

Abstract
The GlmS riboswitch is located in the 5′-untranslated region of the gene encoding glucosamine-6-phosphate (GlcN6P) synthetase. The GlmS riboswitch is a ribozyme with activity triggered by binding of the metabolite GlcN6P. Presented here is the first structure of the GlmS ribozyme (2.5Å resolution) with GlcN6P bound in the active site. The GlmS ribozyme adopts a compact double pseudoknot tertiary structure, with two closely packed helical stacks. Recognition of GlcN6P is achieved through coordination of the phosphate moiety by two hydrated magnesium ions as well as specific nucleobase contacts to the GlcN6P sugar ring. Comparison of this activator bound and the previously published apoenzyme complex supports a model in which GlcN6P does not induce a conformational change in the RNA, as is typical of other riboswitches, but instead functions as a catalytic cofactor for the reaction. This demonstrates that RNA, like protein enzymes, can employ the chemical diversity of small molecules to promote catalytic activity.
Riboswitches are cis-acting structural RNA molecules that directly control gene expression[1]. These biosensors are sensitive to the concentration of a specific small molecule, and control expression through a feedback loop mechanism. Ligand binding causes a structural rearrangement in the RNA that generally leads to the down regulation of the gene, through transcriptional termination or inefficient translational initiation. In many cases, genes controlled by the riboswitch are part of the biosynthetic pathway of the small molecule recognized by the RNA[2]. Riboswitches are commonly found in Gram-positive bacteria, and control over 2% of genes in Bacillus subtillis in response to a variety of cofactors, including guanine, adenine, co-enzyme B12, lysine and even glycine[1].
The GlmS riboswitch is located in the 5′-untranslated region (5′-UTR) of the gene for glucosamine-6-phosphate synthetase[3], which converts glutamine and fructose-6-phosphate to glucosamine-6-phosphate (GlcN6P)[4]. Like many other riboswitches that control downstream gene expression, the GlmS riboswitch negatively regulates GlcN6P synthetase production[3]. High GlcN6P concentrations activate the riboswitch, resulting in reduced synthetase expression. However, unlike every other riboswitch identified to date, the GlmS riboswitch does not appear to undergo any structural rearrangement upon effector binding, suggesting that the binding pocket is pre-formed in the absence of ligand[3, 5]. Instead, GlcN6P binding results in the specific cleavage of the GlmS mRNA at a single site 5′ of the riboswitch sequence. The GlmS riboswitch is a ribozyme and like other autolytic ribozymes, the cleavage reaction involves nucleophilic attack of a vicinal 2′-OH on the scissile phosphate to produce products with a 2′-3′ cyclic phosphate and a 5′-OH[3, 6].
Although no large-scale conformational changes were detected upon GlcN6P binding, the effector is absolutely required for GlmS ribozyme activation[3]. GlcN6P addition increases the cleavage rate 100,000-fold over background hydrolysis with the amino group being the principal determinant of activity. Glucose-6-phosphate (Glc6P), which contains a hydroxyl in place of the GlcN6P amine, is a competitive inhibitor of the reaction[7]. Glucosamine (GlcN), which lacks the phosphate but retains the primary amine, is able to promote the cleavage reaction, albeit 30-fold less efficiently than GlcN6P[3, 7]. Activity has even been reported for other small molecules such as Tris and serinol though they are poor activators presumably due to weak RNA binding[7]. However, they all share the common feature of an ethanolamine moiety, a primary amine and a vicinal hydroxyl group. Additional evidence for the catalytic importance of the amine in the phosphoryltransfer reaction came from biochemical experiments that demonstrated a pH dependence of the reaction as a function of the pKa of the effector[7]. For GlcN6P, GlcN and serinol, the reaction rate went up with increasing pH revealing an apparent pKa that approximately matched the pKa of the amine for each ligand. This suggested that the amino rather than the ammonium form of the metabolite is active in the cleavage reaction[7].
To understand the structural basis of effector mediated riboswitch activation, we determined the crystal structure of the Bacillus anthracis GlmS ribozyme bound to the effector GlcN6P. During the final stages of our structural refinement, Klein and Ferré-D'Amaré reported a series of structures of the Thermoanerobacter tengcongenis GlmS[8]. These included the 2-deoxy and 2′-amino substrate form of the GlmS ribozyme with and without the competitive inhibitor, Glc6P, and the cleaved ribozyme product. However, they were unable to crystallize the GlnN6P bound complex[8]. Comparison of these reaction states with the GlcN6P bound complex reported here, addresses how the RNA recognizes its ligand and provides insight into how GlcN6P, especially the amine and adjacent hydroxyl group, promotes catalysis.
RESULTS AND DISCUSSION
Crystallization and activity of a two part Bacillus anthracis GlmS Ribozyme
The crystallization construct was designed based on the GlmS ribozyme located in the 5′-UTR of the Bacillus anthracis glmS gene[9] (Fig. 1a). The crystallization construct contained all of the nucleotides previously identified as critical for ribozyme activity[3]. Two modifications were made to the ribozyme to promote crystallization. First, the recognition sequence for the RNA binding domain of the U1A protein was appended to the top of helix P1, a loop of variable size in GlmS ribozymes[9], to promote co-crystallization with the U1A protein[10]. Second, the first eleven nucleotides of the GlmS ribozyme were removed from the transcribed RNA sequence and added as a substrate strand in trans. The synthetic oligonucleotide contained two nucleotides 5′ of the cleavage site and the first eleven nucleotides of the GlmS ribozyme. This made it possible to inhibit ribozyme activity by introducing a 2′-O-methyl substitution at the cleavage site. Diffraction quality crystals were obtained of a complex containing equimolar ratios of the GlmS ribozyme RNA, the synthetic oligonucleotide and the U1A protein.
Figure 1.
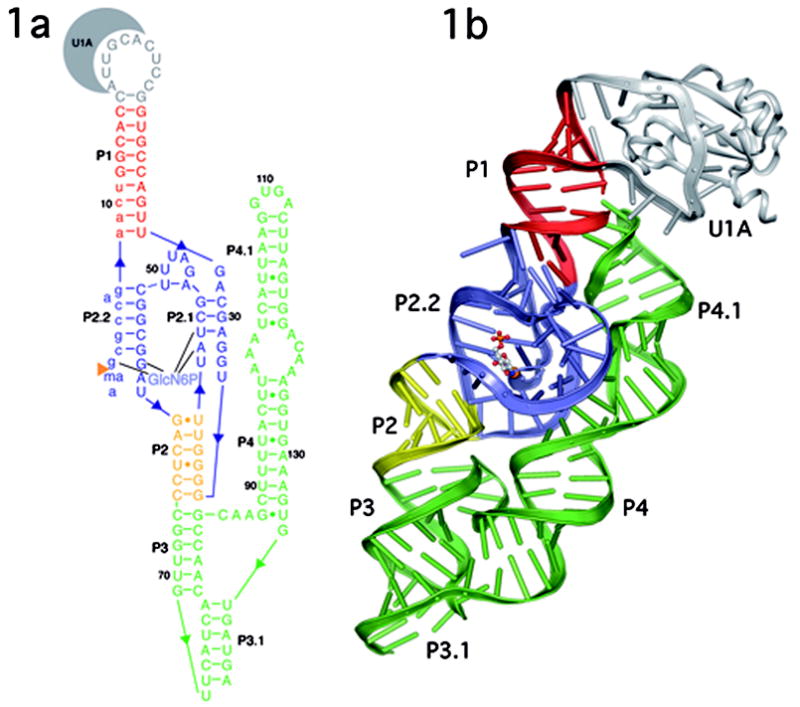
A. Secondary and B. Tertiary structure of the B. anthracis GlmS ribozyme depicted in ribbons format. The P1 helix is shown in red, P2 in yellow, and P3–P4 in green. The double pseudoknot, consisting of helices P2.1, P2.2, P2a and related joiner regions is in blue. GlcN6P is bound in the active site and is shown in ball and stick representation primarily in gray. U1A and the U1A binding loop are in gray.
The GlcN6P dependent reactivity of this GlmS ribozyme construct was confirmed in solution and within the crystals. In solution, the GlmS RNA reacted with an all-ribose substrate at the rate of 6 min−1, and the rate was unaffected by U1A addition (Fig. 2, lanes 2 and 3). Substantially less activity was observed in the absence of GlcN6P (0.0006 min−1) or upon addition of Glu6P instead of GlcN6P (0.0006 min−1) (Fig. 2, lanes 4 and 5). The 2′-O-methyl substitution at the cleavage site resulted in complete inactivation of ribozyme reaction even in the presence of GlcN6P (Fig. 2, lane 6). To determine the activity of the crystallized RNA, the ribozyme was crystallized with an all ribose substrate radiolabeled at its 5′-end in the absence of GlcN6P. The radioactive crystals were harvested, washed and soaked in buffer either with or without 2 mM GlcN6P. RNA in crystals incubated with GlcN6P reacted to completion within 2 hours, while those soaked in buffer alone showed no activity (Fig. 2, lanes 7 and 8). No noticeable change in crystal morphology was evident upon the addition of GlcN6P. These results suggest that the RNA is in a catalytically relevant conformation in the crystalline state.
Figure 2.

Reactivity of the B. anthracis GlmS ribozyme construct under crystallization conditions. Activity assays conditions were similar to those used for the refolding of the GlmS RNA with a radiolabeled synthetic oligonucleotide substrate. The substrate rS has a ribose at position A-1; oMeS has a 2′-O-methyl at A-1. Reactions were performed either in solution or in crystals as indicated. The reagents included in each lane are as indicated above in lane. The mobility of the substrate and product are marked with arrows. The percent cleavage for each reaction after a 2 hour incubation is listed below the autoradiogram.
Overview of the of the B. anthracis GlmS ribozyme
The structure of the GlmS ribozyme in a state prior to cleavage (Fig. 1b) was determined by multiple isomorphous replacement to 2.5Å resolution (Table 1). There are four molecules in the asymmetric unit with intermolecular contacts dominated by the U1A protein, which forms a dimer of dimers. The tertiary structure of the GlmS ribozyme is highly compact; P1, P2, P3 and P3.1 form one helical stack that abuts a second helical stack formed from P4 and P4.1 (Fig. 1b). In all four molecules there is unambiguous density for the entire phosphate backbone and a molecular model for all nucleotides was built and refined to an R-free of 27.2%. Clear density was observed in the active site for glucosamine-6-phosphate (Fig. 3). This is the first GlmS ribozyme structure to include the required cofactor for the reaction.
Figure 3.
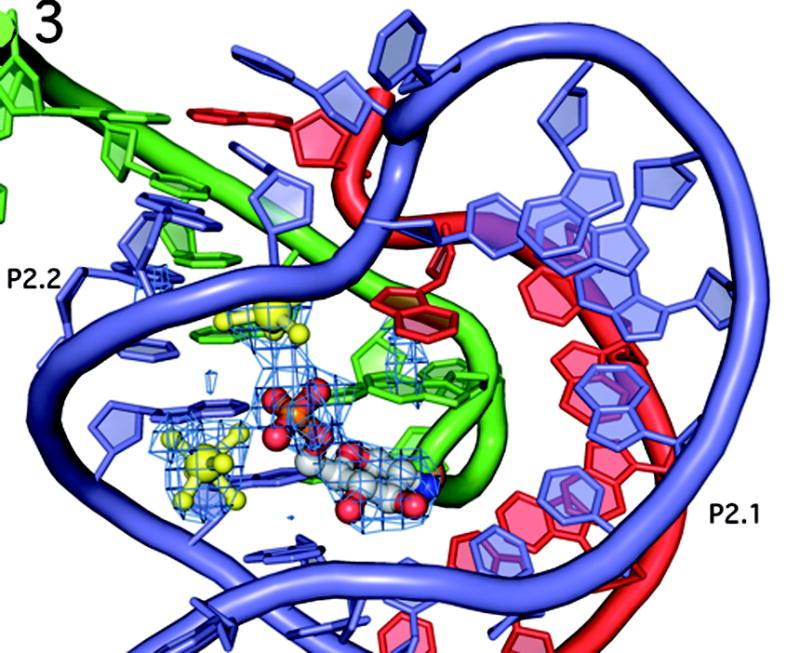
The double pseudoknotted active site of the GlmS ribozyme in the same orientation as in Fig. 1b. The color scheme has been altered to emphasize the contribution of each strand in the active site. The substrate strand (nucleotides −2 to 9) are shown in green, with the scissile phosphate depicted as an orange ball. Nucleotides 27–34 are in red, and nucleotides 41–59, which comprise the P2 loop, are in blue. This color scheme is also used in Figures 4, 5 and 7. The P2.1 pseudoknot is formed from the red and blue strands; the P2.2 pseudoknot is formed from the green and blue strands. GlcN6P is shown primarily in gray and the two hydrated magnesium ions are in yellow. Fo − Fc density is shown in blue mesh, contoured at 2.5σ, calculated before addition of GlcN6P, metal ions or water molecules to the model.
The structures of the B. anthracis and the T. tengcongenis GlmS ribozymes reveal a complex secondary structural fold that differs significantly from that predicted for the GlmS ribozyme in the core region of the molecule[3, 8]. The critical difference is a double pseudoknot formed from nucleotides in the P2 loop (Fig. 3). One pseudoknot, P2.1, is formed from the joiner region between P1 and P2 and the 5′ end of the P2 loop. The second pseudoknot, P2.2, is formed from a base pairing interaction between the 3′ side of the P2 loop and the 5′ end of the ribozyme. This double pseudoknot forms the ribozyme core, including the GlcN6P binding pocket. The bottom of the active site is organized through the non-sequential stacking of unpaired nucleotides. The P2.1 and P2.2 pseudoknots form the sides of an active site that is capped by a single base pair between A46 and U51 that closes the P2 loop. This single base pair establishes the relative orientations of the P2.1 and P2.2 helices. This constrained geometry forces the unpaired A28 in the joiner region between P1 and P2.1 to stack directly on top of G1, the nucleotide 3′ of the scissile phosphate. G1, in turn, stacks on top of the GlcN6P.
The compact core of the ribozyme is buttressed by the functionally dispensable P3 and P4 helices[3]. About ~1200 Å2 of the ribozyme core surface area is buried by P4. Long-range tertiary interactions serve to further solidify this compact structure. The GNRA tetraloop at the top of P4.1 docks into the minor groove of P1, connecting these two disparate regions of the RNA structure. The cross-strand stack of As in the bulge between P4/P4.1 pack obliquely into the minor groove of P2.1. The predicted pseudoknot, P3.1, between the 3′ end of the ribozyme and the P3 loop completes the compact overall fold of this ribozyme[11].
The GlcN6P binding pocket
Binding of GlcN6P by the GlmS ribozyme is achieved through recognition of both the phosphate and sugar moieties (Fig. 4). Two fully hydrated Mg2+ ions contact the phosphate group, while the glucosamine ring makes direct interactions with the RNA. This may be a common recognition mechanism for riboswitches that bind ligands containing phosphate. For example, the thiamine pyrophosphate riboswitch also binds the pyrophosphate using two solvated Mg2+ ions while using nucleotide functional groups for direct interaction with the thiamine[12].
Figure 4.
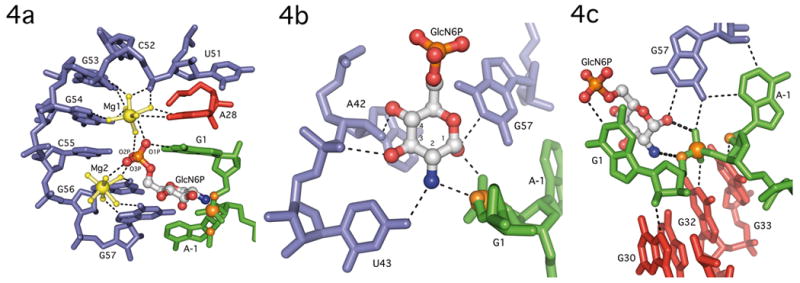
GlcN6P binding by the GlmS ribozyome. A. Phosphate coordination by two magnesium ions in the GlmS ribozyme. Nucleotides in P2.1 (blue), A28 (red) and G1 (green) use water mediated contacts to organize two hydrated magnesium ions (yellow) and orient the GlcN6P (primarily in gray) phosphate oxygens in the active site. B. Recognition of the GlcN6P sugar ring by nucleobase functional groups. The sugar contacts nucleotides A42, U43 and G57 (blue), and the sugar and 3′-phosphate of G1 (green). The guanine base at G1, which stacks on top of GlcN6P (see Fig. 4a), is not shown to allow all hydrogen bonding contacts to be visualized. The scissile phosphate, 5′-O leaving group and 2′-O nucleophile are shown as orange spheres. C. Active site interactions expected to stabilize this conformation are depicted as thin dashed lines. The catalytically critical interactions between the ethanolamine moiety of GlcN6P and the reactive phosphate are shown as thicker dashed lines. The coloring of individual nucleotides follows the convention in Fig. 3. The scissile phosphate, the 5′-O leaving group and the 2′-O nucleophile (which has been methylated) are shown as orange spheres.
The phosphate binding edge of the GlcN6P pocket is solvent exposed. Magnesium ions and water molecules appear to screen the negative charge of the phosphate backbone from the phosphate group of GlcN6P. In the GlmS structure each of the three GlcN6P non-bridging phosphate oxygens are coordinated to at least one of the two hydrated Mg2+ ions (Mg1 and Mg2) (Fig. 4a). The Mg2+ ions are ~7.5Å apart, with the phosphate of GlcN6P positioned equidistant from both. One phosphate oxygen, here designated O2P, hydrogen bonds to waters from both metals, while a second water from Mg2 interacts with the O3P oxygen. The O1P oxygen interacts with a water from Mg1 and makes a direct contact to the N1 of G1.
Both metals are positioned in the major groove of the P2.2 helix in the RNA's effector binding pocket. The vast majority of the contacts are to nucleobase functional groups. An outer sphere interaction between Mg1 and the pro-SP non-bridging phosphate oxygen of C52 is the only contact to an RNA phosphate group. Mg1 makes additional outer sphere contacts with the N7 and O/N6 of A28, G53 and G54. Interference seen with 7-deazaadenosince at A28 demonstrates the importance of this functional group for activity[13]. Mg2 makes water-mediated contacts to the N7 and O6 of G56 and G57. In contrast to this structure, only one metal, Mg1, was observed in the T. tengcongenis structure bound to the inhibitor Glu6P[8]. In that case, Mg1 appears to make direct contact with one phosphate oxygen of Glu6P. This difference may result from alternate binding modes for Glu6P compared to GluN6P or other aspects of the crystallization conditions.
The effector binding pocket in the RNA makes an extensive set of hydrogen bonding interactions with all four of the exocyclic functional groups on the glucosamine ring. Recognition of the ethanolamine moiety of GlcN6P (the C1-OH and adjacent C2-amine), the minimal substrate for the ribozyme reaction[7], explains the specificity of the GlmS ribozyme. The C1-OH accepts a hydrogen bond from the N1 of G57 and donates a hydrogen bond to the pro-SP non-bridging oxygen of the scissile phosphate (Fig. 4b). The C2-amine donates hydrogen bonds to the G1 5′-O leaving group and to the O4 of U43 (Fig. 4b). The contacts to the pro-SP oxygen and the 5′-O are the only interactions between GlcN6P and atoms involved in bond making and bond breaking. Additional binding specificity for the effector is provided by contacts to the C3 and C4 hydroxyl groups. The C3-OH makes a hydrogen bond with the pro-RP oxygen of U43 and the C4-OH makes a hydrogen bond with the 2′-OH of A42 (Fig. 4b).
Subtle, but potentially important, differences were observed in the hydrogen bonding patterns to the sugars of Glu6P and GlcN6P[8]. In the Glu6P inhibitor structure, the C2-OH also hydrogen bonds to the 5′-O leaving group, but because of the C2 hydroxyl substitution, it can donate only one hydrogen bond and does not interact with the O4 of U43 (U51 in T. tengcongenis ). There is an approximately 1Å shift in nucleotides A42 (A50) and U43 (U51) between the GlcN6P bound and Glu6P bound forms of the ribozyme (Fig. 5). This shift leads to changes in the hydrogen bonding network around the C3 and C4 hydroxyls of Glu6P[8]. The C3-OH makes a hydrogen bond to the 2′-OH of A42 (A50), while no hydrogen bonding is observed to the C4-OH. Biochemical studies found that a GlcN6P mimic with inverted stereochemistry at C4-OH does not activate the B. cereus GlmS ribozyme, which suggests that this functional group contacts the ribozyme[14]. The presence of such a contact to the C4-OH in the B. anthracis GlcN6P structure, and its absence in the Glc6P T. tencongenis structure may reflect a subtle, but functionally important, difference between activator and inhibitor binding.
Figure 5.
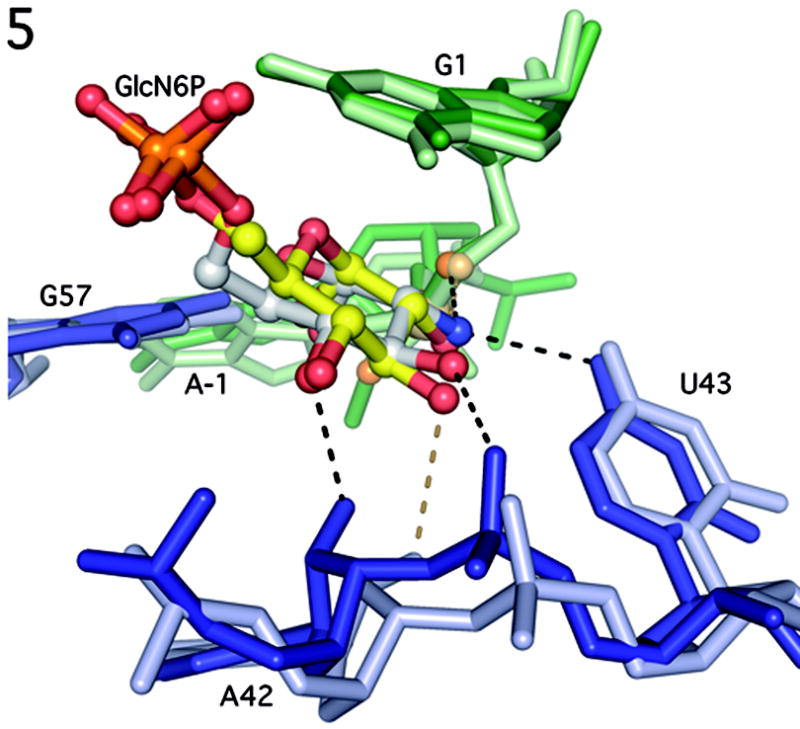
Comparison of the active sites of GlcN6P bound B. anthracis GlmS ribozyme and Glu6P bound T. tengcongenis GlmS ribozyme. B. anthracis nucleotides are in darker shades of blue and green and T. tengcongenis nucleotides are in lighter shades. GlcN6P is shown primarily in gray and Glu6P is shown primarily in yellow. Hydrogen bonds to GlcN6P are show in black, hydrogen bonds to Glu6P are shown in dark yellow.
Catalysis by the GlmS ribozyme
The structure suggests that the GlmS ribozyme is not a metalloenzyme. The active site is devoid of metal ions that could directly participate in the cleavage reaction, which is consistent with biochemical observations[9]. High concentrations of monovalent metal ions or cobalt hexamine, which mimics a hydrated magnesium, are able to fully support activity[9]. The catalytic potential of the GlmS ribozyme appears to be achieved by nucleobases and GlcN6P acting as a catalytic cofactor.
Based on analogy to other phosphorotransferases such as ribonuclease A, there are at least four strategies the GlmS ribozyme might use to promote its autolytic reaction[6]: (i) Alignment of the nucleophile and leaving group, (ii) deprotonation of the 2′-OH nucleophile, (iii) stabilization of the negative charge on the scissile phosphate, and/or (iv) protonation of the 5′-O leaving group. GlcN6P could promote the ribozyme activity of this riboswitch by fulfilling any one or a combination of these roles, through inducing a localized conformational change to create an inline conformation, or by directly participating in the chemical reaction.
The structure of the GlmS ribozyme reveals an active site in which the scissile phosphate and flanking nucleotides are oriented for chemistry (Fig. 4c). The nucleotides immediately 5′ and 3′ of the reactive center are unstacked and splayed away from each other. The nucleotide 5′ of the cleavage site, A-1 is orientated by an interaction with G57. Consistent with an important role in substrate orientation, mutation of G57 severely impairs ribozyme function, leading to minimal (G57C) or no (G57A) activity[15]. The nucleotide 3′ of the cleavage site, G1 stacks underneath A28 and on top of GlcN6P (Fig. 4a). It is positioned through the donation of two hydrogen bonds: one from the N1 of G1 to a phosphate oxygen of GlcN6P and the second from the G1 2′-OH to the N7 of G30. Substitution of G30 with 7-deazaguanosine is detrimental to GlmS ribozyme activity in interference analysis[13]. These interactions confer a 165° torsion angle between the 2′-OH nucleophile and the 5′-O leaving group in the ground state, very close to the 180° angle predicted in the transition state for inline attack on the scissile phosphate.
The active sites of the uncleaved T. tencongenis and the GlcN6P bound B. anthracis GlmS ribozyme structures reveal only small changes in geometry upon effector binding[8]. The core region of the GlcN6P bound ribozyme superimposes with only a 1.5Å deviation on the unliganded form of the T. tengcongenis GlmS ribozyme. Thus, while the GlmS ribozyme aligns the reactive 2′-OH and 5′-O groups for nucleophilic attack, it does so in a manner largely independent of GlcN6P binding. This is contrary to a model in which the GlmS riboswitch functions through a global or local conformational change upon ligand binding, as the active site appears to be largely pre-organized in the absence of GlcN6P[5].
If effector binding does not induce a conformational change, how does the GlcN6P trigger the cleavage activity of this riboswitch? The structure suggests that GlcN6P directly participates in the reaction as a catalytic cofactor using the ethanolamine moiety on the glucosamine sugar. The C1-OH and C2-NH2 groups are in direct hydrogen bonding contact with the scissile phosphate and the 5′-O leaving group, respectively (Fig. 4c). Based upon the following structural and biochemical data, we propose that the C2-amine serves as a general acid to activate the 5′-O leaving group and the C1-OH stabilizes developing charge on the scissile phosphate.
General Acid Catalysis by the GlcN6P Amine
All biochemical data collected to date have shown the amine of GlcN6P to be required for catalysis[3, 7, 14]. The GlmS ribozyme positions the C2-amine of GlcN6P 2.7Å from the 5′-O leaving group of G1 (Fig. 4c). An equivalent interaction with the C2-OH is made in the structure of the Glc6P bound ribozyme[8], which indicates that the chemical properties, not just the physical position, of the amine are critical to its catalytic contribution. The pKa of the amine is 8.2[7], closer to neutrality than any RNA functional group and substantially lower than the pKa of a hydroxyl group at the same position (pKa >12). The structure suggests general acid catalysis with the ammonium form of the GlcN6P donating a proton to the 5′-O leaving group (Fig. 4c).
Initial experiments suggested that the amino rather than the ammonium form of GlcN6P activates the ribozyme[7]. This would be most consistent with GlcN6P acting as a general base to activate the 2′-OH nucleophile. In the T. tengcongenis GlmS structure waters observed in the active site were proposed to mediate C2-amine deprotonation of the A-1 2′-OH nucleophile[8]. We do not observe equivalent water molecules in the active site of the B. anthracis GlcN6P bound ribozyme, which argues against this mechanism. How can the observed pH dependence be reconciled with the structural proximity of GlcN6P to the 5′-O leaving group?
The reported pH dependence was determined at sub-saturating concentrations of effector (10 μM GlcN6P, 10mM alternative ligands)[7]. As a result, the measured kobs values do not distinguish between a pH dependence on the Km or the kcat of the reaction. The pH dependence of the effector Km was tested and found to be minimal, but the experiments were performed across only a narrow pH range (7.5–8.8) near the pKa (8.2) where the predicted effect on Km is expected to be small[7].
Because these biochemical data significantly affect the interpretation of the GlmS structures, we determined the pH dependence of both Km and kcat for the GlcN6P promoted reaction. Consistent with the previous report[7], we found that the reaction rate (kobs) at subsaturating concentrations of effector (10 μM GlcN6P) was highly pH dependent (~50 fold change in rate between pH 6.5 and 9.0), However, at saturating concentrations of GlcN6P (10mM) kcat showed less than a two-fold change from pH 6.5–9.0 (Fig. 6a). By contrast, there was a dramatic GlcN6P Km dependence on pH (Fig. 6b). The Km was about 1000-fold higher at low pH (~65000 μM at pH 5.5) than at high pH (~50 μM at pH 9.0). In fact the Km was so large at low pH (5.5–6.5) that it became impossible to reach saturating concentrations of the ligand (Fig. 6a). This may explain why Klein and Ferré-D'Amaré were unable to observe a GlcN6P bound complex; the pH of their crystals was between 5.2 and 5.8[8]. Although the observed rate of 6 min−1 is faster than the rate reported by Soukup and coworkers, it is likely that it still does not reflect the rate of chemistry (kcat ≠ kchem). For example a rate of 16 min−1 has been reported for another GlmS ribozyme construct[11]. Consequently, the pH dependence of kchem remains to be determined, but the data demonstrate a clear effect of pH on Km. These data obviate a model that invokes GlcN6P as a general base in catalysis and do not preclude the structurally predicted mechanism of GlcN6P as a general acid. Physical organic chemistry implicates stabilization of the 5′-O leaving group as the critical factor in the mechanism of phosphodiester cleavage[16]. In the GlmS ribozyme, leaving group stabilization appears to be achieved by the cofactor.
Figure 6.
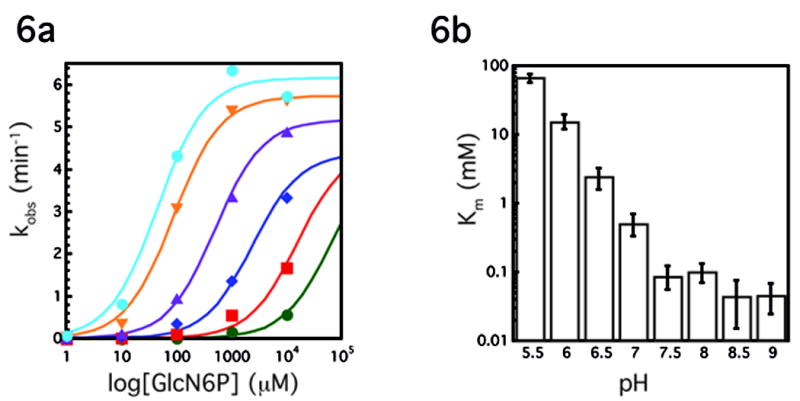
pH dependence of the Glms ribozyme reaction. A. Plot of kobs(min−1) versus log concentration of GlcN6P (μM) at various pHs, each curve represents the average of two experiments. Green circles, pH 5.5; red squares, pH 6.0; blue diamonds, pH 6.5; purple triangles, pH 7.0; orange triangles, pH 7.5; blue circles, pH 8.5. Experiments at pH 8 and pH 9 were omitted for simplicity. Lines represent best fit to the data. At pH 5.5, 6.0 and to a less degree 6.5, saturating GlcN6P concentrations could not be achieved. These data were fit assuming a kcat of 4.5 min−1. Because the reaction is so far from saturation, the resulting fits provide only a rough estimate for the Km at the lowest pHs. B. Km (mM) vs pH, each bar represents the average of two experiments.
Transition State Charge Stabilization
GlcN6P could also provide transition state stabilization through interaction between the C1-OH and the scissile phosphate. In this ground state structure the pro-SP non-bridging oxygen of the scissile phosphate accepts hydrogen bonds from the exocyclic amine of G57 and the C1-OH of GlcN6P (Fig. 4c). Both groups have been shown to contribute significantly to the reaction[7, 13, 14]. The importance of the G57 exocyclic amine is evidenced by interference resulting from inosine or N-methylguanosine substitutions[13]. The importance of the vicinal hydroxyl is further demonstrated in experiments with the two enantiomers of serine[7]. L-serine, which presents the C1-OH and C2-amine functional groups in the same stereochemical orientation as GlcN6P, supports ribozyme activity, but D-serine does not. Introduction of any additional functionalities at the C1 position fully inhibits the GlmS reaction; while removal of the oxygen results in a 70-fold rate reduction[14]. As the reaction proceeds toward the trigonal bipyrimidal transition state, the pro-SP phosphate oxygen will shift still closer to the GlcN6P C1-OH and the G57 amine. Likewise, the pro-RP oxygen could form stronger interactions with the G32 N1, the importance of which is still untested. These interactions would provide selective stabilization of the growing negative charge at the transition state.
Nucleobase contributions to catalysis
GlcN6P does not directly interact with the A-1 2′-O nucleophile, so if nucleophilic activation occurs at all, it is likely to come from another source. The closest functional group to the A-1 2′-O, which is methylated in this structure, is the N1 of G33. G33 is orientated through a hydrogen bond network between its 2′-OH and N3 and the 2′-OH of U59. While, mutation of U59 does not have a significant effect on catalysis[15], interference analysis found that the U59 2′-OH is absolutely required for activity[13]. Given the proximity of G33 to the nucleophile, we tested its importance by mutating it to A, C or U. At saturating GlcN6P concentrations, the G33A mutation resulted in complete inhibition of the reaction (at least a 10,000-fold effect). G33U and G33C ribozymes were over 500-fold reduced in their ability to promote the cleavage reaction. G33 is 100% conserved in GlmS ribozymes[9], while the nucleotide immediately 3′ is less conserved and can be mutated with only a modest (~2 fold) effect on the rate[3].
Catalytically important G residues have been observed in the active sites of other autolytic ribozymes[17, 18]. Structures of the hairpin[18] and hammerhead[17] ribozymes also revealed that the N1 of a G donates a hydrogen bond to the 2′-O. Mutagenesis of these Gs also led to large rate reductions[19, 20]. There are two main hypotheses to explain the mechanistic role of the guanosine in these catalytic RNAs. Nucleobase rescue of an abasic site in the hairpin ribozyme suggested that the G stabilizes the negative charge in the transition state[21]. Alternatively, despite its high pKa, the N1 of G has been proposed to act as the general base to the 2′-OH nucleophile, possibly by adopting an alternative tautomeric form[22, 23]. It is unclear what contribution G33 makes to the GlmS mechanism though geometric constraints favor the possibility of G33 acting as a general base (Fig. 7). Formation of the transition state would appear to move the non-bridging oxygens further from, rather than closer to, the G33 N1, which argues against charge stabilization. A similar orientation of G33 (G40) was observed in the structures from T. tengcongenis[8]. In this 2′-O-methyl inhibited structure, there are no functional groups other than G33 sufficiently close to the nucleophile to activate the 2′-OH.
Figure 7.
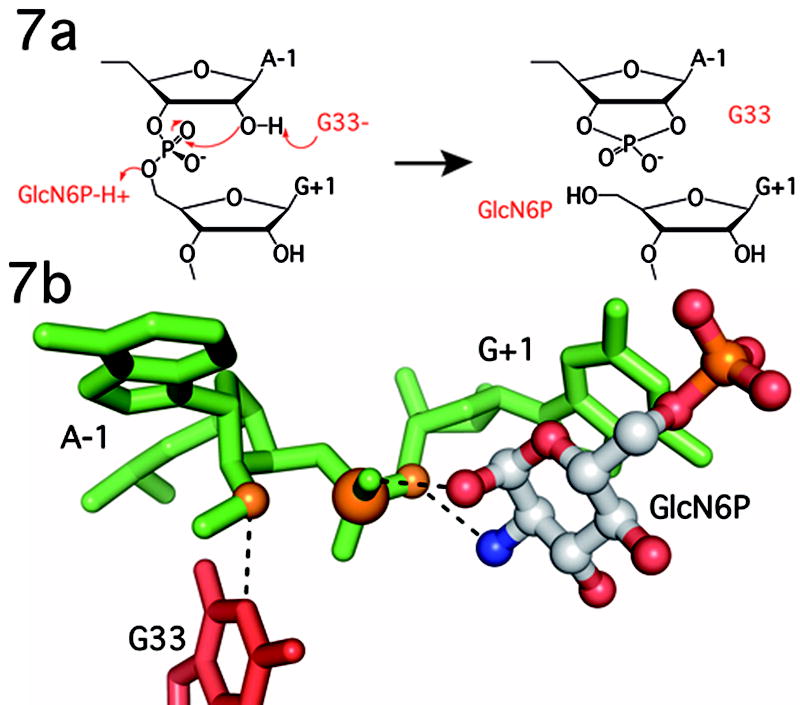
Proposed catalytic mechanism of the GlmS ribozyme. In this mechanism G33 functions as a general base to deprotonate the 2′-OH nucleophile and GlcN6P as a general acid to protonate the 5′-O leaving group. Other functional groups, including the C1-OH of GlcN6P stabilize charge developing on the scissile phosphate. See text for complete discussion and mechanistic alternatives. B. Position of the key functional groups as observed in the ground state structure of the GlcN6P bound GlmS ribozyme.
Key determinants in efficient general acid-base catalysis are the pKa values of the general acid and base and the difference between them[24, 25]. Ideally the pKas are close to neutral pH and separated by less than 2 pH units to allow both protonation states of the acid and the base to exist in reasonable proportions. The pKa of the GlcN6P amine is 8.2, while that of the G33 N1 is between 9.3 and 10.4[26]. At the elevated pH optimum (pH 8–9) of GlmS catalyzed cleavage the GlcN6P and the G33 N1 are predicted to be in the correct protonation state a sufficient fraction of the time for both to contribute significantly to catalysis. At neutral pH, the predominant species of GlcN6P is the ammonium form, capable of protonating the 5′-O leaving group. The G33 N1 would also be protonated and would function as a poor general base. This suggests that at physiological pH, GlcN6P would contribute substantially more catalytic power than G33.
Significance
Unlike all other examples of riboswitches, the GlmS riboswitch is not triggered by a conformational change. Comparison of apo, inhibitor and activator bound GlmS ribozyme structures reveal that the active site is preorganized in the absence of effector[8]. The nucleophile and leaving group are aligned prior to ligand binding, G33 is properly positioned, but cleavage is slow in the absence of GlcN6P. The GlmS ribozyme is activated by GlcN6P binding, which positions a primary amine next to leaving group. This is the first example of an RNA enzyme that uses a cofactor to achieve efficient catalysis. Like many protein enzymes, this ribozyme catalyzes a reaction by relying upon the unique chemical properties of a small molecule cofactor. Such a catalytic strategy broadens the chemical repertoire accessible to an evolving pre-biotic RNA world.
METHODS
Complex formation and crystallization
RNA genes were constructed by fusing the Ban-11U GlmS gene, which has an 11-nucleotide deletion at the 5′ end and the recognition sequence for U1A at the end of P1, and the gene for the antigenomic HDV ribozyme. RNA was prepared by in vitro transcription with T7 polymerase. Post transcriptional processing of the ag-HDV ribozyme generated homogenous 3′-termini containing a 2′-3′ cyclic phosphate. Oligonucleotides containing active site substitutions were purchased from Dharmacon Research (Lafayette, CO), deprotected by manufacturer recommended procedures and used in crystallization trials. The U1A double mutant (Y31H and Q36R) was expressed and purified as described (with personal communication from D. Schechner)[10, 27]. RNA (60 μM), the 5′-oligonucleotide (oMeS, 60μM) and GlcN6P (2mM) were heated to 70° for 3 minutes in folding buffer (10mM sodium cacodylate pH 6.8, 10mM MgCl2, 10mM KCl) and allowed to cool before addition of the U1A protein (65μM). Two volumes of the complex were combined with one volume of 11% polyethylene glycol-8000 (PEG-8000), 9% dimethyl sulfoxide (DMSO), 20mM sodium cacodylate pH 6.8, 20mM MgCl2 and 200mM KCl at 25° C. Crystals were grown by vapor diffusion to a maximum size of 500 X 100 X 100 mm in about 3 days. The crystals were stabilized in a solution containing 35% PEG-8000, 1.5M 1,6-hexanediol, 20mM sodium cacodylate pH 6.8, 20mM MgCl2 and 200mM KCl before flash freezing.
Structure determination
The iridium hexamine derivative was obtained by soaking native crystals in stabilization solution supplemented with 5mM iridium hexamine. A pseudo-native crystal that was isomorphous to the iridium hexamine derivative was obtained by soaking a native crystal in 2mM cobalt hexamine. Additional phasing information was obtained from crystals containing an oligonucleotide iodinated at position +11 and soaked in 1mM cobalt hexamine. The 2.8 Å iridium derivative, 2.75Å iodine derivative and 2.75Å pseudo-native data sets were collected at beamline X26C at the National Synchrotron Light Source (NSLS) at 1.154 Å, 1.6Å and 1.1Å wavelength respectively. A 2.5Å native dataset was collected at beamline X25 at the NSLS at 0.985Å wavelength. Data were processed using HKL2000[28].
To determine the structure of the GlmS ribozyme, 20 iridium hexamine sites were located using single isomorphous replacement and anomalous scattering techniques (SIRAS). The initial sites were found with SHELXD software[29]. SIRAS phasing was performed with SHARP and additional iridium sites (50 total), cobalt hexamine (14) and iodine (4) sites were determined using difference Fourier synthesis. Multiple isomorphous replacement and anomalous scattering (MIRAS) was then used for the final phasing of the experimental data and density modified using SOLOMON and an optimized solvent content of 50%[30]. The initial model was built manually using the MIRAS electron density map and the program O[31]. Strong density was observed for the phosphate backbone. The model was then used in PHASER as a molecular replacement search model for the higher resolution (2.5 Å) native data set[32]. The model was refined against the 2.5Å native data set using the program REFMAC[33]. Figures were made with PyMOL[34].
Kinetic Assays
The ribozyme construct (1μM) was preincubated in a solution containing 25mM acetate, 25mM cacodylate, 25mM HEPES, 25mM TAPSO, 10mM MgCl2 and various concentrations of GlcN6P. This four buffer system was used so a single buffering scheme could be implemented over the entire pH range. Buffers were avoided that either competed with GlcN6P binding (such as MES and MOPS) or that had ethanolamine moieties that could promote chemistry in the absence of GlcN6P (such as Tris and glycine). The pH of the solution was adjusted after the addition of GlcN6P because high sugar concentrations were not adequately buffered. The reaction was started by addition of trace 5′ radiolabeled rS oligonucleotide (Dharmacon, Lafayette, Co). Aliquots were taken at appropriate time points, quenched in formamide loading buffer (95% formamide, 2.5mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol) and analyzed by denaturing polyacrylamide gel electrophoresis. Gels were visualized using a STORM PhosphoImager (GE Healthcare) and the data quantitated using ImageQuant (GE Healthcare). Rates were determined using the equation
and fit using the least squares method implemented in KaleidaGraph (Synergy Software). Km and kcat at each pH were determined using the equation
Supplementary Material
Acknowledgments
We would like to thank M. Becker, H. Robinson, A. Heroux, S. Myers and the beamline staff at X25, X26C and X29 at the NSLS at Brookhaven National Laboratory; M. Strickler and the CSB Core Staff; R. Breaker, Y. Modis, J. Wang, A. Roth, D. Schechner, I. Sudyam, D. Hiller, M. Stahley, R. Voorhees and other members of the Strobel Lab for discussions and comments on the manuscript; Y. Xiong for all of his help; R. Batey for the gift of iridium hexamine; and M. Calabrese for mass spectrometry. This work was supported by a grant from the National Science Foundation (MCB0544255) and the National Institutes of Health (GM022778).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 2.Mandal M, Boese B, Barrick JE, Winkler WC, Breaker RR. Riboswitches control fundamental biochemical pathways in Bacillus subtilis and other bacteria. Cell. 2003;113:577–586. doi: 10.1016/s0092-8674(03)00391-x. [DOI] [PubMed] [Google Scholar]
- 3.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 4.Milewski S. Glucosamine-6-phosphate synthase--the multi-facets enzyme. Biochim Biophys Acta. 2002;1597:173–192. doi: 10.1016/s0167-4838(02)00318-7. [DOI] [PubMed] [Google Scholar]
- 5.Hampel KJ, Tinsley MM. Evidence for preorganization of the glmS ribozyme ligand binding pocket. Biochemistry. 2006;45:7861–7871. doi: 10.1021/bi060337z. [DOI] [PubMed] [Google Scholar]
- 6.Fedor MJ, Williamson JR. The catalytic diversity of RNAs. Nat Rev Mol Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 7.McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, Soukup GA. Ligand requirements for glmS ribozyme self-cleavage. Chemistry & Biology. 2005;12:1221–1226. doi: 10.1016/j.chembiol.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Klein DJ, Ferre-D'Amare AR. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006;313:1752–1756. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 9.Roth A, Nahvi A, Lee M, Jona I, Breaker RR. Characteristics of the glmS ribozyme suggest only structural roles for divalent metal ions. RNA. 2006;12:607–619. doi: 10.1261/rna.2266506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferre-D'Amare AR, Doudna JA. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J Mol Biol. 2000;295:541–556. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson SR, Been MD. A pseudoknot in the 3′ non-core region of the glmS ribozyme enhances self-cleavage activity. RNA. 2005;11:1788–1794. doi: 10.1261/rna.2203605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167–1171. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jansen JA, McCarthy TJ, Soukup GA, Soukup JK. Backbone and nucleobase contacts to glucosamine-6-phosphate in the glmS ribozyme. Nat Struct Mol Biol. 2006;13:517–523. doi: 10.1038/nsmb1094. [DOI] [PubMed] [Google Scholar]
- 14.Lim J, Grove BC, Roth A, Breaker RR. Characteristics of Ligand Recognition by a glmS Self-Cleaving Ribozyme. Angew Chem Int Ed Engl. 2006;45:6689–6693. doi: 10.1002/anie.200602534. [DOI] [PubMed] [Google Scholar]
- 15.Soukup GA. Core requirements for glmS ribozyme self-cleavage reveal a putative pseudoknot structure. Nucleic Acids Research. 2006;34:968–975. doi: 10.1093/nar/gkj497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oivanen M, Kuusela S, Lonnberg H. Kinetics and Mechanisms for the Cleavage and Isomerization of the Phosphodiester Bonds of RNA by Bronsted Acids and Bases. Chem Rev. 1998;98:961–990. doi: 10.1021/cr960425x. [DOI] [PubMed] [Google Scholar]
- 17.Martick M, Scott WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rupert PB, Ferre-D'Amare AR. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410:780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 19.Pinard R, Hampel KJ, Heckman JE, Lambert D, Chan PA, Major F, Burke JM. Functional involvement of G8 in the hairpin ribozyme cleavage mechanism. EMBO J. 2001;20:6434–6442. doi: 10.1093/emboj/20.22.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruffner DE, Stormo GD, Uhlenbeck OC. Sequence requirements of the hammerhead RNA self-cleavage reaction. Biochemistry. 1990;29:10695–10702. doi: 10.1021/bi00499a018. [DOI] [PubMed] [Google Scholar]
- 21.Kuzmin YI, Da Costa CP, Fedor MJ. Role of an active site guanine in hairpin ribozyme catalysis probed by exogenous nucleobase rescue. J Mol Biol. 2004;340:233–251. doi: 10.1016/j.jmb.2004.04.067. [DOI] [PubMed] [Google Scholar]
- 22.Han J, Burke JM. Model for general acid-base catalysis by the hammerhead ribozyme: pH-activity relationships of G8 and G12 variants at the putative active site. Biochemistry. 2005;44:7864–7870. doi: 10.1021/bi047941z. [DOI] [PubMed] [Google Scholar]
- 23.Wilson TJ, Ouellet J, Zhao ZY, Harusawa S, Araki L, Kurihara T, Lilley DM. Nucleobase catalysis in the hairpin ribozyme. RNA. 2006;12:980–987. doi: 10.1261/rna.11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bevilacqua PC. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry. 2003;42:2259–2265. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 25.Cleland WW. Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv Enzymol Relat Areas Mol Biol. 1977;45:273–387. doi: 10.1002/9780470122907.ch4. [DOI] [PubMed] [Google Scholar]
- 26.Da Costa CP, Sigel H. Acid-base and metal ion binding properties of guanylyl(3′->5′)guanosine (GpG(−)) and 2′-deoxyguanylyl(3′-5′)-2′-deoxyguanosine [d(GpG)( −)] in aqueous solution. Inorganic Chemistry. 2003;42:3475–3482. doi: 10.1021/ic020672l. [DOI] [PubMed] [Google Scholar]
- 27.Oubridge C, Ito N, Evans PR, Teo CH, Nagai K. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 30.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D Biol Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 31.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. Pt 2. [DOI] [PubMed] [Google Scholar]
- 32.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 33.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 34.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.