [Google Scholar]
- Debnath J, Brugge JS (2005) Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer 5: 675–688 [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30: 256–268 [DOI] [PubMed] [Google Scholar]
- Donaldson JG (2003) Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem 278: 41573–41576 [DOI] [PubMed] [Google Scholar]
- Frost JA, Xu S, Hutchison MR, Marcus S, Cobb MH (1996) Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol Cell Biol 16: 3707–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AJ, Wireman RS, Hamilton M, Marshall MS (2001) Phosphorylation site specificity of the Pak-mediated regulation of Raf-1 and cooperativity with Src. FEBS Lett 497: 6–14 [DOI] [PubMed] [Google Scholar]
- Kjoller L (2002) The urokinase plasminogen activator receptor in the regulation of the actin cytoskeleton and cell motility. Biol Chem 383: 5–19 [DOI] [PubMed] [Google Scholar]
- Kjoller L, Hall A (2001) Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J Cell Biol 152: 1145–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Ng SS, Wang J, Lai L, Leung SY, Franco M, Peng Y, He ML, Kung HF, Lin MC (2006) EFA6A enhances glioma cell invasion through ADP ribosylation factor 6/extracellular signal-regulated kinase signaling. Cancer Res 66: 1583–1590 [DOI] [PubMed] [Google Scholar]
- Lipschutz JH, Guo W, O'Brien LE, Nguyen YH, Novick P, Mostov KE (2000) Exocyst is involved in cystogenesis and tubulogenesis and acts by modulating synthesis and delivery of basolateral plasma membrane and secretory proteins. Mol Biol Cell 11: 4259–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubarsky B, Krasnow MA (2003) Tube morphogenesis: making and shaping biological tubes. Cell 112: 19–28 [DOI] [PubMed] [Google Scholar]
- Muller SM, Okan E, Jones P (2000) Regulation of urokinase receptor transcription by Ras- and Rho-family GTPases. Biochem Biophys Res Commun 270: 892–898 [DOI] [PubMed] [Google Scholar]
- O'Brien LE, Tang K, Kats ES, Schutz-Geschwender A, Lipschutz JH, Mostov KE (2004) ERK and MMPs sequentially regulate distinct stages of epithelial tubule development. Dev Cell 7: 21–32 [DOI] [PubMed] [Google Scholar]
- O'Brien LE, Zegers MM, Mostov KE (2002) Opinion: building epithelial architecture: insights from three-dimensional culture models. Nat Rev Mol Cell Biol 3: 531–537 [DOI] [PubMed] [Google Scholar]
- Palacios F, D'Souza-Schorey C (2003) Modulation of Rac1 and ARF6 activation during epithelial cell scattering. J Biol Chem 278: 17395–17400 [DOI] [PubMed] [Google Scholar]
- Palacios F, Price L, Schweitzer J, Collard JG, D'Souza-Schorey C (2001) An essential role for ARF6-regulated membrane traffic in adherens junction turnover and epithelial cell migration. EMBO J 20: 4973–4986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios F, Schweitzer JK, Boshans RL, D'Souza-Schorey C (2002) ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly. Nat Cell Biol 4: 929–936 [DOI] [PubMed] [Google Scholar]
- Pollack AL, Runyan RB, Mostov KE (1998) Morphogenetic mechanisms of epithelial tubulogenesis: MDCK cell polarity is transiently rearranged without loss of cell–cell contact during scatter factor/hepatocyte growth factor-induced tubulogenesis. Dev Biol 204: 64–79 [DOI] [PubMed] [Google Scholar]
- Powelka AM, Sun J, Li J, Gao M, Shaw LM, Sonnenberg A, Hsu VW (2004) Stimulation-dependent recycling of integrin beta1 regulated by ARF6 and Rab11. Traffic 5: 20–36 [DOI] [PubMed] [Google Scholar]
- Prigent M, Dubois T, Raposo G, Derrien V, Tenza D, Rosse C, Camonis J, Chavrier P (2003) ARF6 controls post-endocytic recycling through its downstream exocyst complex effector. J Cell Biol 163: 1111–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishna H, Al-Awar O, Khachikian Z, Donaldson JG (1999) ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J Cell Sci 112: 855–866 [DOI] [PubMed] [Google Scholar]
- Robertson SE, Setty SR, Sitaram A, Marks MS, Lewis RE, Chou MM (2006) Extracellular signal-regulated kinase regulates clathrin-independent endosomal trafficking. Mol Biol Cell 17: 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KK, Jou TS, Guo W, Lipschutz JH (2003) The Rho family of small GTPases is involved in epithelial cystogenesis and tubulogenesis. Kidney Int 63: 1632–1644 [DOI] [PubMed] [Google Scholar]
- Rosario M, Birchmeier W (2003) How to make tubes: signaling by the Met receptor tyrosine kinase. Trends Cell Biol 13: 328–335 [DOI] [PubMed] [Google Scholar]
- Santy LC, Casanova JE (2001a) Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J Cell Biol 154: 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santy LC, Casanova JE (2001b) Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J Cell Biol 154: 599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santy LC, Ravichandran KS, Casanova JE (2005) The DOCK180/Elmo complex couples ARNO-mediated Arf6 activation to the downstream activation of Rac1. Curr Biol 15: 1749–1754 [DOI] [PubMed] [Google Scholar]
- Schweitzer JK, D'Souza-Schorey C (2002) Localization and activation of the ARF6 GTPase during cleavage furrow ingression and cytokinesis. J Biol Chem 277: 27210–27216 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Kanai Y, Hara T, Sasaki J, Sasaki T, Kohara M, Maehama T, Taya C, Shitara H, Yonekawa H, Frohman MA, Yokozeki T, Kanaho Y (2006) Crucial role of the small GTPase ARF6 in hepatic cord formation during liver development. Mol Cell Biol 26: 6149–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tague SE, Muralidharan V, D'Souza-Schorey C (2004) ADP-ribosylation factor 6 regulates tumor cell invasion through the activation of the MEK/ERK signaling pathway. Proc Natl Acad Sci USA 101: 9671–9676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vial E, Sahai E, Marshall CJ (2003) ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell 4: 67–79 [DOI] [PubMed] [Google Scholar]
- Webb CP, Hose CD, Koochekpour S, Jeffers M, Oskarsson M, Sausville E, Monks A, Vande Woude GF (2000) The geldanamycins are potent inhibitors of the hepatocyte growth factor/scatter factor–met-urokinase plasminogen activator–plasmin proteolytic network. Cancer Res 60: 342–349 [PubMed] [Google Scholar]
- Xie Q, Gao CF, Shinomiya N, Sausville E, Hay R, Gustafson M, Shen Y, Wenkert D, Vande Woude GF (2005) Geldanamycins exquisitely inhibit HGF/SF-mediated tumor cell invasion. Oncogene 24: 3697–3707 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Calafat J, Janssen H, Greenberg S (1999) ARF6 is required for growth factor- and rac-mediated membrane ruffling in macrophages at a stage distal to rac membrane targeting. Mol Cell Biol 19: 8158–8168 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information and Figures