

Abstract
Long-standing behavioral abnormalities emerge after puberty in rats following neonatal hippocampal lesion, providing a developmental model of abnormal rat behavior that may have predictive validity in identifying compounds effective in treating symptoms of schizophrenia. We sought to test the predictive validity of the neonatal hippocampal lesion model in identifying preventive treatment for first-episode psychosis. We determined the effect of risperidone, recently studied for prevention of first-episode psychosis, on the development of elevated locomotor activity following neonatal hippocampal lesions. Rat pups received hippocampal or sham lesions on postnatal day 7, followed by treatment with risperidone or vehicle from postnatal days 35 to 56. Locomotor activity in response to novelty, amphetamine, and nocturnal locomotion were determined on postnatal day 57. Low-dose risperidone (45 μg/kg) pretreatment prevented elevated locomotor activity in some, but not all, of the behavioral tasks following neonatal hippocampal lesions. In contrast, higher risperidone pretreatment was less effective in preventing elevated locomotor activity following neonatal hippocampal lesions. Because low risperidone dosages were also found to be effective in preventing first-episode psychosis in human studies, these data support the predictive validity of the hippocampal lesion model in identifying medications for prevention of first-episode psychosis. Additionally, these data support the use of low-dose risperidone in psychosis prevention, and suggest the possibility that higher risperidone doses could be less effective in this application.
Keywords: hippocampus, schizophrenia, animal model, first-episode psychosis, prodrome, amphetamine
INTRODUCTION
Although the concept of preventive treatment for schizophrenia is not new (Sullivan, 1927; Meares, 1959), several factors impede the identification of effective primary prevention treatments for first-episode psychosis. First, the most effective primary prevention interventions would target the etiology of the illness. At present, the etiological factors causing schizophrenia and other psychotic disorders are not known, necessitating screening many diverse compounds to identify the most effective candidates for primary prevention. For example, antiviral compounds (Yolken et al, 2000), excitatory amino acid and/or neuroprotective agents (Tsai and Coyle, 2003; Javitt et al, 2004), and D3 receptor antagonists (Richtand et al, 2001) have all been discussed as potential medication classes for primary prevention against psychotic disorders. While it is therefore likely that it will be necessary to test a diverse array of chemical classes to identify an effective primary prevention treatment for first-episode psychosis, in practice the short-term and long-term risks to human subjects, as well as the practical limitations of limited clinical trial enrollment, make it impractical to screen even a small number of compounds in human subjects. Second, medication compliance in any long-term study of adolescents and young adults is typically poor. For example, less than 50% of subjects were fully adherent with medication treatment in the study evaluating risperidone efficacy in first-episode psychosis prevention (McGorry et al, 2002). This confounding variable greatly increases the likelihood of failing to detect beneficial effects of preventive treatment. Third, rather than preventing the development of psychotic symptoms, treatment interventions may be merely suppressing or delaying the appearance of abnormal behavior. Adequately addressing this question requires following a large number of subjects over an extended length of time, a study design that is exceedingly difficult to accomplish in an adolescent or young adult human sample, but can be directly studied in an animal model. And finally, for any drug holding clinical promise for use in primary prevention, dose–response data are needed. Again, limited subject enrollment in primary prevention clinical trials, and the risks of study participation, precludes obtaining this information in human subjects. Because of the tremendous human and economic burden of schizophrenia and other psychotic disorders, primary prevention modalities of even modest impact would likely have significant public health consequence, and there has been a recent growth in studies evaluating preventive treatment for individuals at high risk of developing first-episode psychosis (McGorry et al, 2002; Tsuang et al, 2002; Cornblatt, 2002; Woods et al, 2003; Morrison et al, 2004; Hafner et al, 2004). In summary, there is a compelling need for animal models with predictive validity in the first-episode psychosis prevention field.
The neonatal hippocampal lesion model developed by Lipska and Weinberger is a developmental model of abnormal rat behavior. Long-standing behavioral abnormalities, including increased locomotor responsiveness to stress, novel environment, and amphetamine (Lipska et al, 1993), and deficits in prepulse inhibition of startle (Lipska et al, 1995; Swerdlow et al, 1995) emerge after puberty in rats following neonatal hippocampal lesion. Neonatal hippocampal lesioned rats also exhibit deficits in social behavior (Sams-Dodd et al, 1997) and cognition (Chambers et al, 1996) during the prepubertal period, as well as in adulthood, which may be analogous to the negative and neurocognitive deficits, respectively, observed in schizophrenic patients. Because the time course of development of abnormal behaviors, as well as the behaviors themselves, shares similarities with schizophrenia, treatments inhibiting behavioral alterations using this animal model could have predictive validity in identifying compounds effective in treating a range of symptoms of schizophrenia (Lipska and Weinberger, 2000). Previous studies have described the effectiveness of haloperidol (Lipska et al, 1993; Lipska and Weinberger, 1994b), clozapine (Lipska and Weinberger, 1994b; Rueter et al, 2004), and risperidone (Rueter et al, 2004) in suppressing elevated spontaneous locomotor activity in adult neonatally lesioned rats, which may be analogous to alleviating psychotic symptoms (Lipska and Weinberger, 2000). Monitoring the development and expression of abnormal limbically mediated hyperlocomotor behaviors following neonatal hippocampal lesion may therefore provide a model with predictive validity in identifying primary preventive treatments for schizophrenia and first-episode psychosis (Lipska and Weinberger, 1994b, 2000; Sams-Dodd et al, 1997; Al-Amin et al, 2000).
We determined the effectiveness of subchronic risperidone treatment (postnatal days 35–56) in preventing the appearance of abnormally elevated locomotor behaviors emerging postpuberty following neonatal hippocampal lesion. Our study had two goals. First, we sought to test whether the neonatal hippocampal lesion model has predictive validity in identifying effective treatment strategies for prevention of first-episode psychosis. We therefore evaluated the effect of risperidone, recently found to be effective in preventing first-episode psychosis (McGorry et al, 2002), in preventing the appearance of abnormal locomotor behavior following neonatal hippocampal lesions. Second, we sought to evaluate two different risperidone dosages in our animal studies, since dose–response information is more easily determined in animal than in human studies.
Here we report that the lowest risperidone dose tested in our study prevents the appearance of some of the abnormal locomotor behaviors following neonatal hippocampal lesion. Of interest, a higher risperidone dose was less effective in preventing appearance of behavioral abnormalities. Possible mechanisms underlying these findings are discussed. Because low-dose risperidone was also found to be effective in preventing psychosis in human studies (McGorry et al, 2002), these data support the predictive validity of the hippocampal lesion model in identifying medications for prevention of first-episode psychosis. Additionally, these data support the use of low-dose risperidone in psychosis prevention (McGorry et al, 2002), and suggest the possibility that higher risperidone doses could be less effective in this application.
RESEARCH DESIGN AND METHODS
Subjects
Pregnant Sprague–Dawley rats (Harlan Farms, Indianapolis, IN) were obtained at 12–15 days gestation and housed individually with a 12-h light/dark cycle. Litters of 4–8 male pups were formed from the offspring. On the seventh day postnatally (weight 15–18 g), male pups within each litter were randomized to lesioned (n = 60) or sham-lesioned (n = 63) status. Rats were weaned on postnatal day 21 and housed in groups of three until testing. Animals were maintained at all times on the same 12-h light : dark cycle (0500 on; 1700 off). All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals.
Surgery
Rat pups were lesioned according to protocols developed by Lipska et al (1993). Animals were anesthetized by hypothermia by placement on ice for 10–20 min. An incision was made in the skin overlying the skull and 0.3 μml ibotenic acid or phosphate-buffered saline (PBS) infused bilaterally into the ventral hippocampal formation (AP −3.0 mm, ML ±3.5 mm, VD −5.0 mm relative to bregma) at a rate of 0.15 μl/min. The needle was left in place for 1 min after infusion to facilitate diffusion. Pups were placed under a warming lamp, the scalp sutured, and then returned to their mothers. The overall mortality rate was 15% (9/60) in lesioned animals, and 11 % (7/63) in sham-lesioned animals.
Drug Treatment
Ibotenic acid (Sigma, St Louis, MO) was dissolved in PBS at a final concentration of 10 μg/ml and pH adjusted to 7.4 with NaOH, and then divided into single-use aliquots and frozen on dry ice. Each single-use aliquot was kept frozen until use and discarded if thawed for longer than 10 min. Risperidone, dissolved as an aqueous solution (1 mg/ml), was from Janssen.d-Amphetamine sulfate (Sigma, St Louis, MO) was dissolved in 0.9% saline. Amphetamine concentration is described as free base. All injections were in a final volume of 1 ml/kg.
Risperidone doses of 45 and 85 μg/kg were both well below the ED50 of 1.1 μg/kg for inhibition of spontaneous locomotion in rat (Arnt, 1995). In rat, the ED50 for centrally acting serotonin 5-HT2 antagonism is 14 μg/kg (Megens et al, 1994), while the effects of D2 receptor antagonism first become apparent at 16 μg/kg (Janssen et al, 1988), with an ED50 for D2 antagonism from 56 to 150 μg/kg (Megens et al, 1994). Based on these observations, the risperidone dosages chosen for study have significant serotonin 5-HT2A receptor antagonism, with partial D2 dopamine receptor occupancy, which increases with ascending dose from 45 to 85 μg/kg. It has been previously suggested that the therapeutic effects of risperidone may be mediated by high-affinity serotonin 5-HT2A receptor antagonism, coupled with partial D2 dopamine receptor occupancy. Higher D2 dopamine receptor occupancy, which may induce extrapyramidal side effects, is believed unnecessary for the therapeutic effects of risperidone (Leysen et al, 1993; Schotte et al, 1996).
Amphetamine is used at a dose of 1.5 mg/kg because this low dose minimizes competing stereotyped behaviors elicited with higher amphetamine doses (Segal and Kuczenski, 1994); avoids a ‘ceiling effect’ that could occur if locomotion were unable to increase further; and is identical to the amphetamine dose used by other investigators (Lipska et al, 1993), facilitating comparisons with previous studies.
Rats were injected i.p. once daily between 1000 and 1100 with either saline (lesion n = 22/group, sham lesion n = 21/group), risperidone 45 μg/kg (lesion n = 19/group, sham lesion n = 20/group), or risperidone 85 μg/kg (lesion n = 19/group, sham lesion n = 20/group) from postnatal days 35 to 56. In the rat, postnatal days 29 through approximately 44 are the period of prepuberty, and postnatal days ∼45 through 60–70 represent the pubertal period of sexual maturation (Zicha and Kunes, 1999). Behavioral abnormalities following neonatal hippocampal lesion are not apparent at postnatal day 35, but are manifest when animals are tested at postnatal day 56 (Lipska et al, 1993). The rationale for treating animals during postnatal days 35–56 is that this interval covers the prepubertal and pubertal periods encompassing the period of development of behavioral abnormalities following neonatal hippocampal lesion.
Behavioral Testing
Behavioral testing was performed in 30 residential activity chambers (RACs). Each RAC consisted of a lighted, ventilated, sound-attenuated cabinet (Cline Builders, Covington, KY) housing a 16 × 16 × 15 inches Plexiglas enclosure. Locomotion was monitored with a 16 × 16 photobeam array (San Diego Instruments, San Diego, CA) located 0.5 inches above the floor of the enclosure. Locomotion is expressed as crossovers, defined as entry into any of the active zones of the chamber, as we have previously described (Pritchard et al, 2003). Animals were maintained throughout behavioral testing on the same 12-h light/dark cycle (0500 on; 1700 off). Animals were placed into RACs at 1000 on postnatal day 57 and locomotor response during the first hour in the novel RAC was determined. Following this behavioral observation, animals were injected at 1100 with amphetamine (1.5 mg/kg subcutaneous), and locomotion monitored for 3 h following injection. Animals remained in the RAC for both housing and behavioral observation, and nocturnal locomotion recorded during the first 2-h ‘lights off’ period (1700–1900). Behavioral measures used to evaluate differences between lesioned and sham-lesioned treatment groups included two previously described locomotion measures, response to novel environment and response to amphetamine injection (Lipska et al, 1993; Lipska and Weinberger, 1994a, 1995; Wan et al, 1996; Flores et al, 1996a, b; Swerdlow et al, 2001). Additionally, because the RACs used in our study provide locomotion data on the first evening in a novel environment, we also utilized initial locomotor response to change between light and dark cycle (first 2-h ‘lights off’ period in the RAC) as a third measure to identify behavioral differences between treatment groups.
Lesion Verification
Following behavioral testing, animals were killed by overdose with 50 mg/kg pentobarbital and perfused with 4% paraformaldehyde, and brains removed and cut in 50 μm sections on a freezing microtome. Lesion placement was determined immunocytochemically using neuron-specific nuclear protein antibody. Tissue was blocked in 1% H2O2 in PBS for 10 min at room temperature (RT) and then blocked in PBS + 0.4% Triton-X + 0.1% BSA (PBS + ) for 1 h at RT. Tissue was then incubated with mouse anti-NeuN (Chemicon, Temecula, CA) diluted 1:10 000 in PBS+ 0.4% Triton-X + 0.1% BSA overnight at RT followed by +incubation with biotinylated donkey anti-mouse IgG (Jackson, West Grove, PA) diluted 1 : 400 in PBS + 0.4% Triton-X + 0.1% BSA for 1 h at RT. Signal was amplified using the Vectastain elite ABC kit (Vector, Burlingame, CA) diluted 1 : 800 in PBS + 0.4% Triton-X + 0.1% BSA for 1 h at RT and stained with 3,3′ diaminobenzidine (Sigma, St Louis, MO) for 10 min at RT. Subsequently, representative sections were counter-stained using cresyl violet.
Statistical Analysis
Data were collected in 15-min bins and analyzed by three-way ANOVA with LESION STATUS (lesion, sham) and DRUG DOSE (0 (saline), risperidone 45 or 85 μg/kg) as main factors and 15-min LOCOMOTOR TIME INTERVAL as a repeated measure. Our predicted hypothesis that risperidone pretreatment would reduce the hyperlocomotor effect of hippocampal lesion may be expressed as a statistical hypothesis (H) as follows, where μ is defined as the population mean in each treatment group:
Planned linear contrasts among cell means from a three-way ANOVA were used to test this hypothesis for each of two risperidone doses (45 and 85 μg/kg) over each 15 min observation interval (15–60 min for novelty and amphetamine, 15–120 min for nocturnal locomotion), providing a total of eight or 16 planned comparisons tested. Each test was one-sided, based on the predicted direction of drug effect (Milliken and Johnson, 1992). Since we planned to interpret a significant result for any of these comparisons as support for the experimental hypothesis, the Bonferroni correction for multiple testing was used in order to maintain the overall Type I error rate at α = 0.05. Therefore, for the novelty and amphetamine conditions, the required one-sided p-value for statistical significance was 0.05/8 = 0.00625. The corresponding p-value for the noctural locomotion condition was 0.05/16 = 0.003125. Identical analyses were performed on locomotor activity under three conditions: novelty, amphetamine injection (1.5 mg/kg), and initial response to change between light and dark cycle. Statistical analyses were performed using the MIXED procedure of the SAS System for Windows, version 8.2 (SAS Institute, Cary, NC).
RESULTS
Lesion Placement Verification
Lesion placement and size were verified based on loss of immunostaining for neuronal marker NeuN. Representative sections from lesioned and sham-lesioned animals are shown in Figure 1. The boundaries of lesion placement and size for animals with bilateral hippocampal lesions are shown in Figure 2. As observed in Figures 1b and 2, lesions are characterized by loss of hippocampal tissue secondary to long-term absence of neuronal elements, and range from loss restricted to CA1 and CA3 components of the hippocampus to almost complete loss of all hippocampal tissue. This pattern is characteristic of the hippocampal lesion model, as described previously (Lipska et al, 1993; Swerdlow et al, 2001). Loss of neurons was not observed in neighboring cortical or thalamic areas. Ibotenate-injected animals lacking clear bilateral hippocampal lesions were omitted from the subsequent behavioral analyses. The final group sizes were sham saline, 21 animals; lesion saline, six animals; sham risperidone (45 μg/kg), 20 animals; lesion risperidone (45 μg/kg), eight animals; sham risperidone (85 μg/kg), 20 animals; and lesion risperidone (85 μg/kg), six animals. The large attrition rate in the final number of animals in the lesion group was primarily attributable to animals with a clear unilateral lesion, but for whom clear bilateral lesions could not be confirmed upon histological assessment.
Figure 1.
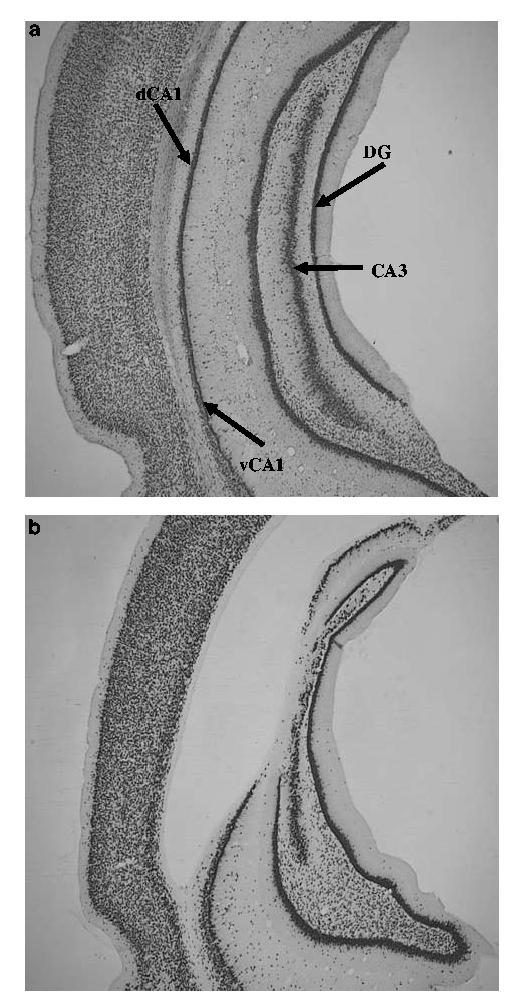
Photomicrographs of representative NeuN-stained sections of adult control (a) and neonatal hippocampal-lesioned (b) rats. Note the neuronal loss in the dorsal CA1, CA3 and dentate gyrus (DG) granule cell layer, and associated ventricular dilation.
Figure 2.
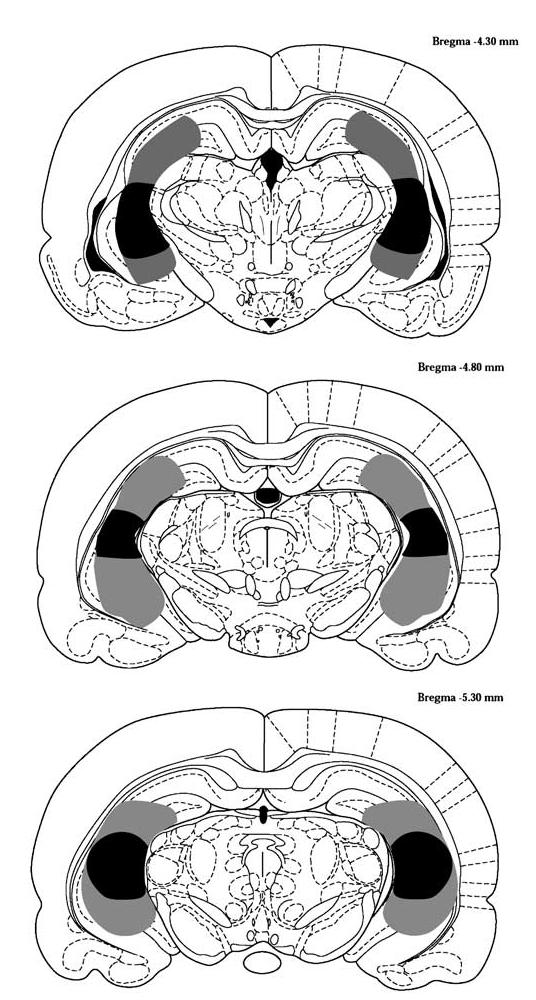
Schematic diagram of lesion boundaries following bilateral ibotenic acid infusion at P7. Lesion boundaries for individual animals were defined by neuronal loss, as determined immunocytochemically by NeuN staining. Shading denotes boundaries of the largest (gray) and smallest (black) lesions. Diagrams are from (Paxinos and Watson, 1986).
Behavioral Response in Adult Rats Following Neonatal Hippocampal Lesion
Behavioral testing was performed on 57-day-old rats treated daily with either saline or risperidone injection (1 ml/kg i.p.) on postnatal days 35–56. Main effects of LESION, DRUG DOSE, and TIME INTERVAL, and interactions between these factors determined by three-way ANOVA are listed in Table 1. Specific findings are described below.
Table 1.
Analysis of Variance (ANOVA) Summary Tables for Novelty-, Amphetamine-, and Transition from Light to Dark Cycle-Stimulated Locomotion
| Condition effect | Numerator DF |
Denominator DF |
F | p |
|---|---|---|---|---|
| Novelty | ||||
| Dose | 2 | 75 | 0.58 | 0.56 |
| Time | 3 | 225 | 137.01 | 0.00 |
| Dose × Time | 6 | 225 | 1.31 | 0.25 |
| Lesion | 1 | 75 | 15.17 | 0.00 |
| Dose × Lesion | 2 | 75 | 0.88 | 0.42 |
| Time × Lesion | 3 | 225 | 1.42 | 0.24 |
| Dose × Time × Lesion | 6 | 225 | 1.69 | 0.13 |
| AMPH | ||||
| Dose | 2 | 75 | 0.61 | 0.55 |
| Time | 3 | 225 | 39.70 | 0.00 |
| Dose × Time | 6 | 225 | 1.01 | 0.42 |
| Lesion | 1 | 75 | 4.73 | 0.03 |
| Dose × Lesion | 2 | 75 | 3.50 | 0.04 |
| Time × Lesion | 3 | 225 | 4.83 | 0.00 |
| Dose × Time × Lesion | 6 | 225 | 2.23 | 0.04 |
| Nocturnal | ||||
| Dose | 2 | 75 | 0.09 | 0.91 |
| Time | 7 | 525 | 30.91 | 0.00 |
| Dose × Time | 14 | 525 | 1.47 | 0.12 |
| Lesion | 1 | 75 | 13.41 | 0.00 |
| Dose × Lesion | 2 | 75 | 1.18 | 0.31 |
| Time × Lesion | 7 | 525 | 3.02 | 0.00 |
| Dose × Time × Lesion | 14 | 525 | 1.95 | 0.02 |
Novelty
As expected based on previous studies (Lipska et al, 1993, 1995; Wan et al, 1996; Flores et al, 1996a, b; Alquicer et al, 2004), locomotion during the first hour following introduction into the RAC on day 57, a novel environment, was increased in lesioned relative to sham-lesioned animals treated with a daily saline injection on postnatal days 35–56 (Figure 3a, left panel). Locomotion was significantly increased at the 15 (p = 0.019), 30 (p = 0.001), and 45 min (p = 0.012) time intervals following introduction into the novel environment. Behavioral response to the same novel environment for lesioned and sham-lesioned rats treated with risperidone 45 and 85 μg/kg on postnatal days 35–56 is shown in Figure 3a (center and right panels). Locomotion was elevated in the risperidone 45 μg/kg lesioned group relative to the sham-lesioned group at the 45 min time interval (p = 0.014). Locomotion was significantly higher in lesioned relative to sham-lesioned animals at the 60 min time interval in the risperidone 85 μg/kg treatment groups (p = 0.005).
Figure 3.

Effect of risperidone pretreatment on locomotor response to novelty. (a) Locomotor responses (crossovers/15 min interval) following introduction into a novel environment (RACs) on postnatal day (PD) 57 of lesioned and sham-lesioned rats treated on PD 35–56 with saline or risperidone (45 or 85 μg/kg). Data are expressed as mean crossovers; error bars are 95% confidence intervals. (b) Risperidone treatment effects (45 or 85 μg/kg) determined from [μ{Lesion/Risp x μg/kg}−μ{Sham/Risp x μg/kg}]−[μ{Lesion/Saline}−μ{Sham/Saline}]. Negative values indicate suppression of the hyperlocomotor effect of hippocampal lesions (0). Data are expressed as mean ± 95% confidence intervals. *P≤0.05, **P≤0.01, ***P≤0.001 vs lesion group.
Examination of locomotion in risperidone-treated, sham-lesioned animals identified a tendency toward higher locomotion in sham-lesioned animals following risperidone pretreatment; however, this trend did not achieve statistical significance (dose effect among sham-lesioned rats, p = 0.054).
In order to test the hypothesis that risperidone pretreatment reduced the hyperlocomotor effect of hippocampal lesion, the difference in population means between lesioned and sham-lesioned groups was examined for each risperidone pretreatment dose relative to the saline pretreatment population mean difference as a function of time following introduction into the novel environment using a three-way ANOVA (Figure 3b). Although unadjusted tests suggest a reduction in the effect of lesion at 30 min for both risperidone 45 and 85 μg/kg pretreatment groups (Figure 3b, 30 min time interval), neither of these comparisons are significant after adjustment for multiple comparisons. In summary, for both the risperidone 45 and 85 μg/kg pretreatment groups [μ{Lesion/Risp x μg/kg}−μ{Sham/Risp x μg/kg}]−[μ{Lesion/Saline}−μ{Sham/Saline}] was not significantly less than zero.
Amphetamine Injection
Locomotion during the first hour following amphetamine injection on day 57 was increased in lesioned relative to sham-lesioned animals treated with a daily saline injection on postnatal days 35–56 (Figure 4a, left panel), consistent with earlier findings from other laboratories (Lipska et al, 1993; Wan et al, 1996; Wan and Corbett, 1997; Lipska and Weinberger, 1994a, 1995; Flores et al, 1996a, b; Swerdlow et al, 2001). Locomotion was significantly increased at the 30 (p = 0.046) and 60 min (p = 0.010) time intervals following amphetamine injection. Behavioral response to amphetamine injection for lesioned and sham-lesioned rats treated with risperidone 45 or 85 μg/kg on postnatal days 35–56 is shown in Figure 4a (center and right panels). Locomotion was significantly higher in lesioned relative to sham-lesioned animals at the 30 (p = 0.009) and 60 min (p = 0.017) time intervals in the risperidone 85 μg/kg treatment groups; however, no differences in locomotion were detected between lesioned and sham-lesioned animals in the risperidone 45 μg/kg pretreatment groups at any of the time intervals tested.
Figure 4.

Effect of risperidone pretreatment on amphetamine-stimulated locomotion. (a) Locomotor responses (crossovers per 15 min interval) following amphetamine (1.5 mg/kg) injection on PD 57 of lesioned and sham-lesioned rats treated on PD 35–56 with saline or risperidone (45 or 85 μg/kg). Data are expressed as mean crossovers; error bars are 95% confidence intervals. (b) Risperidone treatment effects (45 or 85 μg/kg) determined from [μ{Lesion/Risp x μg/kg}−μ{Sham/Risp x μg/kg}]−[μ{Lesion/Saline}−μ{Sham/Saline}]. Negative values indicate suppression of the hyperlocomotor effect of hippocampal lesions (0). Note that the protective effect of 45 μg/kg risperidone emerges over time, becoming statistically significant after 30 min. Data are expressed as mean ±95% confidence intervals. *P ≤0.05, **P≤0.01 vs lesion group.
Risperidone pretreatment did not significantly affect amphetamine-stimulated locomotion in sham-treated animals (dose effect among sham-lesioned rats, p = 0.146).
In order to test the hypothesis that risperidone pretreatment reduced the hyperlocomotor effect of hippocampal lesion, the difference in population means between lesioned and sham-lesioned groups was examined for each risperidone pretreatment dose relative to the saline pretreatment population mean difference as a function of time following amphetamine injection using a three-way ANOVA (Figure 4b). Relative decreases in the hyperlocomotor effect of lesion were observed at 30, 45, and 60 min post injection in the risperidone 45 μg/kg group. Unadjusted tests suggest reductions in the effect of lesion at the 30 (p = 0.049) and 45 min (p = 0.017) time intervals following risperidone 45 μg/kg pretreatment (Figure 4b, 30 and 45 min intervals); however neither of these comparisons achieve statistical significance after adjustment for multiple comparisons. The maximum reduction of the hyperlocomotor effect of lesion following risperidone 45 μg/kg pretreatment is observed at the 60 min time interval (p = 0.003). Following correction for multiple comparisons, this result is statistically significant. In contrast, there was no reduction in the effect of lesion at any time point for the risperidone 85 μg/kg pretreatment group (Figure 4b). In summary, for the risperidone 45 μg/kg pretreatment groups, [μ{Lesion/Risp x μg/kg}−μ{Sham/Risp x μg/kg}]−[μ{Lesion/Saline}−μ{Sham/Saline}] is significantly less than zero at the 60 min time point. In contrast, for the risperidone 85 μg/kg pretreatment group, [μ{Lesion/Risp x μg/kg}−μ{Sham/Risp x μg/kg}]−[μ{Lesion/Saline}−μ{Sham/Saline}] was not significantly less than zero.
Nocturnal Locomotion
Locomotion during the first 2 h time period following lights out during the first nocturnal time period in the RAC on day 57, a novel home environment, was increased in lesioned relative to sham-lesioned animals treated with a daily saline injection on postnatal days 35–56 (Figure 5a, left panel). Locomotion was significantly increased during the 30 (p = 0.029), 60 (p = 0.008), 75 (p = 0.007), 90 (p = 0.001), and 105 min (p = 0.002) time intervals following lights out. Behavioral response to the same nocturnal environment is shown in Figure 5a (center and right panels) for lesioned and sham-lesioned rats treated with risperidone 45 and 85 μg/kg on postnatal days 35–56. Locomotion was elevated in the risperidone 45 μg/kg lesioned group relative to the sham-lesioned group at the 15 (p = 0.015) and 30 min (p = 0.035) time intervals. Locomotion was significantly higher in lesioned relative to sham-lesioned animals at the 30 (p = 0.047), 75 (p = 0.026), and 90 min (p = 0.028) time intervals in the risperidone 85 μg/kg treatment groups.
Figure 5.

Effect of risperidone pretreatment on locomotion following change between light and dark cycle. (a) Locomotor response (crossovers per 15 min interval) during the first 120 min of the ‘lights-out’ period in the RACs on PD 57 of lesioned and sham-lesioned animals following pretreatment with saline or risperidone (45 or 85 μg/kg) on PD 35–56. Data are expressed as mean crossovers; error bars are 95% confidence intervals. (b) Risperi-done treatment effects (45 or 85 μg/kg) determined from [μ{Lesion/Risp x μg/kg}−μ{Sham/Risp x μg/kg}]−[μ{Lesion/Saline}−μ{Sham/Saline}]. Negative values indicate suppression of the hyperlocomotor effect of hippocampal lesions (0). Data are expressed as mean ±95% confidence intervals. *P≤0.05, **P≤0.01, ***P≤0.001 vs lesion group.
Risperidone pretreatment did not significantly affect locomotor response to transition between light and dark cycle in sham-treated animals (dose effect among sham-lesioned rats, p = 0.269).
In order to test the hypothesis that risperidone pretreatment reduced the hyperlocomotor effect of hippocampal lesion, the difference in population means between lesioned and sham-lesioned groups was examined for each risperidone pretreatment dose relative to the saline pretreatment population mean difference as a function of time following change from light to dark cycle using a three-way ANOVA (Figure 5b). Relative decreases in the hyperlocomotor effect of lesion were observed at 60, 90, and 105 min post lights out in the risperidone 45 μg/kg group. Unadjusted tests suggest reductions in the effect of lesion at the 60 (p = 0.037) and 105 min (p = 0.023) time intervals following risperidone 45 μg/kg pretreatment (Figure 5B, 60 and 105 min intervals). The maximum reduction of the hyper-locomotor effect of lesion following risperidone 45 μg/kg pretreatment is observed at the 90 min time interval (p = 0.005). None of these comparisons, however, achieve statistical significance after adjustment for multiple comparisons. There was no detectable reduction in the effect of lesion at any time point for the risperidone 85 μg/kg pretreatment group (Figure 5b). In summary, for initial locomotor response to change between light and dark cycle for both the risperidone 45 and 85 μg/kg pretreatment groups, [μ{Lesion/Risp x μg/kg}− μ{Sham/Risp x μg/kg}]−[μ{Lesion/Saline}−μ{Sham/Saline}] was not significantly less than zero.
DISCUSSION
Adult rats display elevations in locomotor activity in response to novelty and amphetamine following neonatal hippocampal lesions, thereby providing a model of the developmental progression from normal to abnormal behavior. In agreement with earlier studies (Lipska et al, 1993; Wan et al, 1996; Wan and Corbett, 1997; Lipska and Weinberger, 1994a, 1995; Flores et al, 1996a, b; Swerdlow et al, 2001; Alquicer et al, 2004), neonatal hippocampal lesions significantly altered locomotor response to novelty and amphetamine in adult rats. We have also identified increased locomotion during the initial 2-h period following change between light and dark cycle in a novel environment as a newly identified behavior altered in adult rats following neonatal hippocampal lesions.
Our findings support the concept of predictive validity for the neonatal hippocampal lesion model in identifying medications with potential efficacy for primary prevention in schizophrenia and first-episode psychosis. We observed that risperidone, at a dosage of 45 μg/kg per day, had a significant impact on appearance of abnormally elevated locomotor response to amphetamine following neonatal hippocampal lesions. The human behavioral response to amphetamine frequently includes psychotic symptoms of suspiciousness, fear, paranoia, delusions of persecution, and hallucinations (Ellinwood, 1967). The rodent hyperlocomotor response to amphetamine, which evolves from exploration and examination of the environment to more complex patterns of stereotyped behaviors (Ellinwood et al, 1973; Segal and Kuczenski, 1994), has both face and predictive validity as an animal model of psychosis (Lipska and Weinberger, 2000). Significantly, a higher risperidone dose of 85 μg/kg per day was less effective in preventing appearance of hyperlocomotor response to amphetamine in lesioned animals, demonstrating that it is unlikely that the normalization of amphetamine-stimulated locomotion provided by risperidone dosages of 45 μg/kg was the result of residual risperidone on the day of testing (day 57) in animals treated with risperidone on days 35–56. An important limitation of our findings is that we did not distinguish true prevention, vs suppression or postponement, of the emergence of abnormal behaviors following neonatal hippocampal lesions. Additional studies using later time points following treatment cessation will be needed in order to address this critical issue.
In contrast to the protective effect on abnormal amphetamine-stimulated locomotion, pretreatment with risperidone, at 45 and 85 μg/kg, was less effective in protecting against augmented novelty-stimulated and nocturnal locomotion in lesioned animals. This may suggest a normalization of function by risperidone at some, but not all, behaviorally relevant circuits, and may reflect differences in the impaired circuitry underlying increased novelty-stimulated locomotion vs amphetamine-stimulated locomotion. For example, amphetamine-stimulated locomotion is believed to result primarily from increased dopamine release in nucleus accumbens (reviewed in Le Moal and Simon, 1991). In contrast, novelty-stimulated locomotion may be mediated in part via prefrontal cortical input to ventral tegmentum and accumbens. A brief, transient increase in dopamine release in both nucleus accumbens shell and prefrontal cortex accompanies entry into a novel environment (Rebec et al, 1997a, b; Rebec, 1998). Increased dopamine release in medial prefrontal cortex generally results in decreased dopamine activity in nucleus accumbens, however (Louilot et al, 1989; Jaskiw et al, 1991; Kolachana et al, 1995; Tzschentke, 2001), and thus while the ventral tegmental–nucleus accumbens–pallidal circuit plays a critical role in modulation of locomotion response to both novelty and amphetamine-stimulated locomotion (Hooks and Kalivas, 1995), the role of medial prefrontal cortex in these different behavioral measures is likely different. Thus, differences in normalization by risperidone of these two behaviors could reflect differences in the relative importance of cortical and perhaps thalamic inputs to the ventral tegmental–nucleus accumbens–pallidal loop in the regulation of these two different locomotor responses. Further study will be needed to determine whether risperidone pretreatment provides normalization of some but not all of these circuits. Further study is also needed to test whether a wider dose range (ie perhaps lower risperidone dosages) would provide additional protection for normalization of novelty-stimulated locomotion. It is also possible that an increase in stereotyped behaviors, which could compete with and thereby reduce amphetamine-induced locomotor activity, might have reduced locomotion in some of the amphetamine-treated rats. However, a low amphetamine dose was used to minimize this potential (Segal and Kuczenski, 1994), and the pattern of activity did not indicate the occurrence of stereotypy-induced reductions in locomotor activity. Further studies monitoring behavioral stereotypy will be needed to definitively exclude this possibility.
The antipsychotic effects of risperidone are believed to result from high-affinity binding to D2, D3, and D4 dopamine receptors (Strange, 2001; Bymaster et al, 1996). High-affinity binding to serotonin 5-HT2A receptors, and a somewhat lower affinity for serotonin 5-HT2C receptors, is also believed to underlie the antipsychotic effect of risperidone (Richelson and Souder, 2000; Schotte et al, 1996; Leysen et al, 1993; Kongsamut et al, 1996; Bymaster et al, 1996). Additionally, risperidone binds with high affinity to both α1 and α2 adrenergic receptors, and binds with moderate affinity to H1 histamine receptors (Bymaster et al, 1996). While the mechanism(s) underlying the efficacy of risperidone in preventing appearance of abnormal behaviors following neonatal hippocampal lesions is not known, it is reasonable to consider hypotheses for mechanisms of action relating to these high-affinity binding activities at G protein-coupled receptors.
We have previously suggested that tolerance of D3 receptor-mediated inhibitory function may contribute to the development of psychosis (Richtand et al, 2001). Stress activates dopamine release in prefrontal cortex (Ventura et al, 2002; Cabib and Puglisi-Allegra, 1996), and both D3 receptor protein expression and receptor binding have been identified in prefrontal cortex (Diaz et al, 2000; Khan et al, 1998). Because the D3 receptor has the highest dopamine affinity, and is the only dopamine receptor occupied at concentrations in the range of basal dopamine concentrations, the ‘D3 dopamine receptor hypothesis’ of psychosis (Richtand et al, 2001, 2003, 2005) suggests that elevated dopamine concentrations from stress or other environmental factors prior to the development of psychosis result in a compensatory downregulation of prefrontal cortical D3 receptor function, thereby releasing D3 receptor-mediated inhibition of limbically modulated behaviors. The D3 receptor inhibits both novelty-stimulated locomotion (Pritchard et al, 2003; Xu et al, 1997; Menalled et al, 1999; Ekman et al, 1998; Accili et al, 1996) and amphetamine-stimulated locomotion (Waters et al, 1993) in rodents, and may also inhibit expression of analogous limbically modulated behaviors in humans including paranoia, delusions, and hallucinations (Ellinwood et al, 1973; Ellinwood, 1967). The loss of a D3 receptor-mediated ‘brake’ on these behaviors would be expressed as the emergence of augmented locomotor response to amphetamine in rodents and paranoid psychotic symptoms in humans. We (Richtand et al, 2000, 2003) and others (Chiang et al, 2003; Wan and Corbett, 1997; Flores et al, 1996a; Henry et al, 1995; Wallace et al, 1996) have described evidence supporting this hypothesis in rodent behavioral sensitization and other ‘animal models’ of psychosis.
Loss of inhibitory hippocampal input to medial prefrontal cortex following neonatal hippocampal lesions could similarly elevate cortical dopamine levels during early development, thereby resulting in a homeostatic down-regulation of D3 receptor function in prefrontal cortex. This would result in decreased D3 receptor-mediated inhibition of glutamatergic projections from prefrontal cortex to nucleus accumbens and ventral tegmentum, thereby activating these regions. Evidence suggests that the long-term adaptive response may include a decline in prefrontal cortex dopamine release, as stress-induced prefrontal cortical dopamine release is decreased in adult rats following neonatal hippocampal lesion (Laplante et al, 2004). Hippocampal lesions also disrupt excitatory projections from hippocampus to prefrontal cortex (Jay and Witter, 1991). Loss of this excitatory projection could also result in loss of cortical inhibition of projections from prefrontal cortex to accumbens and cortex to ventral tegmentum, similarly increasing accumbens dopamine release or dopamine responsivity. In support of both of these models, intact prefrontal cortical function is required for expression of behavioral abnormalities following neonatal hippocampal lesions (Lipska et al, 1998). Alternatively, excitatory direct glutamatergic projections from ventral hippocampus to nucleus accumbens are believed to interact with dopaminergic innervation of intrinsic cells of the nucleus accumbens (Sesack and Pickel, 1990). Loss of this excitatory input to accumbens could lead directly to increased accumbens dopamine release, perhaps through an inhibitory interneuron, or alternatively to increased dopamine responsivity in accumbens. While previous studies have reported decreased D3 receptor binding in nucleus accumbens following neonatal hippocampal lesions (Flores et al, 1996a), a finding consistent with our hypothesis, to our knowledge D3 receptor function in prefrontal cortex has not been determined following neonatal hippocampal lesions, nor has this hypothesis been widely tested. Further study is needed to test directly the ‘D3 receptor model of psychosis’ in the neonatal hippocampal lesion model, as well as in human psychotic disorders.
Alternative mechanisms to dopamine hypotheses are possible, and multiple mechanisms are not mutually exclusive. For example, repetitive risperidone treatment increases 5-HT but not dopamine release in nucleus accumbens, possibly through interactions with 5-HT2A and 5-HT2C autoreceptors (Broderick et al, 2003), supporting the hypothesis that risperidone effects at serotonin 5-HT2A and 5-HT2C receptors contribute to the findings in our study. Additionally, neuroprotective effects of atypical antipsychotic medications, including risperidone, in protecting against apoptotic cell death resulting from oxidative stress have been previously described (Qing et al, 2003; Bai et al, 2002). Risperidone could protect animals from lesion-induced development of behavioral abnormalities through protection against cell death resulting from oxidative stress secondary to increased catecholamine release in lesioned animals. Alternatively, risperidone could provide neuro-protection from excitotoxic cell death at sites distant from the primary ibotenic acid infusion in lesioned animals. This neuroprotective effect could then result in protection against loss of inhibitory gating of glutamatergic efferents from cortex to ventral tegmentum and nucleus accumbens. In partial support of this mechanism, apoptotic DNA fragmentation was observed in nucleus accumbens and striatum of lesioned animals 1 day post lesion, but not in prefrontal cortex, and not at later time points (Khaing et al, 2000). Further studies are needed to clarify whether such a mechanism contributes to the protective effects of risperidone against abnormal behaviors following neonatal hippocampal lesions.
An additional mechanism that could account for the observed findings might be the antidepressant effects of risperidone, which have been previously described (Myers and Thase, 1903; Dwight et al, 1994), and might also account for the dose differences observed in our study. Antidepressant medications appear to improve outcome for children at high risk of progression to first-episode psychosis (Cornblatt et al, 2002; Cornblatt, 2002). Again, further study is needed to determine if medications with a purely antidepressant profile have any efficacy in preventing behavioral abnormalities in the neonatal hippocampal lesion model. Our findings suggest that such studies may have some predictive validity in informing clinicians whether those drugs would also be of benefit to children at high risk for psychosis.
Possible Mechanisms Underlying Difference in Efficacy of High vs Low-Dose Risperidone
The decreased efficacy of higher risperidone dosages in preventing abnormal behaviors may result from lower affinity interaction of risperidone, or its metabolite(s), at higher dosages with a receptor type opposing the therapeutic action of risperidone. Potential candidates for this effect include 5-HT1A receptors opposing the actions of 5-HT2A receptors (Meltzer, 1999), and D3 receptors opposing the action of D2 receptors (Waters et al, 1993; Xu et al, 1997). Further study is needed to identify potential receptors with lower risperidone binding affinity and identify the underlying mechanism. In light of the potential clinical implications, it may be particularly useful to clearly delineate the dose–response parameters of this effect.
Conclusions
Because risperidone prevents the appearance of some elevated locomotor behaviors following neonatal hippo-campal lesion, and also prevents the appearance of first-episode psychosis in human studies (McGorry et al, 2002), these data provide support for the predictive validity of the hippocampal lesion model in identifying medications for psychosis prevention. Additionally, these data provide theoretical support for the use of low-dose risperidone in prevention of first-episode psychosis (McGorry et al, 2002), and suggest the possibility that higher risperidone doses may be less effective in this application. A limitation of our findings is that we did not distinguish prevention, vs suppression or postponement, of the emergence of abnormal behaviors following neonatal hippocampal lesions. Additional studies using later time points following treatment cessation will be needed in order to address this important issue. These data suggest that further studies with the hippocampal lesion model may also provide insight into pathophysiological mechanisms underlying the development of schizophrenia and other psychotic disorders.
ACKNOWLEDGEMENTS
This work was supported by the Department of Veterans Affairs Medical Research Service, National Institute of Drug Abuse (DA016778-01), Janssen Pharmaceutica Research Foundation, and Scottish Rite Schizophrenia Fellowship Award (LMP).
REFERENCES
- Accili D, Fishburn CS, Drago J, Steiner H, Lachowicz JE, Park BH, et al. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci USA. 1996;93:1945–1949. doi: 10.1073/pnas.93.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Amin HA, Weinberger DR, Lipska BK. Exaggerated MK-801-induced motor hyperactivity in rats with the neonatal lesion of the ventral hippocampus. Behav Pharmacol. 2000;11:269–278. doi: 10.1097/00008877-200006000-00010. [DOI] [PubMed] [Google Scholar]
- Alquicer G, Silva-Gomez AB, Peralta F, Flores G. Neonatal ventral hippocampus lesion alters the dopamine content in the limbic regions in postpubertal rats. Int J Dev Neurosci. 2004;22:103–111. doi: 10.1016/j.ijdevneu.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Arnt J. Differential effects of classical and newer antipsychotics on the hypermotility induced by two dose levels of d-amphetamine. Eur J Pharmacol. 1995;283:55–62. doi: 10.1016/0014-2999(95)00292-s. [DOI] [PubMed] [Google Scholar]
- Bai O, Wei Z, Lu W, Bowen R, Keegan D, Li XM. Protective effects of atypical antipsychotic drugs on PC12 cells after serum withdrawal. J Neurosci Res. 2002;69:278–283. doi: 10.1002/jnr.10290. [DOI] [PubMed] [Google Scholar]
- Broderick PA, Rahni DN, Zhou Y. Acute and subacute effects of risperidone and cocaine on accumbens dopamine and serotonin release using in vivo microvoltammetry on line with open-field behavior. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1037–1054. doi: 10.1016/S0278-5846(03)00176-3. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Calligaro DO, Falcone JF, Marsh RD, Moore NA, Tye NC, et al. Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology. 1996;14:87–96. doi: 10.1016/0893-133X(94)00129-N. [DOI] [PubMed] [Google Scholar]
- Cabib S, Puglisi-Allegra S. Different effects of repeated stressful experiences on mesocortical and mesolimbic dopamine metabolism. Neuroscience. 1996;73:375–380. doi: 10.1016/0306-4522(96)00750-6. [DOI] [PubMed] [Google Scholar]
- Chambers RA, Moore J, McEvoy JP, Levin ED. Cognitive effects of neonatal hippocampal lesions in a rat model of schizophrenia. Neuropsychopharmacology. 1996;15:587–594. doi: 10.1016/S0893-133X(96)00132-7. [DOI] [PubMed] [Google Scholar]
- Chiang YC, Chen PC, Chen JC. D(3) dopamine receptors are down-regulated in amphetamine sensitized rats and their putative antagonists modulate the locomotor sensitization to amphetamine. Brain Res. 2003;972:159–167. doi: 10.1016/s0006-8993(03)02522-8. [DOI] [PubMed] [Google Scholar]
- Cornblatt B, Lencz T, Obuchowski M. The schizophrenia prodrome: treatment and high-risk perspectives. Schizophr Res. 2002;54:177–186. doi: 10.1016/s0920-9964(01)00365-6. [DOI] [PubMed] [Google Scholar]
- Cornblatt BA. The New York high risk project to the Hillside recognition and prevention (RAP) program. Am J Med Genet. 2002;114:956–966. doi: 10.1002/ajmg.b.10520. [DOI] [PubMed] [Google Scholar]
- Diaz J, Pilon C, Le Foll B, Gros C, Triller A, Schwartz JC, et al. Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J Neurosci. 2000;20:8677–8684. doi: 10.1523/JNEUROSCI.20-23-08677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwight MM, Keck PEJ, Stanton SP, Strakowski SM, McElroy SL. Antidepressant activity and mania associated with risperidone treatment of schizoaffective disorder. Lancet. 1994;344:554–555. doi: 10.1016/s0140-6736(94)91952-6. [DOI] [PubMed] [Google Scholar]
- Ekman A, Nissbrandt H, Heilig M, Dijkstra D, Eriksson E. Central administration of dopamine D3 receptor antisense to rat: effects on locomotion, dopamine release and [3H]spiperone binding. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:342–350. doi: 10.1007/pl00005263. [DOI] [PubMed] [Google Scholar]
- Ellinwood EHJ, Sudilovsky A, Nelson LM. Evolving behavior in the clinical and experimental amphetamine (model) psychosis. Am J Psychiatry. 1973;130:1088–1093. doi: 10.1176/ajp.130.10.1088. [DOI] [PubMed] [Google Scholar]
- Ellinwood EH., Jr Amphetamine psychosis: I. Description of the individuals and process. J Nerv Ment Dis. 1967;144:273–283. [Google Scholar]
- Flores G, Barbeau D, Quirion R, Srivastava LK. Decreased binding of dopamine D3 receptors in limbic subregions after neonatal bilateral lesion of rat hippocampus. J Neurosci. 1996a;16:2020–2026. doi: 10.1523/JNEUROSCI.16-06-02020.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores G, Wood GK, Liang JJ, Quirion R, Srivastava LK. Enhanced amphetamine sensitivity and increased expression of dopamine D2 receptors in postpubertal rats after neonatal excitotoxic lesions of the medial prefrontal cortex. J Neurosci. 1996b;16:7366–7375. doi: 10.1523/JNEUROSCI.16-22-07366.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner H, Maurer K, Ruhrmann S, Bechdolf A, Klosterkotter J, Wagner M, et al. Early detection and secondary prevention of psychosis: facts and visions. Eur Arch Psychiatry Clin Neurosci. 2004;254:117–128. doi: 10.1007/s00406-004-0508-z. [DOI] [PubMed] [Google Scholar]
- Henry C, Guegant G, Cador M, Arnauld E, Arsaut J, Le Moal M, et al. Prenatal stress in rats facilitates amphetamine-induced sensitization and induces long-lasting changes in dopamine receptors in the nucleus accumbens. Brain Res. 1995;685:179–186. doi: 10.1016/0006-8993(95)00430-x. [DOI] [PubMed] [Google Scholar]
- Hooks MS, Kalivas PW. The role of mesoaccumbens—pallidal circuitry in novelty-induced behavioral activation. Neuroscience. 1995;64:587–597. doi: 10.1016/0306-4522(94)00409-x. [DOI] [PubMed] [Google Scholar]
- Janssen PA, Niemegeers CJ, Awouters F, Schellekens KH, Megens AA, Meert TF. Pharmacology of risperidone (R 64 766), a new antipsychotic with serotonin-S2 and dopamine-D2 antagonistic properties. J Pharmacol Exp Ther. 1988;244:685–693. [PubMed] [Google Scholar]
- Jaskiw GE, Weinberger DR, Crawley JN. Microinjection of apomorphine into the prefrontal cortex of the rat reduces dopamine metabolite concentrations in microdialysate from the caudate nucleus. Biol Psychiatry. 1991;29:703–706. doi: 10.1016/0006-3223(91)90144-b. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Balla A, Burch S, Suckow R, Xie S, Sershen H. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-d-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29:300–307. doi: 10.1038/sj.npp.1300313. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP. Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol. 1991;313:574–586. doi: 10.1002/cne.903130404. [DOI] [PubMed] [Google Scholar]
- Khaing ZZ, Weickert CS, Weinberger DR, Lipska BK. Differential DNA damage in response to the neonatal and adult excitotoxic hippocampal lesion in rats. Eur J Neurosci. 2000;12:4424–4433. doi: 10.1046/j.0953-816x.2000.01320.x. [DOI] [PubMed] [Google Scholar]
- Khan ZU, Gutierrez A, Martin R, Penafiel A, Rivera A, De La Calle A. Differential regional and cellular distribution of dopamine D2-like receptors: an immunocytochemical study of subtype-specific antibodies in rat and human brain. J Comp Neurol. 1998;402:353–371. doi: 10.1002/(sici)1096-9861(19981221)402:3<353::aid-cne5>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Kolachana BS, Saunders RC, Weinberger DR. Augmentation of prefrontal cortical monoaminergic activity inhibits dopamine release in the caudate nucleus: an in vivo neurochemical assessment in the rhesus monkey. Neuroscience. 1995;69:859–868. doi: 10.1016/0306-4522(95)00246-f. [DOI] [PubMed] [Google Scholar]
- Kongsamut S, Roehr JE, Cai J, Hartman HB, Weissensee P, Kerman LL, et al. Iloperidone binding to human and rat dopamine and 5-HT receptors. Eur J Pharmacol. 1996;317:417–423. doi: 10.1016/s0014-2999(96)00840-0. [DOI] [PubMed] [Google Scholar]
- Laplante F, Stevenson CW, Gratton A, Srivastava LK, Quirion R. Effects of neonatal ventral hippocampal lesion in rats on stress-induced acetylcholine release in the prefrontal cortex. J Neurochem. 2004;91:1473–1482. doi: 10.1111/j.1471-4159.2004.02831.x. [DOI] [PubMed] [Google Scholar]
- Le Moal M, Simon H. Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev. 1991;71:155–234. doi: 10.1152/physrev.1991.71.1.155. [DOI] [PubMed] [Google Scholar]
- Leysen JE, Janssen PM, Schotte A, Luyten WH, Megens AA. Interaction of antipsychotic drugs with neurotransmitter receptor sites in vitro and in vivo in relation to pharmacological and clinical effects: role of 5HT2 receptors. Psychopharmacology (Berl) 1993;112:S40–S54. doi: 10.1007/BF02245006. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Al-Amin HA, Weinberger DR. Excitotoxic lesions of the rat medial prefrontal cortex. Effects on abnormal behaviors associated with neonatal hippocampal damage. Neuropsychopharmacology. 1998;19:451–464. doi: 10.1016/S0893-133X(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Swerdlow NR, Geyer MA, Jaskiw GE, Braff DL, Weinberger DR. Neonatal excitotoxic hippocampal damage in rats causes post-pubertal changes in prepulse inhibition of startle and its disruption by apomorphine. Psychopharmacology (Berl) 1995;122:35–43. doi: 10.1007/BF02246439. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. Gonadectomy does not prevent novelty or drug-induced motor hyperresponsiveness in rats with neonatal hippocampal damage. Brain Res Dev Brain Res. 1994a;78:253–258. doi: 10.1016/0165-3806(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. Subchronic treatment with haloperidol and clozapine in rats with neonatal excitotoxic hippocampal damage. Neuropsychopharmacology. 1994b;10:199–205. doi: 10.1038/npp.1994.22. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. Genetic variation in vulnerability to the behavioral effects of neonatal hippocampal damage in rats. Proc Natl Acad Sci USA. 1995;92:8906–8910. doi: 10.1073/pnas.92.19.8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology. 2000;23:223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Louilot A, Le Moal M, Simon H. Opposite influences of dopaminergic pathways to the prefrontal cortex or the septum on the dopaminergic transmission in the nucleus accumbens. An in vivo voltammetric study. Neuroscience. 1989;29:45–56. doi: 10.1016/0306-4522(89)90331-x. [DOI] [PubMed] [Google Scholar]
- McGorry PD, Yung AR, Phillips LJ, Yuen HP, Francey S, Cosgrave EM, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry. 2002;59:921–928. doi: 10.1001/archpsyc.59.10.921. [DOI] [PubMed] [Google Scholar]
- Meares A. The diagnosis of prepsychotic schizophrenia. Lancet. 1959;1:55–59. doi: 10.1016/s0140-6736(59)91134-1. [DOI] [PubMed] [Google Scholar]
- Megens AA, Awouters FH, Schotte A, Meert TF, Dugovic C, Niemegeers CJ, et al. Survey on the pharmacodynamics of the new antipsychotic risperidone. Psychopharmacology (Berl) 1994;114:9–23. doi: 10.1007/BF02245439. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology. 1999;21:106S–115S. doi: 10.1016/S0893-133X(99)00046-9. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Dziewczapolski G, Garcia MC, Rubinstein M, Gershanik OS. D3 receptor knockdown through antisense oligonucleotide administration supports its inhibitory role in locomotion. Neuroreport. 1999;10:3131–3136. doi: 10.1097/00001756-199910190-00002. [DOI] [PubMed] [Google Scholar]
- Milliken GA, Johnson DE. Analysis of Messy Data. Volume 1: Designed Experiments. Chapman & Hall; London: 1992. [Google Scholar]
- Morrison AP, French P, Walford L, Lewis SW, Kilcommons A, Green J, et al. Cognitive therapy for the prevention of psychosis in people at ultra-high risk: randomised controlled trial. Br J Psychiatry. 2004;185:291–297. doi: 10.1192/bjp.185.4.291. [DOI] [PubMed] [Google Scholar]
- Myers JE, Thase ME. Risperidone: review of its therapeutic utility in depression. Psychopharmacol Bull. 1903;35:109–129. [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1986. [Google Scholar]
- Pritchard LM, Logue AD, Hayes S, Welge JA, Xu M, Zhang J, et al. 7-OH-DPAT and PD 128907 selectively activate the D3 dopamine receptor in a novel environment. Neuropsychopharmacology. 2003;28:100–107. doi: 10.1038/sj.npp.1300018. [DOI] [PubMed] [Google Scholar]
- Qing H, Xu H, Wei Z, Gibson K, Li XM. The ability of atypical antipsychotic drugs vs. haloperidol to protect PC12 cells against MPP+-induced apoptosis. Eur J Neurosci. 2003;17:1563–1570. doi: 10.1046/j.1460-9568.2003.02590.x. [DOI] [PubMed] [Google Scholar]
- Rebec GV. Real-time assessments of dopamine function during behavior: single-unit recording, iontophoresis, and fast-scan cyclic voltammetry in awake, unrestrained rats. Alcohol Clin Exp Res. 1998;22:32–40. doi: 10.1111/j.1530-0277.1998.tb03614.x. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Christensen JR, Guerra C, Bardo MT. Regional and temporal differences in real-time dopamine efflux in the nucleus accumbens during free-choice novelty. Brain Res. 1997a;776:61–67. doi: 10.1016/s0006-8993(97)01004-4. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Grabner CP, Johnson M, Pierce RC, Bardo MT. Transient increases in catecholaminergic activity in medial prefrontal cortex and nucleus accumbens shell during novelty. Neuroscience. 1997b;76:707–714. doi: 10.1016/s0306-4522(96)00382-x. [DOI] [PubMed] [Google Scholar]
- Richelson E, Souder T. Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci. 2000;68:29–39. doi: 10.1016/s0024-3205(00)00911-5. [DOI] [PubMed] [Google Scholar]
- Richtand NM, Logue AD, Welge JA, Tubbs LJ, Spitzer RH, Sethuraman G, et al. The dopamine D3 receptor antagonist nafadotride inhibits development of locomotor sensitization to amphetamine. Brain Res. 2000;867:239–241. doi: 10.1016/s0006-8993(00)02247-2. [DOI] [PubMed] [Google Scholar]
- Richtand NM, Pritchard LM, Coolen LM. Dopamine receptor alternative splicing. In: Schmidt WJ, Reith MEA, editors. Dopamine and Glutamate in Psychiatric Disorders. Humana Press; Totowa, NJ: 2005. pp. 45–61. [Google Scholar]
- Richtand NM, Welge JA, Levant B, Logue AD, Hayes S, Pritchard LM, et al. Altered behavioral response to dopamine D3 receptor agonists 7-OH-DPAT and PD 128907 following repetitive amphetamine administration. Neuropsychopharmacology. 2003;28:1422–1432. doi: 10.1038/sj.npp.1300182. [DOI] [PubMed] [Google Scholar]
- Richtand NM, Woods SC, Berger SP, Strakowski SM. D3 dopamine receptor, behavioral sensitization, and psychosis. Neurosci Biobehav Rev. 2001;25:427–443. doi: 10.1016/s0149-7634(01)00023-9. [DOI] [PubMed] [Google Scholar]
- Rueter LE, Ballard ME, Gallagher KB, Basso AM, Curzon P, Kohlhaas KL. Chronic low dose risperidone and clozapine alleviate positive but not negative symptoms in the rat neonatal ventral hippocampal lesion model of schizophrenia. Psycho-pharmacology (Berl) 2004;176:312–319. doi: 10.1007/s00213-004-1897-4. [DOI] [PubMed] [Google Scholar]
- Sams-Dodd F, Lipska BK, Weinberger DR. Neonatal lesions of the rat ventral hippocampus result in hyperlocomotion and deficits in social behaviour in adulthood. Psychopharmacology (Berl) 1997;132:303–310. doi: 10.1007/s002130050349. [DOI] [PubMed] [Google Scholar]
- Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) 1996;124:57–73. doi: 10.1007/BF02245606. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R. Behavioral pharmacology of amphetamine. In: Cho AK, Segal DS, editors. Amphetamine and Its Analogues: Pharmacology, Toxicology, and Abuse. Academic Press; Orlando, FL: 1994. pp. 115–150. [Google Scholar]
- Sesack SR, Pickel VM. In the rat medial nucleus accumbens, hippocampal and catecholaminergic terminals converge on spiny neurons and are in apposition to each other. Brain Res. 1990;527:266–279. doi: 10.1016/0006-8993(90)91146-8. [DOI] [PubMed] [Google Scholar]
- Strange PG. Antipsychotic drugs: importance of dopamine receptors for mechanisms of therapeutic actions and side effects. Pharmacol Rev. 2001;53:119–133. [PubMed] [Google Scholar]
- Sullivan HS. The onset of schizophrenia. Am J Psychiatry. 1927;6:105–134. doi: 10.1176/ajp.151.6.134. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Halim N, Hanlon FM, Platten A, Auerbach PP. Lesion size and amphetamine hyperlocomotion after neonatal ventral hippocampal lesions: more is less. Brain Res Bull. 2001;55:71–77. doi: 10.1016/s0361-9230(01)00492-0. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Lipska BK, Weinberger DR, Braff DL, Jaskiw GE, Geyer MA. Increased sensitivity to the sensorimotor gating-disruptive effects of apomorphine after lesions of medial prefrontal cortex or ventral hippocampus in adult rats. Psychopharmacology (Berl) 1995;122:27–34. doi: 10.1007/BF02246438. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2003;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Tsuang MT, Stone WS, Tarbox SI, Faraone SV. Treatment of nonpsychotic relatives of patients with schizophrenia: six case studies. Am J Med Genet. 2002;114:943–948. doi: 10.1002/ajmg.10363. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol. 2001;63:241–320. doi: 10.1016/s0301-0082(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Ventura R, Cabib S, Puglisi-Allegra S. Genetic susceptibility of mesocortical dopamine to stress determines liability to inhibition of mesoaccumbens dopamine and to behavioral ‘despair’ in a mouse model of depression. Neuroscience. 2002;115:999–1007. doi: 10.1016/s0306-4522(02)00581-x. [DOI] [PubMed] [Google Scholar]
- Wallace DR, Mactutus CF, Booze RM. Repeated intravenous cocaine administration: locomotor activity and dopamine D2/D3 receptors. Synapse. 1996;23:152–163. doi: 10.1002/(SICI)1098-2396(199607)23:3<152::AID-SYN4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Wan RQ, Corbett R. Enhancement of postsynaptic sensitivity to dopaminergic agonists induced by neonatal hippocampal lesions. Neuropsychopharmacology. 1997;16:259–268. doi: 10.1016/S0893-133X(96)00217-5. [DOI] [PubMed] [Google Scholar]
- Wan RQ, Giovanni A, Kafka SH, Corbett R. Neonatal hippocampal lesions induced hyperresponsiveness to amphetamine: behavioral and in vivo microdialysis studies. Behav Brain Res. 1996;78:211–223. doi: 10.1016/0166-4328(95)00251-0. [DOI] [PubMed] [Google Scholar]
- Waters N, Svensson K, Haadsma-Svensson SR, Smith MW, Carlsson A. The dopamine D3-receptor: a postsynaptic receptor inhibitory on rat locomotor activity. J Neural Transm Gen Sect. 1993;94:11–19. doi: 10.1007/BF01244979. [DOI] [PubMed] [Google Scholar]
- Woods SW, Breier A, Zipursky RB, Perkins DO, Addington J, Miller TJ, et al. Randomized trial of olanzapine versus placebo in the symptomatic acute treatment of the schizophrenic prodrome. Biol Psychiatry. 2003;54:453–464. doi: 10.1016/s0006-3223(03)00321-4. [DOI] [PubMed] [Google Scholar]
- Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu XT, et al. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron. 1997;19:837–848. doi: 10.1016/s0896-6273(00)80965-4. [DOI] [PubMed] [Google Scholar]
- Yolken RH, Karlsson H, Yee F, Johnston-Wilson NL, Torrey EF. Endogenous retroviruses and schizophrenia. Brain Res Brain Res Rev. 2000;31:193–199. doi: 10.1016/s0165-0173(99)00037-5. [DOI] [PubMed] [Google Scholar]
- Zicha J, Kunes J. Ontogenetic aspects of hypertension development: analysis in the rat. Physiol Rev. 1999;79:1227–1282. doi: 10.1152/physrev.1999.79.4.1227. [DOI] [PubMed] [Google Scholar]