

Abstract
Vascular smooth muscle cells can perform both contractile and synthetic functions, which are associated with and characterised by changes in morphology, proliferation and migration rates, and the expression of different marker proteins. The resulting phenotypic diversity of smooth muscle cells appears to be a function of innate genetic programmes and environmental cues, which include biochemical factors, extracellular matrix components, and physical factors such as stretch and shear stress. Because of the diversity among smooth muscle cells, blood vessels attain the flexibility that is necessary to perform efficiently under different physiological and pathological conditions. In this review, we discuss recent literature demonstrating the extent and nature of smooth muscle cell diversity in the vascular wall and address the factors that affect smooth muscle cell phenotype. (Neth Heart J 2007;15:100-8.)
Keywords: muscle (smooth) , muscle (vascular) , cell differentiation, phenotype, diversity, synthetic, contractile
Smooth muscle cells (SMCs) are essential for good performance of the vasculature. By contraction and relaxation, they alter the luminal diameter, which enables blood vessels to maintain an appropriate blood pressure. However, vascular SMCs also perform other functions, which become progressively more important during vessel remodelling in physiological conditions such as pregnancy and exercise, or after vascular injury.1 In these cases, SMCs synthesise large amounts of extracellular matrix (ECM) components and increase proliferation and migration. Because of these properties, SMCs are fit not only for short-term regulation of the vessel diameter, but also for long-term adaptation, via structural remodelling by changing cell number and connective tissue composition.
The different functions that SMCs can exert translate into a diversity of SMC phenotypes, ranging from contractile to synthetic. The diversity becomes apparent in morphology, expression levels of SMC marker genes, proliferative potential and migration properties. These differences are observed between SMCs of different vessels as well as amongst SMCs within the same vessel. Part of the variation in SMC populations can be explained by the diverse embryological origins of SMCs, reviewed by Gittenberger-de Groot et al.2 However, the remarkable capability of SMCs to shuttle between different phenotypes, referred to as phenotypic modulation,3 may be more important, as it is superimposed on the origin-related phenotype.
The mechanisms by which SMCs acquire a particular phenotype are the subject of intense research. This review will focus on the diversity of SMC phenotypes in the vessel wall and the factors and conditions that control it. We start with a brief description of the characteristics of different vascular SMC phenotypes and discuss the lack of definition of those phenotypes. Subsequently, the nature of SMC diversity in the vessel wall will be addressed before describing the factors and conditions that control it.
Characteristics of vascular smooth muscle cell diversity
Contractile and synthetic SMCs, which represent the two ends of a spectrum of SMCs with intermediate phenotypes, have clearly different morphologies. Consequently, morphology is still an important parameter for the definition of SMC phenotypes, although the use of marker proteins for this purpose has become customary. Contractile SMCs are elongated, spindleshaped cells, whereas synthetic SMCs are less elongated and have a cobblestone morphology which is referred to as epithelioid or rhomboid (figure 1).3,4 Synthetic SMCs contain a high number of organelles involved in protein synthesis, whereas these are largely replaced by contractile filaments in contractile SMCs. Moreover, synthetic and contractile SMCs have different proliferative and migratory characteristics. Generally, synthetic SMCs exhibit higher growth rates and higher migratory activity than contractile SMCs.3
Figure 1.
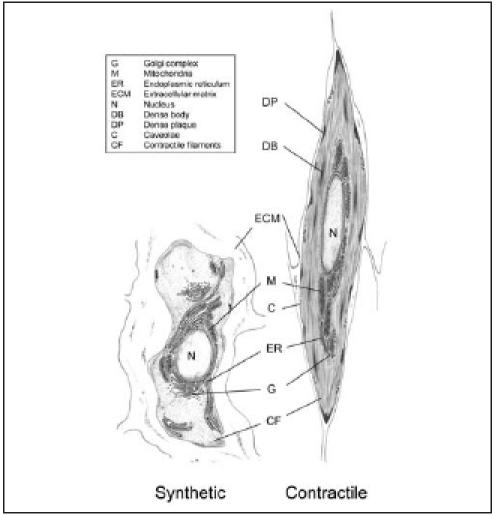
Ultrastructural characteristics of contractile and synthetic SMCs.
Protein markers of vascular smooth muscle cell phenotypes
The SMC marker proteins that are commonly used to define SMC phenotypes have recently been extensively reviewed.1,5 The markers that are most relevant to this review (α-smooth muscle actin (α-SMA), smooth muscle-myosin heavy chain (SM-MHC), smoothelin- A/B, SMemb/non-muscle MHC isoform-B and cellular retinol binding protein (CRBP)-1) and some additional markers that are often used are presented in table 1. Many of these proteins are involved in SMC contraction, either as a structural component of the contractile apparatus or as a regulator of contraction.
Table 1.
Characteristics of widely used SMC marker proteins.
| Marker protein | SMC specificity | Phenotype specificity | Subcellular localisation | Onset of expression | Function |
|---|---|---|---|---|---|
| α-smooth muscle actin Smooth muscle-myosin heavy chain | Embryo -Adult +/-Embryo + Adult + | c>s SM1 c>s SM2 c | Contractile filaments Contractile filaments | E9 heart/ somites/SMCs E10.5 SMCs | Contraction Contraction |
| SM22α | Embryo -Adult + | c>s | Actin-associated | E8 heart/ E9.5 SMCs | Regulation contraction |
| SM-calponin H-caldesmon | Embryo -Adult + Embryo + Adult + | c>s c | Actin-associated/ cytoskeleton Actin-associated | E8.5 heart / E13.5 SMCs WK 10 human | Regulation contraction/ signal transduction Regulation contraction |
| Smoothelin | Embryo -Adult + | c | Actin-associated | E13 | Regulation contraction |
| Telokin | Embryo+/? Adult + | c>s | Cytoplasmic/ membrane | E11.5 gut | Regulation contraction |
| Meta-vinculin | Embryo -Adult - | c>s | Cytoskeleton | WK 24 human | Anchoring cell-ECM |
| Desmin CRBP-1 | Embryo -Adult -Embryo -Adult - | c>s s>c | Cytoskeleton Cytoplasm | E9.5 myotome E10 | Structural mechanical integrity Retinoid transport and metabolism |
| Smemb | Embryo -Adult - | s>c | Contractile filaments | ND | Contraction |
Compiled from Owens et al. and Miano.1,5
c=contractile, s=synthetic, E=mouse embryonic day, WK 24=week 24 of human development, ND=not determined.
The expression levels of these contractile marker proteins gradually decrease when SMCs are cultured, although the extent to which their expression is downregulated differs among the markers.6 Markers that are upregulated in the synthetic phenotype are rare. Instead, disappearance of proteins associated with the contractile phenotype is generally taken as characteristic of the synthetic phenotype (figure 2). SMCs with different phenotypes express varying levels of the marker proteins rather than completely different marker proteins. Therefore, information on the expression of at least two proteins that are associated with a particular phenotype is required to distinguish a contractile from a synthetic SMC, preferably complemented with data on morphology, proliferation and migration characteristics.
Figure 2.
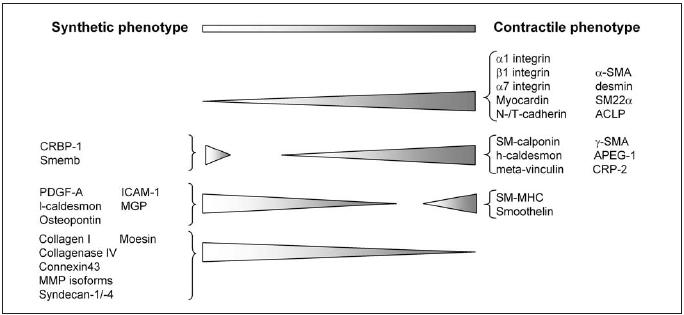
Schematic representation of expression levels of genes associated with a particular SMC phenotype. Note that most of the indicated proteins are not SMC specific.
At present, the two marker proteins that provide the best definition of a mature contractile SMC phenotype are SM-MHC and smoothelin. SM-MHC expression has never been detected in non-SMCs in vivo, and is the only marker protein that is also SMC specific during embryogenesis.7 Smoothelin complements SM-MHC as a contractile SMC marker in that it appears to be more sensitive. Its expression is more uniformly and more rapidly downregulated in cultured vascular SMCs modulating towards a synthetic phenotype.6 Both SMMHC and smoothelin have been shown to be absent in myofibroblasts in models of arterial injury.8
SMemb/non-muscle MHC isoform-B and, in rodents, CRBP-1 represent suitable synthetic SMC markers, since these proteins are quickly and markedly upregulated in proliferating SMCs.9,10 In addition, changes in ratios of specific splice variants of contractile proteins (h-caldesmon, meta-vinculin) can be used to indicate a synthetic phenotype.11
Occurrence of vascular SMC diversity
The nature of SMC diversity in the vessel wall is well illustrated by immunohistochemical staining patterns of contractile SMC phenotype markers. These, almost invariably, reveal highly heterogeneous staining patterns and intensities between adjacent SMCs.6,12 These differences become more distinct after vascular injury. Although the overall expression level of contractile SMC phenotype markers after vascular injury initially goes down, the response is remarkably heterogeneous between the different SMCs in the vessel wall.8 Furthermore, re-expression of contractile SMC markers at later time points does not occur uniformly but, rather, first in subpopulations of cells.13 More detailed information on the nature of SMC diversity has been obtained by analyses of primary cultures.6,14,15 These studies show that immediately after enzymatic digestion, SMCs contain not only different amounts of contractile proteins, but also that some SMCs do not express particular contractile markers at all.
Not only contractile SMC marker proteins reflect SMC diversity in the arterial wall. Gap junctional proteins and adhesion molecules are also differentially expressed. A complex differential expression of integrin subunits in different SMC subsets has been described.16 Ncadherin and T-cadherin are also differentially expressed in aortic SMC layers, with higher levels in SMCs adjacent to endothelial cells. In addition, SMCs of elastic neural crest derived arteries express connexin43, in contrast to SMCs of muscular arteries.
The human internal mammary artery, which has both elastic and muscular segments, shows inversely correlated levels of connexin43 and desmin in SMCs of these different parts.17
The variations between SMCs within a particular vessel, both with respect to marker gene expression and functional and morphological characteristics, suggest that there may be a genetic basis for SMC diversity. After all, such SMCs share similar embryological origins and experience comparable local conditions. This concept is substantiated by several studies reporting the persistence of in vivo phenotypes in culture, despite changed conditions. For example, within the bovine pulmonary artery, four SMC phenotypes with distinct marker protein expression profiles and different morphologies have been described.18 The existence of distinct SMC populations within the same artery has also been demonstrated in the rat,14 the pig,6,15 and in humans.19 In all these studies, in vivo differences were maintained in vitro. SMCs isolated from the human internal thoracic artery form a particularly illustrative example.19 These SMCs were cloned after enzymatic digestion, producing epithelioid as well as spindleshaped cell types. Both had a contractile gene expression profile, but only spindle-shaped cells expressed meta-vinculin. They also had higher SM-MHC and SM-calponin levels and a higher h-caldesmon/lcaldesmon ratio. Apart from these differences, the two types of clones differed with respect to proliferation rate, ECM build-up and responses to various growth factors and hormones. One typical spindle-shaped clone, designated HITB5, was able to adopt either a synthetic or a contractile phenotype, depending on serum concentrations.20 This shows that even though SMC phenotypes can be stable in culture, they can also be manipulated to adopt a certain phenotype, allowing the study of agents that modulate phenotypes. SMC clones that are capable of reversible modulation to both ends of the phenotype spectrum have also been derived from porcine coronary artery SMCs.15 These cells displayed phenotypic modulation after fibroblast growth factor (FGF)-2 or platelet-derived growth factor (PDGF)-B treatment or withdrawal. PDGF-B drove spindle-shaped SMC clones towards the rhomboid phenotype. Concomitantly, proliferation and migration increased and SM-MHC and smoothelin expression greatly diminished.
The studies summarised above indicate that although SMC phenotype appears to be genetically programmed, local environmental cues can still modulate the characteristics of the SMCs. This raises the question of the relative importance of the local environment versus genetic programming. An elegant study in which cultured arterial SMCs with different phenotypes were implanted into rat carotid arteries provided an indication that genetic programming may be at least as important as the local environment.21 The implanted cells, either spindle-shaped cells from newborn rats or epithelioid cells derived from old rats, retained their specific expression pattern of α-SMA and SM-MHC in vivo. Moreover, the epithelioid SMCs maintained expression of CRBP-1 for at least 20 days. Thus, there is also in vivo evidence that diversity is an intrinsic quality of SMCs.
Epigenetic programmes, which differ for various SMC populations, may determine the extremes of the phenotype range that a particular SMC can adopt. Modulation of the phenotype is only possible between these phenotype boundaries and governed by environmental conditions (figure 3). The challenge for future studies will be to show in how far these conditions can lead to genetic reprogramming, thereby shifting the phenotype boundaries.
Figure 3.
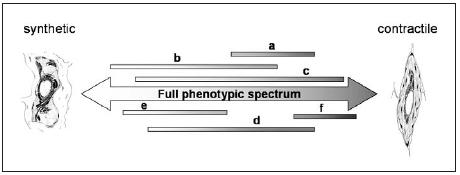
Whereas the SMCs in a vessel can collectively cover the whole spectrum of phenotypes, a given population of SMCs (indicated by a to f, respectively) can only cover a limited area of this spectrum. The boundaries of the spectrum for any given SMC population are defined by (epi)genetic programmes. SMCs can modulate their phenotype within the boundaries, a process which is controlled by the integration of environmental factors.
Determinants of vascular SMC diversity
Much progress has been made in recent years concerning the identification of transcription factors that regulate SMC differentiation and phenotype. The important roles of serum response factor (SRF) and its co-activator myocardin in this respect have been recently reviewed1,5 and will not be discussed here. Relevant to this review is that concentration gradients and alternatively spliced isoforms of SRF have specific effects on SMC gene transcription and consequently may contribute to SMC diversity.22 In addition, myocardin has a heterogeneous expression pattern in SMCs of different tissues,23 which may contribute to variations in marker gene expression and consequently SMC function. Whereas the transcription factors that regulate SMC phenotype are more and more defined, the list of factors that can activate or inhibit them continues to expand. These factors constitute the local environmental cues that, within the genetic predisposition, determine the phenotype of a given SMC. They are very diverse in nature and include a variety of soluble biocompounds, ECM proteins, as well as physical parameters (figure 4).
Figure 4.
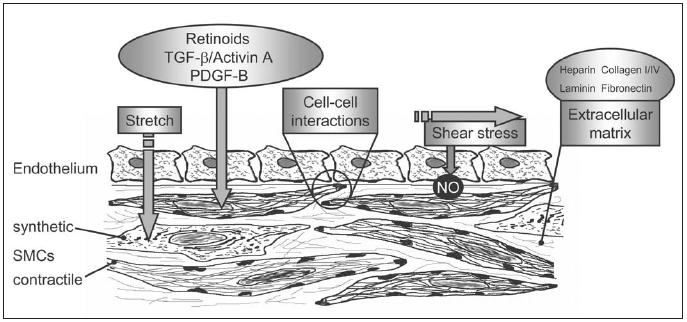
Factors involved in vascular SMC development, differentiation and phenotypic modulation.
Biochemical factors
Many biocompounds have been reported to affect expression of SMC phenotype markers, some of which even have phenotype-dependent effects. In this review, we will focus on a selection of factors that are supported by a substantial amount of experimental data in this context. These factors include PDGF, transforming growth factor (TGF)-β, activin A, retinoids, angiotensin II, and tumour necrosis factor-α (TNF- α). Besides these, compounds such as FGF, insulin-growth factor (IGF)-I and -II, endothelin-1, nitric oxide (NO), reactive oxygen species, peroxisome proliferatoractivated receptor-gamma ligands and complement 3 protein have been shown to affect SMC phenotype.
Two PDGF molecules, PDGF-A and PDGF-B, are important for the initial stages of SMC differentiation during vascular development, which are characterised by recruitment of mesenchymal cells and subsequent proliferation.24,25 In adult SMCs, these PDGF isoforms generally induce a more synthetic phenotype. For example, PDGF-B downregulates α-SMA expression in rat aortic SMCs.26 Similarly, PDGF-B treatment caused contractile pig coronary artery SMC clones to adopt a rhomboid morphology, and increased proliferation and migration rates.15 These in vitro results have been confirmed by in vivo studies showing that inhibition of either PDGF-A or PDGF-B reduced SMC proliferation and migration after arterial injury in adults, leading to reduced neointima formation.27,28
TGF-β isoforms, on the other hand, appear to be essential for the induction of the contractile SMC phenotype. Just as PDGF isoforms, they play a role in the migration of SMC precursors towards endothelial cells and the subsequent interactions between them, as revealed by TGF-β knockout mice.29 Cultured neural crest cells differentiate towards spindle-shaped SMCs when treated with TGF-β.30 Moreover, TGF-β1 signalling has recently been shown to be required for SMCspecific gene expression in embryoid bodies.31 In contrast to PDGF, TGF-β isoforms clearly promote the contractile phenotype in adult SMCs. For example, TGF-β1 increases α-SMA, SM-MHC and SM-calponin levels in cultured SMCs,32 and TGF-β2 increases α-SMA and desmin levels in cultured porcine SMCs.15 In line with this, TGF-β induces cell cycle proteins such as p21, which suppress cell division, and it suppresses the degradation of matrix proteins as reviewed elsewhere.33,34 However, TGF-β2 does not affect expression of more advanced contractile phenotype markers such as SM-MHC and smoothelin.15 The TGF-β-like factor activin A also stimulates expression of contractile marker proteins such as α-SMA and SM22α.35
Just as PDGF and TGF-β, retinoic acid (RA) is required for early SMC differentiation, particularly for neural crest-derived SMCs.36 RA treatment of embryonic stem cells promotes their differentiation towards SMCs.37 Interestingly, in adult SMCs, RA can promote either the synthetic or the contractile SMC phenotype, though its contractile phenotype promoting effects are dominant.38 For example, expression of contractile SMC markers including smoothelin and SM-MHC is increased upon RA treatment,6 and most studies indicate that retinoids decrease SMC proliferation.39 In addition, RA lowers SMC migration through induction of specific ECM proteins.40 However, CRBP-1, one of the best markers of synthetic SMCs, is also increased upon RA treatment, and it has been reported that retinoids stimulate SMC proliferation. 10,39 The differential effects of RA on the SMC phenotype are likely to be related to the initial phenotype of the SMCs tested.
Several other proteins have been reported to have differential effects on SMC phenotype, depending on the initial phenotype. This has led to contradictory findings concerning, for instance, the effects of TNF-α treatment on SMC proliferation. Recently, TNF-α was shown to have a different effect on spindle-shaped versus epithelioid SMCs isolated from the human saphenous vein.41 Whereas spindle-shaped SMCs showed increased proliferation after TNF-α treatment, epithelioid SMCs were induced to undergo apoptosis. Similarly, angiotensin II has been shown to induce apoptosis in epithelioid but not in spindle-shaped SMCs isolated from the rat aorta.42 In addition, IGF-I appears to have differential effects on SMC proliferation and differentiation, which depend on SMC phenotype. 43 Spindle-shaped SMCs require IGF-I signalling for maintenance of the contractile phenotype, whereas epithelioid SMCs increase migration and proliferation upon IGF-I treatment. Thus, besides RA, TNF-α, angiotensin II and IGF-I also contribute to SMC diversity.
Extracellular matrix components
Although the effects of soluble (growth) factors on SMC phenotype regulation have been the focus of the majority of studies in the past, it now seems that the ECM in which SMCs are embedded is at least equally important. Modulation of the SMC phenotype by ECM components is thought to be mediated by their binding to specific integrin receptors. The specific integrin combinations that are important in this respect have been reviewed by Moiseeva.16
Most of the medial ECM is made up of collagen isoforms (mostly type I and III), elastin, and proteoglycans. Among those, the proteoglycan heparin has proven to be an important ECM component for the regulation of SMC phenotype. Heparin generally promotes the maintenance of a contractile phenotype and slows down SMC proliferation. For example, heparin treatment of porcine SMCs inhibits proliferation irrespective of their phenotype and increases desmin levels of spindle-shaped porcine SMCs.15 However heparin decreases proliferation of bovine rhomboid SMCs only,18 suggesting that its effects may be influenced by innate SMC phenotype characteristics. Perlecan, another proteoglycan, appears to inhibit SMC proliferation through its heparan sulphate side chains, which have been suggested to sequester FGF-2.44 Interestingly, the expression of perlecan is negatively regulated by PDGF isoforms, which affects SMC migration.45
Other ECM components also provide good examples of the complex relation between ECM composition and SMC phenotype. Fibrillar collagen type I has been shown to promote the contractile phenotype of SMCs, whereas monomeric collagen type I activates SMC proliferation. When comparing SMCs cultured on either monomeric or polymerised collagen, completely different gene expression profiles are observed.46 Many of these differentially expressed genes code for ECM or cytoskeletal proteins, and several of these genes are also up- or downregulated after balloon injury of the rat carotid artery. Also, the different forms of collagen type I modulate the responsiveness of SMCs to PDGF-B.47 Moreover, the composition of the collagen fibrils influences the migration properties of the SMCs, which is associated with a different focal adhesion composition and integrin function.48
Just as fibrillar collagen type I, collagen type IV and laminin have been shown to promote the contractile phenotype.49 However, laminin-5 was also reported to enhance PDGF-B-stimulated SMC proliferation and migration,50 providing another example of a protein with ambiguous effects on SMC phenotype. Interestingly, expression of this protein is upregulated by both TGF- β and PDGF-B,50 which may explain its variable effects.
Whereas most ECM proteins seem to be required for induction or maintenance of the contractile phenotype, fibronectin, another major ECM protein, stimulates modulation towards the synthetic phenotype. 51 Hyaluronan, a glycosaminoglycan which is present in the medial ECM, has also been shown to enhance proliferation and migration of SMCs.52 Moreover, overexpression of hyaluronan by mouse SMCs accelerates the progression of atherosclerosis,53 which is associated with SMC phenotypic modulation towards the synthetic phenotype.
Not only the composition, but also the organisation of the ECM modulates SMC phenotype. This becomes clear in 3D culture systems, which are a better representation of the in vivo situation than conventional 2D systems. SMCs in a 3D collagen matrix are less proliferative compared with SMCs cultured on a 2D collagen matrix, and have increased TGF- β1 expression.54 On the other hand, α-SMA levels are slightly diminished and collagen I expression is higher in the 3D system, indicating a less contractile phenotype and illustrating the complexity of the effects of the ECM on SMC phenotype. In addition, SMC responses to PDGF-B, TGF-β1 and heparin are affected by the organisation of the ECM.55
Taken together, the summarised studies indicate that both composition and organisation of the ECM have major consequences for SMC phenotype. Because of the great number of ECM compounds and the various integrin combinations that mediate the phenotypic changes, much work remains to be done to understand the complex effects of the ECM on SMC phenotype in vivo. Moreover, it should be considered that the ECM is a reservoir of cytokines and growth factors that are bound to specific ECM components. As a consequence, changes in the ECM composition or volume directly affect the bioavailability of these factors and add another dimension to the interaction between biochemical and extracellular matrix components in the regulation of SMC phenotype.
Physical factors
Vascular SMCs continuously encounter mechanical stimuli that have important effects on their phenotype, for example by changing the nature of cell-cell interactions. Pressure causes stretch (tensile stress) and flow causes shear stress, which both induce vessel wall remodelling by changing SMC characteristics.
The effects of shear stress are mediated by the endothelium, which coordinates the response of SMCs to this type of mechanical stress. This occurs not only through NO release, but likely also via direct cell-cell interactions. It has been shown that upon coculturing adult endothelial cells with adult spindle-shaped porcine SMCs, the latter modulate towards a synthetic phenotype. They acquire a rhomboid morphology, reduce α-SMA and SM-MHC expression, and lose smoothelin expression.15 However, it is obvious that the normal physical and biochemical interactions between SMCs and endothelial cells are difficult to mimic in culture.
In contrast to endothelium-modulated shear stress, stretch acts directly on the SMCs. Mechanical forces appear to enhance the expression of both ECM and contractile proteins by SMCs in the vessel wall. Upon application of physiological mechanical strain on adult aortic SMCs, collagen and fibronectin synthesis as well as matrix degrading enzyme levels increase, eventually resulting in build-up of ECM.56 This indicates that mechanical strain is a causal factor in active remodelling of the vessel wall by SMCs.
It is clear that through their function in the transduction of mechanical stress, integrins are important integrators of the physical factors that affect SMC phenotype. Considering the importance of matrixintegrin interactions for the activation of signal transduction pathways that regulate SMC phenotype, changes in ECM composition are likely to be of major significance for the SMC response to mechanical stimuli. Indeed, the application of cyclic mechanical strain on neonatal rat SMCs had differential effects on, for example, SM-MHC expression, depending on the type of ECM substrate on which they were cultured.57 The response of adult SMCs was, however, not dependent on the type of matrix. In another study, mechanical strain applied to SMCs grown on laminin caused a greater increase in h-caldesmon when compared with SMCs grown on collagen type I or IV.58 The signal transduction pathways that are activated by stretch and shear stress59 have also been shown to increase expression of SM-MHC.60
Similar to the various effects reported on SMC marker gene expression, conflicting data exist on the growth response of SMCs to mechanical strain in culture. Both increased and reduced proliferation have been reported.61,62 This can probably be attributed to differences in the degree and characteristics of the stretch that was applied and the initial phenotype of the cells. Thus, these contradictory findings may actually reflect both the intrinsic SMC diversity as well as the diversity caused by a different local environment.
All in all, mechanical stress clearly has a major impact on SMC phenotype. It appears that physiological stress promotes the contractile SMC phenotype, whereas over- or under-stressing provokes modulation towards the synthetic phenotype. However, the in vitro studies that are required to test this hypothesis are hampered by the lack of a defined system that mimics the stretch encountered in vivo.
Conclusion
Although it is often assumed that SMCs uniformly differentiate to a contractile phenotype, the evidence summarised above clearly shows that SMC differentiation can produce various phenotypes intermediate to the synthetic and contractile ones. Whereas contractile SMCs are in the majority, synthetic SMCs in the normal vessel wall are necessary for maintenance and physiological remodelling. Both contractile and synthetic SMCs can thus be considered differentiated cells, albeit that they have distinct functions. Consequently, the traditional model depicting SMC differentiation as a process in which a SMC precursor first becomes a synthetic SMC before turning into a contractile SMC needs complementation.
We suggest that a basal differentiation programme is responsible for the development of a ‘naive SMC’, which is characterised by the expression of α-SMA, the least phenotype-specific SMC marker. Further differentiation, which is also (epi)genetically programmed, can bring about synthetic or contractile, as well as intermediate phenotypes (figure 5). These initial phenotypes are subjected to influences (forces or factors) of the local environment, which then determine modulation within the range set by the initial (epi)genetic programming (figure 3).
Figure 5.
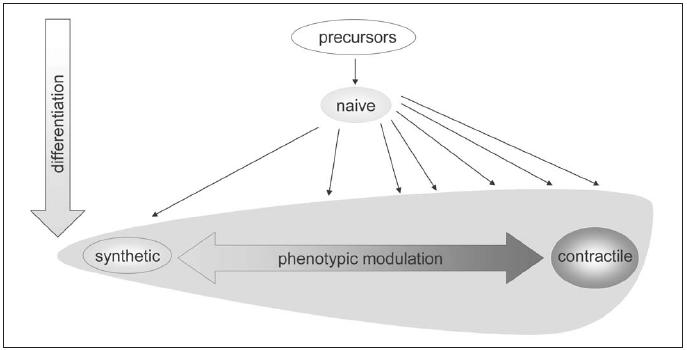
SMC differentiation pathways. SMC precursors first differentiate towards naive SMCs, which have a synthetic phenotype. The large majority of SMCs subsequently acquire a more contractile phenotype, although the extent of contractile differentiation differs and many cells retain a phenotype between synthetic and contractile. Both differentiated synthetic and differentiated contractile SMCs can reversibly change their phenotype via phenotypic modulation.
As a result, every blood vessel has a different composition, consisting of SMCs that are able to fulfil distinct complementary functions. The ability of those SMCs to change their properties by phenotypic modulation assures that each vessel can adapt to chronic changes in the local conditions. As such, SMC diversity does not compromise the performance of the vessel but rather gives it the flexibility that is needed to perform in different physiological or pathological situations.
Acknowledgements
The authors would like to thank Petra Niessen for critically reading the manuscript.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004;84:767-801. [DOI] [PubMed] [Google Scholar]
- 2.Gittenberger-de Groot AC, DeRuiter MC, Bergwerff M, et al. Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler Thromb Vasc Biol 1999;19: 1589-94. [DOI] [PubMed] [Google Scholar]
- 3.Hao H, Gabbiani G, Bochaton-Piallat ML. Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol 2003;23:1510-0. [DOI] [PubMed] [Google Scholar]
- 4.Chamley-Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev 1979;59:1-61. [DOI] [PubMed] [Google Scholar]
- 5.Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol 2003;35:577-93. [DOI] [PubMed] [Google Scholar]
- 6.Christen T, Bochaton-Piallat ML, Neuville P, et al. Cultured porcine coronary artery smooth muscle cells. A new model with advanced differentiation. Circ Res 1999;85:99-107. [DOI] [PubMed] [Google Scholar]
- 7.Miano JM, Cserjesi P, Ligon KL, et al. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ Res 1994;75:803-12. [DOI] [PubMed] [Google Scholar]
- 8.Christen T, Verin V, Bochaton-Piallat M, et al. Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation 2001;103:882-8. [DOI] [PubMed] [Google Scholar]
- 9.Kuro-o M, Nagai R, Nakahara K, et al. cDNA cloning of a myosin heavy chain isoform in embryonic smooth muscle and its expression during vascular development and in arteriosclerosis. J Biol Chem 1991;266:3768-73. [PubMed] [Google Scholar]
- 10.Neuville P, Geinoz A, Benzonana G, et al. Cellular retinol-binding protein-1 is expressed by distinct subsets of rat arterial smooth muscle cells in vitro and in vivo. Am J Pathol 1997;150:509-21. [PMC free article] [PubMed] [Google Scholar]
- 11.Glukhova MA, Kabakov AE, Frid MG, et al. Modulation of human aorta smooth muscle cell phenotype: a study of muscle-specific variants of vinculin, caldesmon, and actin expression. Proc Natl Acad Sci USA 1988;85:9542-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frid MG, Moiseeva EP, Stenmark KR. Multiple phenotypically distinct smooth muscle cell populations exist in the adult and developing bovine pulmonary arterial media in vivo. Circ Res 1994;75:669-81. [DOI] [PubMed] [Google Scholar]
- 13.Regan CP, Adam PJ, Madsen CS, et al. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest 2000;106:1139-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bochaton-Piallat ML, Ropraz P, Gabbiani F, et al. Phenotypic heterogeneity of rat arterial smooth muscle cell clones. Implications for the development of experimental intimal thickening. Arterioscler Thromb Vasc Biol 1996;16:815-20. [DOI] [PubMed] [Google Scholar]
- 15.Hao H, Ropraz P, Verin V, et al. Heterogeneity of smooth muscle cell populations cultured from pig coronary artery. Arterioscler Thromb Vasc Biol 2002;22:1093-9. [DOI] [PubMed] [Google Scholar]
- 16.Moiseeva EP. Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res 2001;52:372-86. [DOI] [PubMed] [Google Scholar]
- 17.Ko YS, Yeh HI, Haw M, et al. Differential expression of connexin43 and desmin defines two subpopulations of medial smooth muscle cells in the human internal mammary artery. Arterioscler Thromb Vasc Biol 1999;19:1669-80. [DOI] [PubMed] [Google Scholar]
- 18.Frid MG, Aldashev AA, Dempsey EC, et al. Smooth muscle cells isolated from discrete compartments of the mature vascular media exhibit unique phenotypes and distinct growth capabilities. Circ Res 1997;81:940-52. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Fan YS, Chow LH, et al. Innate diversity of adult human arterial smooth muscle cells: cloning of distinct subtypes from the internal thoracic artery. Circ Res 2001;89:517-25. [DOI] [PubMed] [Google Scholar]
- 20.Li S, Sims S, Jiao Y, et al. Evidence from a novel human cell clone that adult vascular smooth muscle cells can convert reversibly between noncontractile and contractile phenotypes. Circ Res 1999; 85:338-48. [DOI] [PubMed] [Google Scholar]
- 21.Bochaton-Piallat ML, Clowes AW, Clowes MM, et al. Cultured arterial smooth muscle cells maintain distinct phenotypes when implanted into carotid artery. Arterioscler Thromb Vasc Biol 2001; 21:949-54. [DOI] [PubMed] [Google Scholar]
- 22.Belaguli NS, Zhou W, Trinh TH, et al. Dominant negative murine serum response factor: alternative splicing within the activation domain inhibits transactivation of serum response factor binding targets. Mol Cell Biol 1999;19:4582-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D, Chang PS, Wang Z, et al. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 2001;105:851-62. [DOI] [PubMed] [Google Scholar]
- 24.Hellstrom M, Kalen M, Lindahl P, et al. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999;126:3047-55. [DOI] [PubMed] [Google Scholar]
- 25.Schattemann GC, Loushin C, Li T, et al. PDGF-A is required for normal murine cardiovascular development. Dev Biol 1996;176:133-42. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Van Putten V, Zarinetchi F, et al. Suppression of smoothmuscle alpha-actin expression by platelet-derived growth factor in vascular smooth-muscle cells involves Ras and cytosolic phospholipase A2. Biochem J 1997;327(Pt 3):709-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotani M, Fukuda N, Ando H, et al. Chimeric DNA-RNA hammerhead ribozyme targeting PDGF A-chain mRNA specifically inhibits neointima formation in rat carotid artery after balloon injury. Cardiovasc Res 2003;57:265-76. [DOI] [PubMed] [Google Scholar]
- 28.Deguchi J, Namba T, Hamada H, et al. Targeting endogenous platelet-derived growth factor B-chain by adenovirus-mediated gene transfer potently inhibits in vivo smooth muscle proliferation after arterial injury. Gene Ther 1999;6:956-65. [DOI] [PubMed] [Google Scholar]
- 29.Sanford LP, Ormsby I, Gittenberger-de Groot AC, et al. TGFbeta2 knockout mice have multiple developmental defects that are nonoverlapping with other TGFbeta knockout phenotypes. Development 1997;124:2659-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah NM, Groves AK, Anderson DJ. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell 199 6;85:331-43. [DOI] [PubMed] [Google Scholar]
- 31.Sinha S, Hoofnagle MH, Kingston PA, et al. Transforming growth factor-{beta}1 signaling contributes to development of smooth muscle cells from embryonic stem cells. Am J Physiol Cell Physiol 2004;287:C1560-8. [DOI] [PubMed] [Google Scholar]
- 32.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem 1997;272:10948-56. [DOI] [PubMed] [Google Scholar]
- 33.Moustakas A, Pardali K, Gaal A, et al. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol Lett 2002;82:85-91. [DOI] [PubMed] [Google Scholar]
- 34.Hocevar BA, Howe PH. Analysis of TGF-beta-mediated synthesis of extracellular matrix components. Methods Mol Biol 2000;142: 55-65. [DOI] [PubMed] [Google Scholar]
- 35.Engelse MA, Lardenoye JH, Neele JM, et al. Adenoviral activin a expression prevents intimal hyperplasia in human and murine blood vessels by maintaining the contractile smooth muscle cell phenotype. Circ Res 2002;90:1128-34. [DOI] [PubMed] [Google Scholar]
- 36.Colbert MC, Kirby ML, Robbins J. Endogenous retinoic acid signaling colocalizes with advanced expression of the adult smooth muscle myosin heavy chain isoform during development of the ductus arteriosus. Circ Res 1996;78:790-8. [DOI] [PubMed] [Google Scholar]
- 37.Drab M, Haller H, Bychkov R, et al. From totipotent embryonic stem cells to spontaneously contracting smooth muscle cells: a retinoic acid and db-cAMP in vitro differentiation model. Faseb J 1997;11:905-15. [DOI] [PubMed] [Google Scholar]
- 38.Miano JM, Kelly LA, Artacho CA, et al. all-Trans-retinoic acid reduces neointimal formation and promotes favorable geometric remodeling of the rat carotid artery after balloon withdrawal injury. Circulation 1998;98:1219-27. [DOI] [PubMed] [Google Scholar]
- 39.Chen S, Gardner DG. Retinoic acid uses divergent mechanisms to activate or suppress mitogenesis in rat aortic smooth muscle cells. J Clin Invest 1998;102:653-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johst U, Betsch A, Wiskirchen J, et al. All-Trans and 9-cis Retinoid Acids Inhibit Proliferation, Migration, and Synthesis of Extracellular Matrix of Human Vascular Smooth Muscle Cells by Inducing Differentiation In Vitro. J Cardiovasc Pharmacol 2003;41:526-35. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Rao PJ, Castresana MR, et al. TNF-alpha induces proliferation or apoptosis in human saphenous vein smooth muscle cells depending on phenotype. Am J Physiol Heart Circ Physiol 2005;288:H293-301. [DOI] [PubMed] [Google Scholar]
- 42.Bascands JL, Girolami JP, Troly M, et al. Angiotensin II induces phenotype-dependent apoptosis in vascular smooth muscle cells. Hypertension 2001;38:1294-9. [DOI] [PubMed] [Google Scholar]
- 43.Hayashi K, Shibata K, Morita T, et al. Insulin receptor substrate- 1/SHP-2 interaction, a phenotype-dependent switching machinery of insulin-like growth factor-I signaling in vascular smooth muscle cells. J Biol Chem 2004;279:40807-18. [DOI] [PubMed] [Google Scholar]
- 44.Tran PK, Tran-Lundmark K, Soininen R, et al. Increased intimal hyperplasia and smooth muscle cell proliferation in transgenic mice with heparan sulfate-deficient perlecan. Circ Res 2004;94:550-8. [DOI] [PubMed] [Google Scholar]
- 45.Koyama N, Kinsella MG, Wight TN, et al. Heparan sulfate proteoglycans mediate a potent inhibitory signal for migration of vascular smooth muscle cells. Circ Res 1998;83:305-13. [DOI] [PubMed] [Google Scholar]
- 46.Ichii T, Koyama H, Tanaka S, et al. Fibrillar collagen specifically regulates human vascular smooth muscle cell genes involved in cellular responses and the pericellular matrix environment. Circ Res 2001;88:460-7. [DOI] [PubMed] [Google Scholar]
- 47.Raines EW, Koyama H, Carragher NO. The extracellular matrix dynamically regulates smooth muscle cell responsiveness to PDGF. Ann N Y Acad Sci 2000;902:39-51; discussion 51-2. [DOI] [PubMed] [Google Scholar]
- 48.Li S, Chow LH, Pickering JG. Cell surface-bound collagenase-1 and focal substrate degradation stimulate the rear release of motile vascular smooth muscle cells. J Biol Chem 2000;275:35384-92. [DOI] [PubMed] [Google Scholar]
- 49.Thyberg J, Hultgardh-Nilsson A. Fibronectin and the basement membrane components laminin and collagen type IV influence the phenotypic properties of subcultured rat aortic smooth muscle cells differently. Cell Tissue Res 1994;276:263-71. [DOI] [PubMed] [Google Scholar]
- 50.Kingsley K, Huff JL, Rust WL, et al. ERK1/2 mediates PDGF-BB stimulated vascular smooth muscle cell proliferation and migration on laminin-5. Biochem Biophys Res Commun 2002;293:1000-6. [DOI] [PubMed] [Google Scholar]
- 51.Hedin U, Bottger BA, Forsberg E, et al. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J Cell Biol 1988;107:307-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1999;19:1004-13. [DOI] [PubMed] [Google Scholar]
- 53.Chai S, Chai Q, Danielsen CC, et al. Overexpression of hyaluronan in the tunica media promotes the development of atherosclerosis. Circ Res 2005;96:583-91. [DOI] [PubMed] [Google Scholar]
- 54.Li S, Lao J, Chen BP, et al. Genomic analysis of smooth muscle cells in 3-dimensional collagen matrix. Faseb J 2003;17:97-9. [DOI] [PubMed] [Google Scholar]
- 55.Stegemann JP, Nerem RM. Altered response of vascular smooth muscle cells to exogenous biochemical stimulation in two- and three-dimensional culture. Exp Cell Res 2003;283:146-55. [DOI] [PubMed] [Google Scholar]
- 56.O’Callaghan CJ, Williams B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells: role of TGF-beta(1). Hypertension 2000;36:319-24. [DOI] [PubMed] [Google Scholar]
- 57.Reusch HP, Chan G, Ives HE, et al. Activation of JNK/SAPK and ERK by mechanical strain in vascular smooth muscle cells depends on extracellular matrix composition. Biochem Biophys Res Commun 1997;237:239-44. [DOI] [PubMed] [Google Scholar]
- 58.Birukov KG, Bardy N, Lehoux S, et al. Intraluminal pressure is essential for the maintenance of smooth muscle caldesmon and filamin content in aortic organ culture. Arterioscler Thromb Vasc Biol 1998;18:922-7. [DOI] [PubMed] [Google Scholar]
- 59.Lehoux S, Tedgui A. Signal transduction of mechanical stresses in the vascular wall. Hypertension 1998;32:338-45. [DOI] [PubMed] [Google Scholar]
- 60.Reusch P, Wagdy H, Reusch R, et al. Mechanical strain increases smooth muscle and decreases nonmuscle myosin expression in rat vascular smooth muscle cells. Circ Res 1996;79:1046-53. [DOI] [PubMed] [Google Scholar]
- 61.Hipper A, Isenberg G. Cyclic mechanical strain decreases the DNA synthesis of vascular smooth muscle cells. Pflugers Arch May 2000; 440:19-27. [DOI] [PubMed] [Google Scholar]
- 62.Birukov KG, Shirinsky VP, Stepanova OV, et al. Stretch affects phenotype and proliferation of vascular smooth muscle cells. Mol Cell Biochem 1995;144:131-9. [DOI] [PubMed] [Google Scholar]