

Abstract
Glutamine has been shown to influence endothelial-dependent relaxation and nitric oxide production in vitro, possibly by limiting arginine availability, but its effects in vivo have not been well studied. Hyperammonemia is a pathophysiological condition in which glutamine is elevated and contributes to depressed CO2 reactivity of cerebral arterioles. We tested thehypothesis that acute hyperammonemia decreases pial arteriolar dilation to acetylcholine in vivo and that this decrease could be prevented by inhibiting glutamine synthetase with L-methionine-S-sulfoximine (MSO) or by intravenous infusion of L-arginine. Pial arteriolar diameter responses to topical superfusion of acetylcholine were measured in anesthetized rats before and at 6 h of infusion of either sodium or ammonium acetate. Ammonium acetate infusion increased plasma ammonia concentration from ~ 30 to ~ 600 μM and increased cerebral glutamine concentration fourfold. Arteriolar dilation to acetylcholine was intact after infusion of sodium acetate in groups pretreated with vehicle or with MSO plus methionine, which was coadministered to prevent MSO-induced seizures. In contrast, dilation in response to acetylcholine was completely blocked in hyperammonemic groups pretreated with vehicle or methionine alone. However, MSO plus methionine administration before hyperammonemia, which maintained cerebral glutamine concentration at control values, preserved acetylcholine dilation. Intravenous infusion of L-arginine during the last 2 h of the ammonium acetate infusion partially restored dilation to acetylcholine without reducing cerebral glutamine accumulation. Superfusion of 1 or 2 mM L-glutamine through the cranial window for 1 h in the absence of hyperammonemia attenuated acetylcholine dilation but had no effect on endothelial-independent dilation to nitroprusside. We conclude that 1) hyperammonemia reduces acetylcholine-evoked dilation in cerebral arterioles, 2) this reduction depends on increased glutamine rather than ammonium ions, and 3) increasing arginine partially overcomes the inhibitory effect of glutamine.
Keywords: arginine, cerebral circulation, endothelial-dependent dilation, hepatic encephalopathy, hyperammonemia, nitric oxide
Patients with hepatic failure have been reported to have a depressed cerebrovascular response to hypercapnia (9). Depressed cerebral vasodilation to hypercapnia also has been observed in experimental models of acute hyperammonemia in a variety of species, including monkey, dog, cat, and rat (2, 6, 15, 16, 33). This depressed vasodilatory response to hypercapnia does not represent a generalized vasoparalysis because vasodilation to hypoxia and to arterial hypotension remains intact (14, 15).
Hyperammonemia produces substantial increases in brain glutamine concentration (7). Inhibition of glutamine synthetase preserves cerebrovascular reactivity to hypercapnia and hypocapnia during acute hyperammonemia (13, 15). Thus the loss of CO2 reactivity is related to glutamine accumulation rather than to direct vascular effects of ammonium ions. Moreover, intravenous infusion of glutamine in the absence of hyperammonemia decreases hypercapnic vasodilation, although not to the same extent as hyperammonemia (22). Therefore, glutamine can exert a modulatory role in cerebrovascular CO2 reactivity in vivo.
The mechanism by which glutamine modulates CO2 reactivity is not precisely known but may be related to decreased nitric oxide (NO) production because infusion of arginine during either hyperammonemia or glutamine infusion improves hypercapnic reactivity (22). In isolated cerebral arteries, NO-dependent vasorelaxation evoked by transmural nerve stimulation is inhibited by glutamine (18). Studies in aortic and venular endothelial cell cultures and isolated aortic rings indicate that elevation of glutamine concentration in the physiological range limits NO production and endothelial-dependent vasorelaxation (1, 11, 20, 31). However, the extent to which glutamine modulates endothelial-dependent vasodilation in vivo has not been well studied.
In the present study, we evaluated whether endogenous increases in brain tissue glutamine concentration associated with hyperammonemia or exogenous increases in glutamine bathing pial arterioles cause impaired vasodilation in vivo to the endothelial-dependent dilator acetylcholine (ACh). We tested the hypotheses that 1) acute hyperammonemia decreases pial arteriolar dilation to ACh, 2) this decrease could be prevented by inhibiting glutamine accumulation with the glutamine synthetase inhibitor L-methionine-S-sulfoximine (MSO) or by intravenous infusion of L-arginine, and 3) abluminal increases in glutamine concentration produced by superfusion of a cranial window with artificial cerebrospinal fluid (CSF) containing elevated L-glutamine attenuates dilation to ACh but not to the endothelial-independent dilator sodium nitroprusside.
METHODS
All procedures were approved by the Institutional Animal Care and Use Committee. Male Wistar rats (~350 g) were anesthetized with pentobarbital (65 mg/kg ip). Anesthesia was maintained throughout the experiment by additional intraperitoneal injections of pentobarbital (20 mg/kg every 90 min). The lungs were mechanically ventilated with O2-enriched air to maintain the arterial partial pressure of O2 in the range of 140–190 Torr and the arterial partial pressure of CO2 (PaCO2) in the range of 35–45 Torr. A femoral artery and tail artery were catheterized for monitoring arterial blood pressure and sampling arterial blood. Rectal temperature was maintained at ~ 37°C with a warming blanket.
A closed cranial window was constructed over the parietal cortex for measuring pial arteriolar diameter by intravital microscopy (15). A plastic ring was cemented to the skull around a ~ 4-mm craniotomy. The dura mater was carefully incised and retracted. A glass coverslip was cemented to the plastic ring to seal the window filled with artificial CSF containing (in mM) 151 Na+, 3 K+, 1.3 Ca2+, 0.6 Mg2+, 134 Cl−, 24.6 , 6 urea, and 3.7 glucose (19). The window was equipped with an inflow port and an outflow port for superfusing artificial CSF and ACh, with a port for measuring fluid pressure, and a thermistor for monitoring fluid temperature. The diameter of pial arterioles was measured with a microscope equipped with a video recording system.
The first experimental protocol involved measuring pial arteriolar responses to ACh before and after a 6-h infusion of sodium acetate (NaAc) or ammonium acetate (NH4Ac). Baseline measurements of pial arteriolar diameter and arterial blood analysis were made 45 min after completion of the surgical procedures. The cranial window was superfused with artificial CSF for 3 min followed by 30 μM ACh for 5 min at a rate of 250 μl/min. After the change in diameter was recorded and the ACh was washed out, a continuous intravenous infusion of either NaAc or NH4Ac was started (50 μmol·kg−1·min−1, 0.1 ml/min). Arterial blood analysis was repeated at 2 and 6 h of infusion, and changes in baseline diameter were measured hourly. At 6 h of infusion, the change in diameter during superfusing of 30 μM ACh was measured.
To inhibit glutamine synthesis, 150 mg/kg (0.83 mmol/kg) of MSO (Sigma, St. Louis, MO) was infused intravenously 3 h before the baseline measurements were obtained. Control groups received saline vehicle (3 ml/kg). MSO can induce seizures, but seizures are prevented by coadministration of L-methionine without altering the effect of MSO on glutamine synthetase activity (27). Groups receiving MSO also received an intraperitoneal injection of 6.7 mmol/kg of L-methionine. To control for a possible influence of L-methionine on the ACh response during hyperammonemia, a group receiving NH4Ac was pretreated with L-methionine alone. To test whether L-arginine could reverse the effect of hyperammonemia on the ACh response, 2 mmol·kg−1·h−1 of L-arginine was infused intravenously for the last 2 h of NH4Ac infusion. This infusion rate of L-arginine has been shown to restore hypercapnic dilation during NH4Ac infusion (22). Thus six groups of rats were studied in the first experiment: 1) vehicle pretreatment and NaAc infusion (n = 8); 2) MSO/methionine pretreatment and NaAc infusion (n = 8); 3) vehicle pretreatment and NH4Ac infusion (n = 8); 4) L-methionine pretreatment and NH4Ac infusion (n = 6); 5) MSO/methionine pretreatment and NH4Ac infusion (n = 8); and 6) vehicle pretreatment and NH4Ac infusion with subsequent L-arginine infusion (n = 8).
Arterial blood analysis included measurements of plasma ammonium concentration by a cation-exchange and spectrophotometer analysis previously described (3), plasma osmolarity by freezing-point depression (Advanced Instruments, Norwood, MA), hemoglobin concentration (OSM3 Hemoximeter, Radiometer, Copenhagen), and pH, PaCO2, and arterial partial pressure of O2 (Chiron Diagnostics Blood Gas Analyzer, Halstead, Essex, UK). At the end of the experiment, the cerebral hemispheres were rapidly harvested and frozen in subgroups (n = 5–6). The cerebral tissue concentration of glutamine, glutamate, and arginine was measured by high-performance liquid chromotography with fluorescent detection as described (17, 32).
In the second experimental protocol, the dilator response to ACh was measured in the presence of exogenously increased glutamine in CSF surrounding pial arterioles without hyperammonemia. In four groups of rats, the cranial window was continuously superfused for 1 h at a rate of 0.1 ml/min with artificial CSF containing either 0 (n = 7), 0.3 (n = 6), 1 (n = 7), or 2 mM (n = 7) L-glutamine. The 0 and 0.3 mM concentrations were chosen to bracket the normal glutamine concentration of 0.25 mM previously measured in the CSF sampled from rat cranial windows (22). The concentration of glutamine was measured in the effluent CSF collected between 50 and 60 min of superfusion. At 60 min of glutamine superfusion, 10 μM ACh was added to the glutamine-containing superfusate, and the percent change in pial arteriolar diameter was measured. The dilator response to 3 μM sodium nitroprusside superfusion was also tested in the presence of the different glutamine concentrations.
Biochemical and arterial blood gas measurements were compared among groups by one-way ANOVA and the Newman-Keuls multiple range test. In the first experiment, percent changes in baseline pial arteriolar diameter during salt infusion and tissue amino acid concentration were compared among groups by ANOVA and the Newman-Keuls multiple range test. Within each group, the percent changes in pial arteriolar diameter, measured at 5 min of ACh superfusion, were compared before and at 6 h of salt infusion by paired t-test with n equaling the number of rats per group. Arterial blood measurements and mean arterial blood pressure at 6 h of salt infusion were also compared with baseline values by paired t-test. In the second experiment, the change in baseline diameter during glutamine superfusion was analyzed by paired t-test. The percent dilation to ACh and nitroprusside was compared among the four groups with different glutamine concentrations by one-way ANOVA and the Newman-Keuls multiple range test. The significance level was set at 0.05 for all tests. All values are expressed as means ± SD.
RESULTS
Hyperammonemia experiment
Infusion of NH4Ac increased plasma ammonium concentration from 36 ± 11 to 483 ± 120 μM by 2 h and to 552 ± 182 μM by 6 h of infusion. Similar increases were observed in the other groups infused with NH4Ac (Table 1). Pretreatment with MSO/methionine modestly elevated plasma ammonium before salt infusion. Plasma osmolarity was unchanged during the salt infusion. Arterial pH was slightly lower in the groups treated with MSO/methionine or L-arginine. No differences were present in PaCO2 or hemoglobin concentration among groups, although PaCO2 increased slightly and hemoglobin decreased by 10–15% at the end of the salt infusion in some of the groups (Table 1).
Table 1.
Arterial blood analysis before and at 6 h of sodium acetate or ammonium acetate infusion
| Sodium Acetate
|
Ammonium Acetate
|
|||||
|---|---|---|---|---|---|---|
| Vehicle | MSO + methionine | Vehicle | Methionine | MSO + methionine | Arginine | |
| Ammonium, μM | ||||||
| 0 h | 35±15 | 127±30† | 34±10 | 26±10 | 159±58† | 35±9 |
| 6 h | 45±33 | 92±35* | 563±165*† | 625±94*† | 607±220*† | 665±222*† |
| Osmolarity, mosmol/l | ||||||
| 0 h | 305±15 | 311±11 | 307±16 | 313±17 | 299±22 | 307±17 |
| 6 h | 309±11 | 314±6 | 306±13 | 301±9 | 302±16 | 303±18 |
| pH | ||||||
| 0 h | 7.42±0.03 | 7.35±0.02† | 7.40±0.04 | 7.40±0.04 | 7.35±0.02† | 7.38±0.01 |
| 6 h | 7.43±0.04 | 7.36±0.07† | 7.44±0.04* | 7.34±0.06*† | 7.30±0.03*† | 7.34±0.07† |
| Pco2, Torr | ||||||
| 0 h | 39±2 | 39±2 | 38±2 | 38±2 | 41±4 | 39±3 |
| 6 h | 40±3 | 43±3* | 41±3* | 39±3 | 43±4 | 38±3 |
| Hemoglobin, g/dl | ||||||
| 0 h | 13.6±2.0 | 13.6±1.3 | 12.4±1.6 | 13.2±2.0 | 13.4±1.9 | 14.5±1.2 |
| 6 h | 11.9±1.1* | 11.2±1.1* | 11.9±1.0 | 12.8±0.9 | 10.6±1.1* | 12.2±1.3* |
Values are means ± SD. MSO, L-methionine-S-sulfoximine.
P <0.05 from corresponding value at 0 h by paired t-test.
P <0.05 from corresponding value in vehicle + sodium acetate group by ANOVA and Newman-Keuls test.
The cerebral tissue concentration of glutamine was reduced by ~ 50% in the NaAc group pretreated with MSO/methionine compared with vehicle (Fig. 1). Infusion of NH4Ac increased cerebral glutamine concentration fourfold in the group pretreated with vehicle and twofold in the group pretreated with methionine. Coinfusion of L-arginine during the last 2 h of NH4Ac infusion did not attenuate the increase in tissue glutamine concentration. In contrast, tissue glutamine concentration in the NH4Ac group pretreated with MSO/methionine was similar to that in the NaAc group pretreated with vehicle.
Fig. 1.

Cerebral tissue concentration (±SD) of glutamine (top), glutamate (middle), and arginine (bottom) after a 6-h infusion of sodium acetate in groups pretreated with vehicle (n = 5) and L-methionine-S-sulfoximine (MSO) + L-methionine (Met; n = 6) and after a 6-h infusion of ammonium acetate in groups pretreated with vehicle (n = 5), Met (n = 6), MSO/Met (n = 6), and L-arginine (Arg; n = 5). *P < 0.05 from vehicle-sodium acetate group; +P < 0.05 from MSO/Met-ammonium acetate group. Arginine concentration in the L-arginine group was greater than in the Met-ammonium acetate group but was not significant from the other groups.
Cerebral glutamate concentration was not significantly changed in the hyperammonemic groups pretreated with vehicle or coadministered L-arginine, but decreases occurred in the hyperammonemic groups pretreated with methionine or MSO/methionine (Fig. 1). Analysis of cerebral arginine concentration by ANOVA indicated a significant intergroup effect, but the only difference indicated by the Newman-Keuls multiple range test was a greater arginine concentration in the NH4Ac group receiving L-arginine compared with the NH4Ac group pretreated with L-methionine. In a separate group of nonhyperammonemic rats (n = 5), infusion of L-arginine at a rate of 2 mmol·kg−1·h−1 for 2 h was found to increase plasma arginine concentration sevenfold (98 ± 20 to 740 ± 569 μM).
Mean arterial blood pressure was maintained at an average of 95–110 mmHg during the NaAc infusion and during the NH4Ac infusion with vehicle pretreatment (Fig. 2). However, during the NH4Ac infusion, mean arterial pressure decreased to 73–75 mmHg in groups pretreated with L-methionine and MSO/methionine and to 84 mmHg in the group with L-arginine coinfusion. Intracranial pressure measured in the closed cranial window increased during NH4Ac infusion (Fig. 2). The increase was markedly attenuated in the group pretreated with MSO/methionine but not in the groups treated with L-methionine or L-arginine. Cerebral perfusion pressure, calculated as the difference between arterial blood pressure and intracranial pressure, decreased at 6 h of NH4Ac infusion in the groups treated with L-methionine, MSO/methionine, and arginine, and the levels were significantly lower than that of the NH4Ac group treated with vehicle (Fig. 2).
Fig. 2.
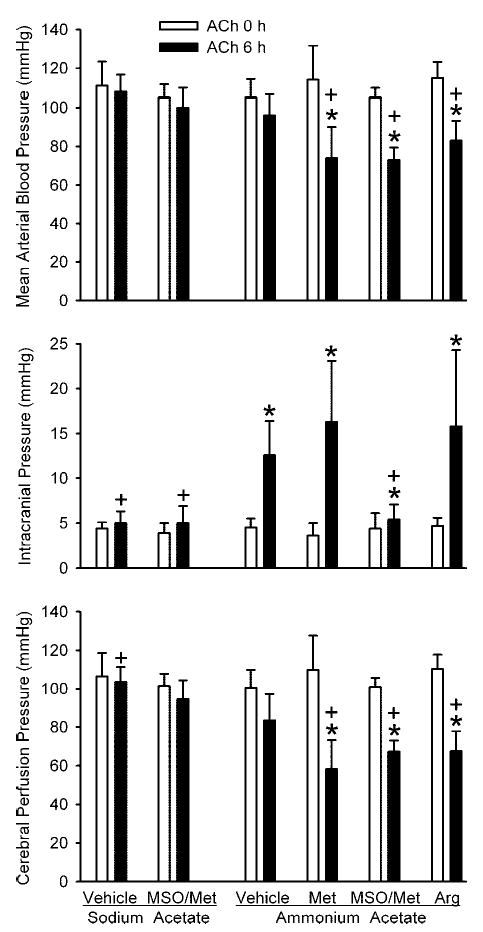
Mean arterial blood pressure (top), intracranial pressure (middle), and cerebral perfusion pressure (bottom) (±SD) during ACh superfusion before (0 h) and at 6 h of infusion of sodium acetate in groups pretreated with vehicle (n = 8) and MSO/Met (n = 8) and after 6 h of infusion of ammonium acetate in groups pretreated with vehicle (n = 8), Met (n = 6), MSO/Met (n = 8), and Arg (n = 8). *P < 0.05 from 0-h value within group; +P < 0.05 from vehicle-ammonium acetate group at same time point.
Baseline diameter of pial arterioles was not significantly changed over the 6-h infusion of NaAc in the group pretreated with vehicle but increased in the NaAc group pretreated with MSO/methionine (Fig. 3). Similar increases occurred in the NH4Ac-infused groups pretreated with vehicle, MSO/methionine, and coinfusion of L-arginine. The increase in baseline diameter was enhanced by 6 h of NH4Ac infusion in the group pretreated with L-methionine. The latter group had the lowest cerebral perfusion pressure (58 3 15 mmHg).
Fig. 3.

Percent change in baseline pial arteriolar diameter (3SD) after 6 h of infusion of sodium acetate in groups pretreated with vehicle (n = 8) and MSO/Met (n = 8) and after 6 h infusion of ammonium acetate in groups pretreated with vehicle (n = 8), Met (n = 6), MSO/Met (n = 8), and Arg (n = 8). *P < 0.05 from vehicle-sodium acetate group; +P < 0.05 from vehicle-ammonium acetate group.
The pial arteriolar dilator response to ACh after 6 h of NaAc infusion was similar to the dilator response before the infusion was started in both the group pretreated with vehicle and the group pretreated with MSO/methionine (Fig. 4). In contrast, the ACh dilator response was completely abolished by 6 h of NH4Ac infusion in the group pretreated with vehicle. Also, no significant dilation was observed in the NH4Ac group pretreated with L-methionine alone. However, pretreatment with MSO/methionine preserved ACh dilation during NH4Ac infusion at the baseline ACh response. Moreover, in the hyperammonemic group receiving L-arginine during the last 2 h of NH4Ac infusion, significant dilation to ACh occurred, although the response was less than the baseline ACh response.
Fig. 4.

Percent change in pial arteriolar diameter (±SD) during 30 M ACh superfusion before (0 h) and at 6 h of infusion of sodium acetate in groups pretreated with vehicle (n = 8) and MSO/Met (n = 8) and after 6 h of infusion of ammonium acetate in groups pretreated with vehicle (n = 8), Met (n = 6), MSO/Met (n = 8), and Arg (n = 8). *P < 0.05 from 0-h value within group;+P < 0.05 from vehicle-ammonium acetate group at same time point.
Glutamine superfusion experiment
Superfusion of the cranial window with artifical CSF containing 0, 0.3, 1, and 2 mM L-glutamine for 1 h resulted in graded increases in glutamine concentrations in the CSF effluent (Table 2). Baseline diameter of pial arterioles remained unchanged during the 1-h superfusion period (Table 2). There were no differences among groups in mean arterial blood pressure or PaCO2. The percent dilation to 10 μM ACh was influenced by the glutamine concentration as assessed by ANOVA (P < 0.01). The ACh response was significantly reduced in the group superfused with 1 mM L-glutamine compared with the group superfused with 0.3 mM L-glutamine (Fig. 5). The difference between the groups superfused with 0 and 1 mM glutamine was of marginal significance (P < 0.06), as determined by the Newman-Keuls multiple range test. Furthermore, the response to ACh in the group superfused with 2 mM L-glutamine was significantly reduced compared with the groups superfused with either 0 or 0.3 mM L-glutamine. In contrast, the dilator response to sodium nitroprusside was not significantly different among the groups superfused with the different L-glutamine concentrations (Fig. 6).
Table 2.
Effects of glutamine superfusion in cranial window
| Inflow Glutamine Concentration, mM
|
||||
|---|---|---|---|---|
| 0 | 0.3 | 1 | 2 | |
| Outflow glutamine, μM | 240±35 | 547±23 | 1325±298 | 1986±87 |
| Baseline arteriolar diameter, %change | −0.1±3.9 | −2.6±2.9 | 4.2±5.8 | −0.4±3.1 |
| Arterial Pco2, Torr | 39±2 | 39±3 | 39±1 | 38±3 |
| MABP at 60 min of glutamine superfusion, mmHg | 104±12 | 109±10 | 114±13 | 113±9 |
| MABP during acetylcholine superfusion, mmHg | 105±15 | 113±9 | 115±12 | 116±8 |
Values are means ± SD. MABP, mean arterial blood pressure.
Fig. 5.

Percent change in pial arteriolar diameter (±SD) during 10 μM ACh superfusion vs. glutamine concentration in outflow of cranial window after 1 h of superfusion with 0 (n = 7), 0.3 (n = 6), 1 (n = 7), and 2 mM (n = 7) glutamine in inflow. *P < 0.05 from diameter at 0 mM inflow glutamine; P < 0.05 from diameter at 0.3 mM inflow glutamine.
Fig. 6.

Percent change in pial arteriolar diameter (±SD) during 3 μM sodium nitroprusside superfusion vs. glutamine concentration in outflow of cranial window after 1 h of superfusion with 0 (n = 7), 0.3 (n = 6), 1 (n = 7), and 2 mM (n = 7) glutamine in inflow. There was no significant effect of glutamine.
DISCUSSION
The major findings of this study are 1) acute hyperammonemia in rats sufficient to increase fourfold the cortical glutamine concentration abolished pial arteriolar dilation to ACh; 2) preventing the increase in tissue glutamine by inhibiting glutamine synthetase preserved the dilatory response to ACh; 3) intravenous infusion of L-arginine during hyperammonemia partially restored ACh dilation; and 4) elevation of CSF glutamine concentration in the absence of hyperammonemia attenuated dilation to ACh but not to the endothelial-independent dilator nitroprusside. These results indicate that glutamine accumulation in the pathophysiological range, such as that associated with hyperammonemia, is sufficient to impair an endothelial-dependent response in cerebral resistance blood vessels in vivo.
Neither ammonia nor glutamine directly inhibit NO synthase (NOS) enzymatic activity when substrate and cofactors are present (20). Arginine availability for NOS activity is determined by recycling of citrulline to arginine (5, 12), cleavage of peptides (12), cellular uptake of arginine and citrulline (37), and metabolism by arginase (39), which has a higher Km for arginine than NOS (4). Glutamine has been reported to limit recycling of citrulline to arginine by inhibiting arginosuccinate synthetase activity in aortic endothelial cells (29), although others report that the primary effect of glutamine is on citrulline uptake (20, 37). Physiological concentrations of glutamine can inhibit release of endothelial-derived relaxing factor in cultured cells (11) and isolated aorta (31), but the inhibitory effect of glutamine on agonist-evoked NO production appears to be independent of arginine availability (1). Alternative mechanisms include glutamine metabolism to glucosamine, which decreases pentose cycle activity and depletes NADPH necessary for NOS activity (36). In addition to endothelial cells, perivascular nerves surrounding cerebral arteries can recycle citrulline to arginine, and glutamine attenuates arterial relaxation evoked by transmural nerve stimulation (5, 18). These effects of glutamine were attributed, in part, to inhibition of citrulline uptake by glutamine. The results of the present study extend these previous in vitro studies by demonstrating that pathophysiological accumulation of glutamine during hyperammonemia is capable of blocking ACh-evoked dilation in vivo and that exogenous application of glutamine can attenuate ACh-evoked dilation. Furthermore, the finding that hyperammonemia in the absence of glutamine accumulation did not block ACh dilation indicates that ammonium ions do not block this NO-dependent response, consistent with the lack of effect of ammonia on arginine synthesis (29) and on NOS-dependent neurogenic vasorelaxation (18). Infusion of L-arginine did not fully restore dilation to ACh during hyperammonemia. Although L-arginine infusion did not significantly increase the overall tissue concentration of L-arginine, this dose of L-arginine increased plasma arginine concentration sevenfold and, therefore, was presumed to increase L-arginine availability within the endothelium. Nonetheless, one possible explanation for the incomplete restoration of the ACh response is that the infusion did not increase arginine concentration sufficiently in the subcellular compartment where NOS is localized and that higher infusion doses are required to fully restore the dilator response.
Hyperammonemia produced two- to fourfold increases in cerebral glutamine concentration. Glutamine synthetase is enriched in astrocytes (21), and much of the increase in glutamine is presumed to occur within astrocytes. However, hyperammonemia also produces increases in interstitial fluid, CSF, and plasma concentrations of glutamine (23, 34). Therefore, the endothelium is expected to be exposed to increased glutamine during hyperammonemia.
The glutamine concentration was previously determined to be 0.25 mM in the CSF sampled from the cranial window of the rat (22). When the CSF concentration of glutamine was increased above 1 mM for 1 h without hyperammonemia, the ACh-evoked dilation was attenuated but was not reduced to the same extent as with hyperammonemia. Perhaps longer exposure to elevated glutamine is necessary to completely block the ACh response. Alternatively, other effects of hyperammonemia related to glutamine accumulation may have contributed indirectly to the impaired response.
In this regard, previous studies of this acute model of hyperammonemia have demonstrated that astrocyte swelling and increases in water content and ICP are ameliorated by MSO (32, 33, 35). The present results showing that ICP measured in the cranial window increased in the hyperammonemic groups treated with vehicle, L-methionine, or L-arginine and that the increase was blunted by MSO/methionine treatment also support these earlier findings that cerebral edema is related to glutamine accumulation in astrocytes enriched in glutamine synthetase. Although the pial arteriolar response to ACh is endothelial dependent (25), loss of astrocyte function can impair endothelial-dependent responses under specific circumstances (38). Therefore, one needs to consider the possibilities that 1) astrocyte dysfunction associated with glutamine-dependent swelling might have secondary effects on endothelial-dependent responses distinct from direct effects of glutamine on the endothelium, and 2) increases in ICP may limit vasodilation.
The first possibility is supported by observations that extracellular K+ activity, which is known to be regulated by astrocytes, increases to 12 mM in this model of hyperammonemia and that this increase is prevented by MSO treatment (30). Elevated extracellular K+ activity might interfere with ACh vasodilation. In this case, part of the restoration of ACh dilation by MSO may be attributed to restoring astrocyte regulation of extracellular K+ and part may be attributed to preventing increases in glutamine that affect endothelial function.
The second possibility, that mechanical effects of ICP will reduce cerebral perfusion pressure and increase baseline arteriolar diameter to an extent that limits ACh-evoked dilation, is not fully supported by the data. Increases in baseline diameter occurred over the 6-h salt infusion in all groups except the control group receiving NaAc after vehicle pretreatment. Similar increases in diameter have been noted previously in hyperammonemic rats (13, 15). In the case of hyperammonemic rats treated with methionine, MSO/methionine, or arginine, the decrease in cerebral perfusion pressure in these groups probably contributed to the increase in baseline pial arteriolar diameter as an autoregulatory response. However, the dilator response to ACh was restored in the MSO/methionine group with NH4Ac, which had a lower cerebral perfusion pressure (67 ± 6 mmHg) than the vehicle group with NH4Ac (84 ± 14 mmHg). Therefore, the low perfusion pressure was not completely responsible for the impaired dilatory response in the other groups. Likewise, the ACh response was partially restored in the L-arginine-transfused group in which the cerebral perfusion pressure was also low (68 ± 10 mmHg).
Increases in glutamine and extracellular K+ activity associated with acute hyperammonemia do not affect all aspects of cerebral vascular reactivity. For example, pial arterioles still dilate in response to hypoxia (15) and constrict in response to the thromboxane agonist U46619 (13). The cerebral blood flow response to hypoxia is intact, and blood flow remains well autoregulated during arterial hypotension (14). Thus the effects on ACh reactivity are specific. However, hyperammonemia also reduces CO2 reactivity and, like ACh reactivity, this reduction depends on increased glutamine and is partially restored by L-arginine infusion (15, 22). Because the CO2 response is modulated by neuronal NOS activity and the ACh response is dependent on endothelial NOS activity, some commonalities may exist in the effect of glutamine on these responses. However, differences also exist in that the CO2 response is determined primarily by extracellular pH and the role of neuronally derived NO is that of a modulator in which neuronal NOS inhibition can attenuate but not abolish CO2 reactivity. Thus factors in addition to suppressing NOS activity by glutamine may come into play in completely abolishing CO2 reactivity in this model.
In addition to inhibiting glutamine synthetase, MSO increases methylation flux and consequently decreases S-adenosylmethionine (28). Seizures associated with MSO have been attributed to altered methylation because coadministration of L-methionine prevented the decrease in S-adenosylmethionine (26) and in the occurrence of seizures (10) without decreasing glutamine synthetase activity (27). In our experiments, L-methionine was coadministered with MSO to avoid the potential confounding influence of seizure activity. Our observation that the increase in cerebral glutamine concentration during hyperammonemia was prevented when L-methionine was coadministered with MSO indicates that MSO was still effective in reducing glutamine synthesis and accumulation. Increasing S-adenosylmethionine by injection of L-methionine can also lead to the formation of homocysteine, which, in turn, is capable of attenuating cerebrovascular dilation to ACh by a superoxide-dependent mechanism (8, 40). Therefore, the improved ACh response in hyperammonemic rats pretreated with MSO/methionine, compared with vehicle treatment, cannot be readily explained by a secondary effect of L-methionine on increasing homocysteine. Nevertheless, the dilator response to ACh was moderately reduced 3 h after MSO/methionine treatment (before the NH4Ac infusion began). This lower baseline ACh response may have been the result of increased homocysteine.
In conclusion, the results of the present experiment indicate that cerebrovascular dilation to ACh becomes impaired during acute hyperammonemia. This impairment is related to glutamine accumulation rather than to ammonium ions because preventing glutamine accumulation during hyperammonemia restored ACh vasodilation and elevation of glutamine alone attenuated ACh vasodilation. Therefore, these results represent the first findings that pathophysiological alterations in glutamine concentration can influence an endothelial-dependent response in vivo. Improved vascular function with L-arginine infusion may help explain an early report showing that arginine administration to rats receiving lethal injections of ammonium sulfate reduced mortality (24).
Acknowledgments
The authors thank Ellen Gordes for technical assistance in these experiments and Tzipora Sofare for editorial assistance.
Present address of R. J. Traystman: Mackenzie Hall 2170E, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd., L335, Portland, OR 97239.
Footnotes
GRANTS
This work was supported by a grant from the National Institute of Neurological Disorders and Stroke (NS-25275).
DISCLOSURES
S. W. Brusilow, R. J. Traystman, and R. C. Koehler have a pending patent entitled, “Novel dosage form of L-methionine-S-sulfoximine.”
References
- 1.Arnal JF, Munzel T, Venema RC, James NL, Bai CI, Mitch WE, Harrison DG. Interactions between L-arginine and L-glutamine change endothelial NO production. J Clin Invest. 1995;95:2565–2572. doi: 10.1172/JCI117957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barzilay Z, Britten AG, Koehler RC, Dean JM, Traystman RJ. Interaction of CO2 and ammonia on cerebral blood flow and O2 consumption in dogs. Am J Physiol Heart Circ Physiol. 1985;248:H500–H507. doi: 10.1152/ajpheart.1985.248.4.H500. [DOI] [PubMed] [Google Scholar]
- 3.Brusilow SW. Determination of urine orotate and orotidine and plasma ammonium. In: Hommes FA, editor. Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual. New York: Wiley-Liss; 1991. pp. 345–357. [Google Scholar]
- 4.Buga GM, Singh R, Pervin S, Rogers NE, Schmitz DA, Jenkinson CP, Cederbaum SD, Ignarro LJ. Arginase activity in endothelial cells: inhibition by NG-hydroxy-L-arginine during high-output NO production. Am J Physiol Heart Circ Physiol. 1996;271:H1988–H1998. doi: 10.1152/ajpheart.1996.271.5.H1988. [DOI] [PubMed] [Google Scholar]
- 5.Chen FY, Lee TJF. Arginine synthesis from citrulline in perivascular nerves of cerebral artery. J Pharmacol Exp Ther. 1995;273:895–901. [PubMed] [Google Scholar]
- 6.Chodobski A, Szmydynger-Chodobska J, Skolasinska K. Effect of ammonia intoxication on cerebral blood flow, its autoregulation and responsiveness to carbon dioxide and papaverine. J Neurol Neurosurg Psychiatry. 1986;49:302–309. doi: 10.1136/jnnp.49.3.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper AJL, Plum F. Biochemistry and physiology of brain ammonia. Physiol Rev. 1987;67:440–519. doi: 10.1152/physrev.1987.67.2.440. [DOI] [PubMed] [Google Scholar]
- 8.Dayal S, Arning E, Bottiglieri T, Boger RH, Sigmund CD, Faraci FM, Lentz SR. Cerebral vascular dysfunction mediated by superoxide in hyperhomocysteinemic mice. Stroke. 2004;35:1957–1962. doi: 10.1161/01.STR.0000131749.81508.18. [DOI] [PubMed] [Google Scholar]
- 9.Durham S, Yonas H, Aggarwal S, Darby J, Kramer D. Regional cerebral blood flow and CO2 reactivity in fulminant hepatic failure. J Cereb Blood Flow Metab. 1995;15:329–335. doi: 10.1038/jcbfm.1995.38. [DOI] [PubMed] [Google Scholar]
- 10.Folbergrova J. Glycogen and glycogen phosphorylase in the cerebral cortex of mice under the influence of methionine sulphoximine. J Neurochem. 1973;20:547–557. doi: 10.1111/j.1471-4159.1973.tb12154.x. [DOI] [PubMed] [Google Scholar]
- 11.Hecker M, Mitchell JA, Swierkosz TA, Sessa WC, Vane JR. Inhibition by L-glutamine of the release of endothelium-derived relaxing factor from cultured endothelial cells. Br J Pharmacol. 1990;101:237–239. doi: 10.1111/j.1476-5381.1990.tb12693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hecker M, Sessa WC, Harris HJ, Anggard EE, Vane JR. The metabolism of L-arginine and its significance for the biosynthesis of endothelium-derived relaxing factor: cultured endothelial cells recycle L-citrulline to L-arginine. Proc Natl Acad Sci USA. 1990;87:8612–8616. doi: 10.1073/pnas.87.21.8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirata T, Kawaguchi T, Brusilow SW, Traystman RJ, Koehler RC. Preserved hypocapnic pial arteriolar constriction during hyperammonemia by glutamine synthetase inhibition. Am J Physiol Heart Circ Physiol. 1999;276:H456–H463. doi: 10.1152/ajpheart.1999.276.2.H456. [DOI] [PubMed] [Google Scholar]
- 14.Hirata T, Koehler RC, Brusilow SW, Traystman RJ. Preservation of cerebral blood flow responses to hypoxia and arterial pressure alterations in hyperammonemic rats. J Cereb Blood Flow Metab. 1995;15:835–844. doi: 10.1038/jcbfm.1995.104. [DOI] [PubMed] [Google Scholar]
- 15.Hirata T, Koehler RC, Kawaguchi T, Brusilow SW, Traystman RJ. Impaired pial arteriolar reactivity to hypercapnia during hyperammonemia depends on glutamine synthesis. Stroke. 1996;27:729–736. doi: 10.1161/01.str.27.4.729. [DOI] [PubMed] [Google Scholar]
- 16.Kindt GW, Altenau LL. Primary dilation of the cerebral resistance vessels as a cause of increased intracranial pressure. Adv Neurol. 1978;20:315–320. [PubMed] [Google Scholar]
- 17.Lavoie J, Giguere JF, Layrargues GP, Butterworth RF. Amino acid changes in autopsied brain tissue from cirrhotic patients with hepatic encephalopathy. J Neurochem. 1987;49:692–697. doi: 10.1111/j.1471-4159.1987.tb00949.x. [DOI] [PubMed] [Google Scholar]
- 18.Lee TJF, Sarwinski S, Ishine T, Lai CC, Chen FY. Inhibition of cerebral neurogenic vasodilation by L-glutamine and nitric oxide synthase inhibitors and its reversal by L-citrulline. J Pharmacol Exp Ther. 1996;276:353–358. [PubMed] [Google Scholar]
- 19.Levasseur JE, Wei EP, Raper AJ, Kontos HA, Patterson JL. Detailed description of a cranial window technique for acute and chronic experiments. Stroke. 1975;6:308–317. doi: 10.1161/01.str.6.3.308. [DOI] [PubMed] [Google Scholar]
- 20.Meininger CJ, Wu G. L-Glutamine inhibits nitric oxide synthesis in bovine venular endothelial cells. J Pharmacol Exp Ther. 1997;281:448–453. [PubMed] [Google Scholar]
- 21.Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979;161:303–310. doi: 10.1016/0006-8993(79)90071-4. [DOI] [PubMed] [Google Scholar]
- 22.Okada T, Watanabe Y, Brusilow SW, Traystman RJ, Koehler RC. Interaction of glutamine and arginine on cerebrovascular reactivity to hypercapnia. Am J Physiol Heart Circ Physiol. 2000;278:H1577–H1584. doi: 10.1152/ajpheart.2000.278.5.H1577. [DOI] [PubMed] [Google Scholar]
- 23.Rao VL, Audet RM, Butterworth RF. Selective alterations of extracellular brain amino acids in relation to function in experimental portal-systemic encephalopathy: results of an in vivo microdialysis study. J Neurochem. 1995;65:1221–1228. doi: 10.1046/j.1471-4159.1995.65031221.x. [DOI] [PubMed] [Google Scholar]
- 24.Roberge A, Charbonneau R. Protective effects of arginine against hepatic coma, insulin shock and epilepsy. Life Sci. 1969;8:369–377. doi: 10.1016/0024-3205(69)90061-7. [DOI] [PubMed] [Google Scholar]
- 25.Rosenblum WI. Endothelial dependent relaxation demonstrated in vivo in cerebral arterioles. Stroke. 1986;17:494–497. doi: 10.1161/01.str.17.3.494. [DOI] [PubMed] [Google Scholar]
- 26.Schatz RA, Sellinger OZ. Effect of methionine and methionine sulphoximine on rat brain S-adenosyl methionine levels. J Neurochem. 1975;24:63–66. doi: 10.1111/j.1471-4159.1975.tb07628.x. [DOI] [PubMed] [Google Scholar]
- 27.Sellinger OZ, Azcurra JM, Ohlsson WG. Methionine sulfoximine seizures. VIII. The dissociation of the convulsant and glutamine synthetase inhibitory effects. J Pharmacol Exp Ther. 1968;164:212–222. [PubMed] [Google Scholar]
- 28.Sellinger OZ, Schatz RA, Porta R, Wilens TE. Brain methylation and epileptogenesis: the case of methionine sulfoximine. Ann Neurol. 1984;16:S115–S120. doi: 10.1002/ana.410160717. [DOI] [PubMed] [Google Scholar]
- 29.Sessa WC, Hecker M, Mitchell JA, Vane JR. The metabolism of L-arginine and its significance for the biosynthesis of endothelium-derived relaxing factor: L-glutamine inhibits the generation of L-arginine by cultured endothelial cells. Proc Natl Acad Sci USA. 1990;87:8607–8611. doi: 10.1073/pnas.87.21.8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugimoto H, Koehler RC, Wilson DA, Brusilow SW, Traystman RJ. Methionine sulfoximine, a glutamine synthetase inhibitor, attenuates increased extracellular potassium activity during acute hyperammonemia. J Cereb Blood Flow Metab. 1997;17:44–49. doi: 10.1097/00004647-199701000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Swierkosz TA, Mitchell JA, Sessa WC, Hecker M, Vane JR. L-Glutamine inhibits the release of endothelium-derived relaxing factor from the rabbit aorta. Biochem Biophys Res Commun. 1990;172:143–148. doi: 10.1016/s0006-291x(05)80184-6. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi H, Koehler RC, Brusilow SW, Traystman RJ. Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats. Am J Physiol Heart Circ Physiol. 1991;261:H825–H829. doi: 10.1152/ajpheart.1991.261.3.H825. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi H, Koehler RC, Hirata T, Brusilow SW, Traystman RJ. Restoration of cerebrovascular CO2 responsivity by glutamine synthesis inhibition in hyperammonemic rats. Circ Res. 1992;71:1220–1230. doi: 10.1161/01.res.71.5.1220. [DOI] [PubMed] [Google Scholar]
- 34.Weigle CG, Koehler RC, Brusilow SW, Traystman RJ. Arterial pH modulation of regional cerebral blood flow during hyperammonemia in dogs. Am J Physiol Heart Circ Physiol. 1990;259:H34–H41. doi: 10.1152/ajpheart.1990.259.1.H34. [DOI] [PubMed] [Google Scholar]
- 35.Willard-Mack CL, Koehler RC, Hirata T, Cork LC, Takahashi H, Traystman RJ, Brusilow SW. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience. 1996;71:589–599. doi: 10.1016/0306-4522(95)00462-9. [DOI] [PubMed] [Google Scholar]
- 36.Wu G, Haynes TE, Li H, Yan W, Meininger CJ. Glutamine metabolism to glucosamine is necessary for glutamine inhibition of endothelial nitric oxide synthesis. Biochem J. 2001;353:245–252. doi: 10.1042/0264-6021:3530245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu G, Meininger CJ. Regulation of L-arginine synthesis from L-citrulline by L-glutamine in endothelial cells. Am J Physiol Heart Circ Physiol. 1993;265:H1965–H1971. doi: 10.1152/ajpheart.1993.265.6.H1965. [DOI] [PubMed] [Google Scholar]
- 38.Xu HL, Ye S, Baughman VL, Feinstein DL, Pelligrino DA. The role of the glia limitans in ADP-induced pial arteriolar relaxation in intact and ovariectomized female rats. Am J Physiol Heart Circ Physiol. 2005;288:H382–H388. doi: 10.1152/ajpheart.00727.2004. [DOI] [PubMed] [Google Scholar]
- 39.Zhang C, Hein TW, Wang W, Chang CI, Kuo L. Constitutive expression of arginase in microvascular endothelial cells counteracts nitric oxide-mediated vasodilatory function. FASEB J. 2001;15:1264–1266. doi: 10.1096/fj.00-0681fje. [DOI] [PubMed] [Google Scholar]
- 40.Zhang F, Slungaard A, Vercellotti GM, Iadecola C. Superoxide-dependent cerebrovascular effects of homocysteine. Am J Physiol Regul Integr Comp Physiol. 1998;274:R1704–R1711. doi: 10.1152/ajpregu.1998.274.6.R1704. [DOI] [PubMed] [Google Scholar]