

Abstract
Systemic infections can trigger heart attacks. We conducted an autopsy study to investigate the pathologic effect of systemic infections on coronary artery inflammation.
We studied 14 atherosclerotic patients diagnosed with an acute systemic infection. Our control group (n=13) had atherosclerosis without infection. The groups were similar in luminal stenosis and age. Coronary artery sections were stained with H&E and markers for macrophages (CD68), T cells (CD3), and dendritic cells (S100).
On pathologic examination, 5 infected patients had acute myocardial infarction with thrombosis. Macrophage density in plaques and in periadventitial fat was higher in the infected group (NS). The infected patients' adventitia had significantly more macrophages (1,577 ± 1,872 vs 265 ± 185 per mm2; P=0.047). The macrophage density, similar in the control group's adventitia and plaque, was significantly greater in the infected group's adventitia than in the plaque. The adventitia and periadventitial fat of the infected group had more T cells than did samples from the control group (48.4 ± 45.0 vs 14.1 ± 6.3 per mm2; P=0.002). The groups exhibited similar plaque T-cell density. The infected patients' plaques, but not the adventitia and periadventitial fat, had more dendritic cells than did the controls' (3.2 ± 2.5 vs 0.3 ± 0.5 per mm2; P=0.022).
To our knowledge, this is the 1st report to establish a connection between acute systemic infections and significant increases in inflammatory cells in the atherosclerotic coronary arteries of human beings. This offers a new therapeutic target for preventing heart attacks in high-risk patients.
Key words: Arteriosclerosis/complications, cardiovascular diseases/diagnosis/prevention & control, coronary arteriosclerosis/pathology, coronary disease/pathology, inflammation/complications/etiology/pathology, coronary vessels/pathology, myocardial infarction/etiology/pathology, retrospective studies, risk assessment, sepsis/complications
Inflammation plays a major part in the initiation and progression of atherosclerosis and in the development of its acute clinical manifestations.1,2 Acute infections, with their consequent inflammation, may affect atherosclerotic disease. This relationship was first proposed by William Osler at the beginning of the 20th century.
The following infectious agents have been linked to atherosclerosis: cytomegalovirus, Chlamydia pneumoniae, herpes simplex viruses 1 and 2, Helicobacter pylori, Mycoplasma pneumoniae, Porphyromonas gingivalis, enterovirus, and, more recently, the influenza virus.3–5 A series of acute and chronic infections, occurring alone or in combination, may lead to the development and progression of atherosclerosis. By rapidly increasing inflammation in the coronary arteries, acute infections may trigger destabilization and possible rupture of vulnerable plaques.6 Given the central role of inflammation in atherosclerosis, we investigated whether a wide range of systemic infections might exacerbate local inflammation in coronary arteries.
Materials and Methods
Study Population
We reviewed the pathology files of a large teaching hospital from 1991 through 2002. The study protocols and procedures were approved by the institutional review boards, and approvals from the treating physicians were obtained before the medical records were released. After excluding patients who might have had an inadequate or altered inflammatory response to infections (for example, persons with human immunodeficiency virus or cancer, or those who were on a regimen involving immunosuppressive or corticosteroid drugs), we identified 14 patients who had clinical and pathologic evidence of coronary artery disease (atherosclerosis) at autopsy and an acute systemic infection within 2 weeks of death. The study group comprised 11 men and 3 women, aged 64 ± 14 yr. This group was compared with 13 control patients (8 men and 5 women; mean age, 65 ± 11 yr) who had died with coronary artery disease but without infection. There were no significant differences between the groups in age (P = 0.82) or sex (P = 0.41). Table I lists the acute infectious agents in the study group.
TABLE I. Acute Infections in the Study Group

Sepsis was defined as the presence of systemic infection as determined by the treating physician or from laboratory data. Twelve study-group patients had upper or lower respiratory infections; 2 had urinary tract infections. Most of the control patients had died abruptly due to pulmonary embolism (2 cases), postoperative decompensation or complications (3), aortic aneurysm (3), acute myocardial infarction (AMI) (1), aortic dissection and AMI (1), asthma (1), airway obstruction (1), and acute respiratory distress syndrome (1).
Histopathologic Examination
The coronary arteries obtained at autopsy were formalin-fixed, paraffin-embedded, and cut into 4-μm-thick serial sections. One to 4 sections per patient (mean, 1.2 sections) were immunohistochemically stained with all 3 of these antibodies: the macrophage marker CD68 (all markers were from DAKO; Carpenteria, Calif), the CD3 marker for T cells, and the S100 protein for dendritic cells. Quantitative morphometric evaluation(blinded) was performed by 2 separate observers who used the Olympus MicroSuite Software™ B3SV on an Olympus BX61 microscope (Olympus America Inc.;Center Valley, Pa). Cell counts were performed in the intimal plaque, adventitia, and periadventitial fat at ×200 magnification for the entire circumference of each coronary artery. The results were presented as the number of cells per mm2.
Histologic Definitions
The plaque area was defined as the area inside the internal elastic lamina (IEL). The adventitia was defined as the region extending from the external elastic lamina to the beginning of the periadventitial fat. The periadventitial fat was considered to extend from the adventitia to 250 μm beyond. Macrophage density was the number of macrophages per mm2. Stenosis denoted the (IEL area – lumen area)/IEL area, expressed as a percentage. We quantified all the slides that were available.
Statistical Tests
Results are expressed as mean ± standard deviation. Because of the small sample size and nonnormal distribution of the data, we used the Mann-Whitney U test and the Wilcoxon signed rank test to study the significance of our findings. An α level of 0.05 was considered the threshold for statistical significance. The Fisher exact test was used to evaluate categorical variables, such as the sex of the patients. The software used for statistical analysis was the Statistical Package for the Social Sciences, version 9 (SPSS Inc.; Chicago, Ill).
Results
The 2 groups had similar percentages of luminal stenosis (67% ± 14% vs 55% ± 25%; P = 0.23). Subocclusive luminal thrombi were seen in 4 infected patients, and a small luminal thrombus was present in a 5th; all 5 had histologic evidence of AMI. Only 1 organized thrombus was seen in the control group. Of the 5 infected patients who had AMI on pathologic examination, only 2 had been diagnosed clinically to have AMI.
In the plaques, the macrophage density showed a nonsignificant trend toward higher levels in the patients with a systemic infection (582 ± 774 vs 281 ± 321 per mm2; P = 0.41) (Table II). However, in the adventitia of the infected patients, there was a significantly greater number of macrophages than in the adventitia of the control patients (1,577 ± 1,872 vs 265 ± 185 per mm2; P = 0.047) (Figs. 1 and 2).
TABLE II. Densities of Inflammatory Cells in the Plaque and Adventitia


Fig. 1 Section of coronary artery from an infected patient. A) Low-power view (H&E, orig. ×4). B) CD68 staining shows a substantial presence of macrophages that heavily infiltrate the plaque, adventitia, and periadventitial fat, mostly sparing the media. C, D) Increased magnification (×10) of the respective stained sections.
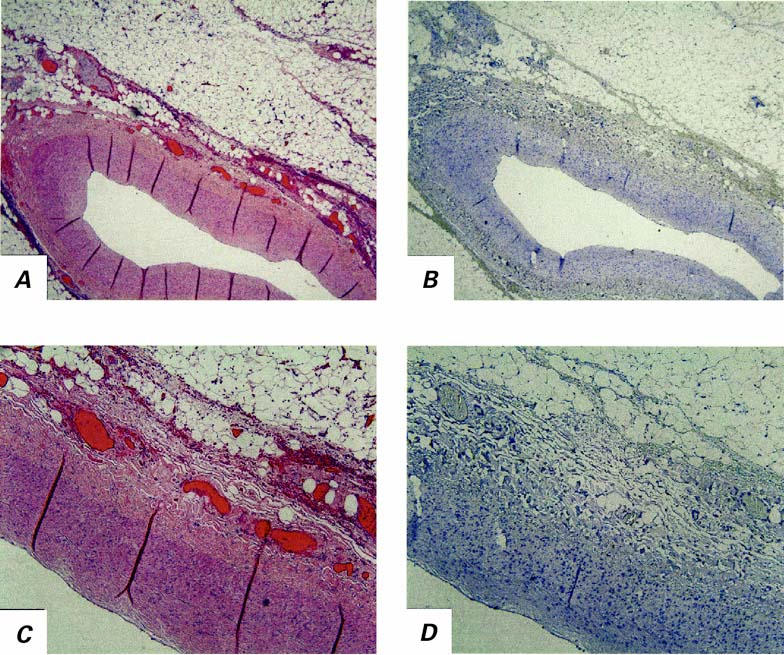
Fig. 2 Section of coronary artery from a control patient. A) Low-power view (H&E, orig. ×4). B) CD68 staining shows an absence of macrophages in comparison with the Figure 1 images of the infected patient's artery. C, D) Increased magnification (orig. ×10) of the respective stained sections.
The macrophage density in the periadventitial fat showed a nonsignificant trend toward higher levels in the infected patients (776 ± 821 vs 212 ± 219 per mm2; P = 0.085). In the control patients, the macrophage density was similar in the adventitia and the plaque (281 ± 321 vs 265 ± 185 per mm2; P = 0.85); however, in the infected patients, the density was significantly higher in the adventitia than in the plaque (1,577 ± 1,872 vs 582 ± 774 per mm2; P = 0.047).
Significantly more T cells were observed in the adventitia and periadventitial fat of the infected patients than in the control patients (48.4 ± 45.0 vs 14.1 ± 6.3 per mm2; P = 0.002). In the intima, the number of T cells did not differ significantly (infected, 14.0 ± 13.2 per mm2; control, 14.3 ± 20.5 per mm2; P = 0.49).
Similarly, the dendritic cell counts were not significantly different in the adventitia and periadventitial fat (infected, 34.1 ± 53.7 per mm2; control, 21.8 ± 15.3 per mm2; P = 0.87). On the other hand, significantly more dendritic cells were seen in the plaque of the infected patients (3.2 ± 2.5 per mm2; control, 0.3 ± 0.5 per mm2; P = 0.022) (Table II; Figs. 3 and 4).
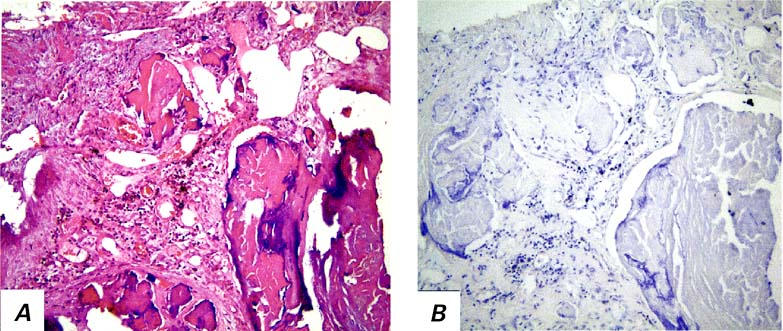
Fig. 3 Section of coronary artery from an infected patient. A) Calcified plaque with substantial neovascularization (H&E, orig. ×20). B) S100-positive dendritic cells are seen mainly around neovascularization (CD68, orig. ×20).

Fig. 4 Higher numbers of macrophages (upper graph) and dendritic cells and T cells (lower graph) were observed in different layers of the atherosclerotic coronary lesions of infected patients versus control patients.
In summary, we found a significantly higher number of macrophages in the coronary adventitia of the infected patients than of the control patients, who were atherosclerotic without infection. The macrophage density tended to be higher in the plaque and periadventitial fat of the infected patients, but this difference was not significant. The infected patients also had more T cells in their adventitia and periadventitial fat, and more dendritic cells in their intima and media. The higher number of inflammatory cells was associated with an increase in the incidence of myocardial infarctions and luminal thrombosis. Interestingly, the percentage of stenosis was not significantly different between the 2 groups.
Discussion
To our knowledge, this is the 1st report to establish a connection between acute systemic infections and significant increases in inflammatory cells—known to play a major role in AMI—in the atherosclerotic coronary arteries of human beings. This discovery suggests a mechanism for the triggering of AMIs after acute infections, and it offers a new therapeutic target for the prevention of heart attacks.
How Might Acute Infections Trigger Acute Myocardial Infarction?
Over several decades, scattered clinical reports have noted that up to one third of myocardial infarctions are preceded by an upper respiratory infection.6–12 The occurrence of AMI undergoes seasonal variation, having its highest incidence in the winter months, when the incidence of upper respiratory infections is also highest.13–15 Influenza epidemics have been associated with a significant increase in overall cardiovascular deaths,16 a spike usually attributed to deaths of patients with known heart failure. In 2000, we described an association between influenza vaccination and a reduced incidence of winter myocardial infarctions,6,17 a finding later confirmed by others.18–21 In addition, a study of 75 patients who had severe acute respiratory syndrome (SARS) found that 2 out of 5 deaths were due to AMI22—an occurrence still overlooked by many.
Infectious agents have several potential effects on the pathophysiology of atherosclerosis and its clinical complications.5,23 Whereas most suspected infectious agents initiate or aggravate a chronic vascular or systemic inflammatory process, acute systemic infections may, instead, destabilize existing vulnerable plaques. For example, we have shown that inoculating atherosclerotic, apolipoprotein-E–deficient mice with the influenza A virus leads to a marked increase in inflammation and thrombosis in murine atherosclerotic plaques but not in normal regions of the aorta.24 Pro-atherosclerotic changes in mice have also been reported after acute infection with cytomegalovirus and P. gingivalis.25
Systemic infections can exert acute and chronic influence on vascular walls. The effects are either direct (through seeding of the microbe in the vascular wall) or indirect (through release of inflammatory cytokines and other systemic effects) (Fig. 5).5,6,26,27
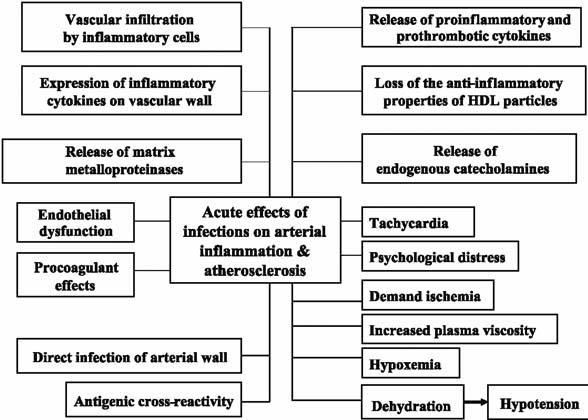
Fig. 5 Various mechanisms by which acute infections may affect atherosclerotic plaques.
Adventitial Inflammation in Atherosclerosis
Evidence increasingly suggests an important role of adventitia in the inflammation that is present in atherosclerotic plaques. In 2004, we reported that human atherosclerotic coronary plaques with large lipid cores have a significantly greater number of macrophages in their periadventitial fat than do fibrocalcific and nonatherosclerotic arterial segments, which suggests the involvement of adventitial inflammatory cells in plaque vulnerability.28 Our previous findings suggested that the adventitia and periadventitial fat may function as a unit.28 The lack of a functional border between the adventitia and the periadventitial fat has been noted by others after balloon angioplasty.29
Dendritic Cells and Atherosclerosis
An immune reaction against certain antigens may play an important role in the development of atherosclerosis.30,31 Dendritic cells—antigen-presenting cells that have important functions in immune responses—have been observed in atherosclerotic plaques. In advanced atherosclerotic lesions, clusters of dendritic cells have been described as part of the inflammatory infiltrates that resemble mucosa-associated lymphoid tissue, where they intermingle closely with B lymphocytes.32 Dendritic cells also interact closely with CD4 helper and CD8 cytotoxic cells.33 In our study, dendritic cells were more numerous in the plaques of the infected group than in those of the control group. We infer that acute systemic infections, by increasing the number of dendritic cells, may enhance the immunologic reactions in the plaques and exacerbate the atherosclerotic process. Further studies are needed to evaluate this hypothesis.
Limitations
Although our study's low sample size might have decreased our power to detect some of the associations at an α level of 0.05, our main findings were statistically significant. At a higher sample size, our significant results would still be valid, and the observed trends—which are already biologically relevant—might also reach levels of significance.
Unfortunately, our autopsy study, by its very nature, limited the number of cases that could be studied. The small sample size did not enable us to differentiate between pathologic responses to different infections, such as gram-negative versus gram-positive infections and those at different loci. Nevertheless, given the long list of infectious agents linked to atherosclerosis and the central role of inflammation as a common final pathway for these agents' effects, we conclude that most acute systemic infections have similar nonspecific, injurious effects on the coronary arteries. In fact, the burden of infection itself may increase the risk of AMI.4 More severe infections, such as influenza and SARS, may have a more profound effect on atherosclerotic disease and could warrant separate studies.6
Recent reports have suggested that statins can decrease morbidity and mortality rates in atherosclerotic patients who have sepsis.34,35 Most of our patients had died in the early 1990s, when statin use was very low. In fact, of the patients in our study, only 1 had received lovastatin. Future studies should seek to determine whether statin use can decrease the recruitment of inflammatory cells to the coronary arteries. Statins may be especially useful in lowering the morbidity and mortality rates among people with and without atherosclerotic disease during a future influenza pandemic. In the event of such a disaster, we foresee a large rise in cardiovascular deaths because of the triggering effect of influenza on AMI.36,37
Implications
If further studies confirm that sepsis promotes the infiltration or survival of inflammatory cells in atherosclerotic arteries, many questions meriting further investigation will be suggested: Why are T cells and dendritic cells drawn to plaque, and macrophages to the adventitia? Is this also seen in atherosclerotic carotid arteries and the aorta? Are some microbes more likely than others to increase plaque inflammation? Does increased plaque inflammation increase the risk of rupture, erosion, or thrombosis? Does adventitial inflammation accelerate the progression of aneurysms? Are some arteries themselves infected? If so, is the inflammatory response beneficial or harmful? Although our study falls short of proving a causal relationship between the preceding systemic infection and the coronary inflammation observed at autopsy, our findings are supported by the growing body of evidence concerning the pivotal role of inflammatory cells in coronary events,38 and these findings deserve further evaluation.
We noted 5 cases of AMI in the infected group, compared with only 1 old myocardial infarction in the control group. However, only 2 of these 5 AMIs had been detected clinically before the patients died—raising the possibility that, in patients with a systemic infection, AMI may easily be missed despite the increased chance of plaque destabilization. Our report highlights the need for clinicians to consider possible myocardial infarction when caring for septic patients.
Inflammatory mediators are central to the pathogenesis of septic shock and multiorgan failure, because sepsis results from an exaggerated systemic inflammatory host response induced by infectious organisms.39 Clearly, the use of anti-inflammatory drugs and statins should be evaluated in patients who have coronary artery disease. These drugs may attenuate the dangerous effects of acute infections on atherosclerotic plaques. The severity of sepsis may be decreased by the administration of statin therapy before the onset of an acute bacterial infection.40
Aggressive anti-inflammatory treatment may be indicated for patients who develop an acute infection anywhere in the body. Our findings may also help explain why the risk of myocardial infarction is increased after surgical interventions (which may be associated with bacteremia), and why—if tooth-brushing leads to bacteremia—patients with periodontal disease have an increased rate of coronary and cerebral events.41
Therapeutic and prophylactic implications for clinicians and patients include oral hygiene; hand-washing; avoidance of infected persons; vaccinations (for example, for influenza, Streptococcus pneumoniae, and Haemophilus influenzae); new indications for antimicrobial, anti-inflammatory, and antithrombotic medications; and closer monitoring of at-risk patients.
Acknowledgments
The authors wish to thank Tomas Klima, MD; Tommy Reese, HTL; and Mr. John Ryan for their help in conducting this study; and Ms Virginia D. Fairchild for preparing this manuscript.
Footnotes
Address for reprints: Mohammad Madjid, MD, Atherosclerosis Research Laboratory, Texas Heart Institute, 6770 Bertner, MC 2-255, Houston, TX 77030. E-mail: Mohammad.Madjid@uth.tmc.edu
Dr. Litovsky is now with the Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama.
Supported in part by U.S. Department of Defense Grant #DAMD 17-01-2-0047.
References
- 1.Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med 1999;340:115–26. [DOI] [PubMed]
- 2.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2000;20:1262–75. [DOI] [PubMed]
- 3.Libby P, Egan D, Skarlatos S. Roles of infectious agents in atherosclerosis and restenosis: an assessment of the evidence and need for future research. Circulation 1997;96:4095–103. [DOI] [PubMed]
- 4.Epstein SE, Zhou YF, Zhu J. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation 1999;100:e20–8. [DOI] [PubMed]
- 5.Madjid M, Aboshady I, Awan I, Litovsky S, Casscells SW. Influenza and cardiovascular disease: is there a causal relationship? Tex Heart Inst J 2004;31:4–13. [PMC free article] [PubMed]
- 6.Madjid M, Naghavi M, Litovsky S, Casscells SW. Influenza and cardiovascular disease: a new opportunity for prevention and the need for further studies. Circulation 2003;108:2730–6. [DOI] [PubMed]
- 7.Pesonen E, Siitonen O. Acute myocardial infarction precipitated by infectious disease. Am Heart J 1981;101:512–3. [DOI] [PubMed]
- 8.Abinader EG, Sharif DS, Omary M. Inferior wall myocardial infarction preceded by acute exudative pharyngitis in young males. Isr J Med Sci 1993;29:764–9. [PubMed]
- 9.Bourne G, Wedgwood J. Heart-disease and influenza. Lancet 1959;1:1226–8. [DOI] [PubMed]
- 10.Penttinen J, Valonen P. The risk of myocardial infarction among Finnish farmers seeking medical care for an infection. Am J Public Health 1996;86:1440–2. [DOI] [PMC free article] [PubMed]
- 11.Zheng ZJ, Mittleman MA, Tofler GH, Pomeroy C, Dampier C, Wides B, Muller JE. Infections prior to acute myocardial infarction onset [abstract]. J Am Coll Cardiol 1998;31(Suppl A):132A.
- 12.Meier CR, Jick SS, Derby LE, Vasilakis C, Jick H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet 1998;351:1467–71. [DOI] [PubMed]
- 13.Spencer FA, Goldberg RJ, Becker RC, Gore JM. Seasonal distribution of acute myocardial infarction in the second National Registry of Myocardial Infarction. J Am Coll Cardiol 1998;31:1226–33. [DOI] [PubMed]
- 14.Sheth T, Nair C, Muller J, Yusuf S. Increased winter mortality from acute myocardial infarction and stroke: the effect of age. J Am Coll Cardiol 1999;33:1916–9. [DOI] [PubMed]
- 15.Jakovljevic D, Salomaa V, Sivenius J, Tamminen M, Sarti C, Salmi K, et al. Seasonal variation in the occurrence of stroke in a Finnish adult population. The FINMONICA Stroke Register. Finnish Monitoring Trends and Determinants in Cardiovascular Disease. Stroke 1996;27:1774–9. [DOI] [PubMed]
- 16.Collins SD. Excess mortality from causes other than influenza and pneumonia during influenza epidemics. Pub Health Rep 1932;47:2159–79.
- 17.Naghavi M, Barlas Z, Siadaty S, Naguib S, Madjid M, Casscells W. Association of influenza vaccination and reduced risk of recurrent myocardial infarction. Circulation 2000; 102:3039–45. [DOI] [PubMed]
- 18.Gurfinkel EP, de la Fuente RL, Mendiz O, Mautner B. Influenza vaccine pilot study in acute coronary syndromes and planned percutaneous coronary interventions: the FLU Vaccination Acute Coronary Syndromes (FLUVACS) Study. Circulation 2002;105:2143–7. [DOI] [PubMed]
- 19.Lavallee P, Perchaud V, Gautier-Bertrand M, Grabli D, Amarenco P. Association between influenza vaccination and reduced risk of brain infarction [published erratum appears in Stroke 2002;33:1171]. Stroke 2002;33:513–8. [DOI] [PubMed]
- 20.Nichol KL, Nordin J, Mullooly J, Lask R, Fillbrandt K, Iwane M. Influenza vaccination and reduction in hospitalizations for cardiac disease and stroke among the elderly. N Engl J Med 2003;348:1322–32. [DOI] [PubMed]
- 21.Siscovick DS, Raghunathan TE, Lin D, Weinmann S, Arbogast P, Lemaitre RN, et al. Influenza vaccination and the risk of primary cardiac arrest. Am J Epidemiol 2000;152:674–7. [DOI] [PubMed]
- 22.Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 2003;361:1767–72. [DOI] [PMC free article] [PubMed]
- 23.Kol A, Libby P. Molecular mediators of arterial inflammation: a role for microbial products? Am Heart J 1999;138(5 Pt 2):S450–2. [DOI] [PubMed]
- 24.Naghavi M, Wyde P, Litovsky S, Madjid M, Akhtar A, Naguib S, et al. Influenza infection exerts prominent inflammatory and thrombotic effects on the atherosclerotic plaques of apolipoprotein E-deficient mice. Circulation 2003;107:762–8. [DOI] [PubMed]
- 25.Li L, Messas E, Batista EL Jr, Levine RA, Amar S. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model [published erratum appears in Circulation 2002;105:1617]. Circulation 2002;105:861–7. [DOI] [PubMed]
- 26.Van Lenten BJ, Wagner AC, Anantharamaiah GM, Garber DW, Fishbein MC, Adhikary L, et al. Influenza infection promotes macrophage traffic into arteries of mice that is prevented by D-4F, an apolipoprotein A-I mimetic peptide. Circulation 2002;106:1127–32. [DOI] [PubMed]
- 27.Van Lenten BJ, Wagner AC, Nayak DP, Hama S, Navab M, Fogelman AM. High-density lipoprotein loses its anti-inflammatory properties during acute influenza A infection. Circulation 2001;103:2283–8. [DOI] [PubMed]
- 28.Vela D, Buja LM, Madjid M, Burke A, Naghavi M, Willerson JT, et al. The role of periadventitial fat in atherosclerosis: an adipose subset with potential diagnostic and therapeutic implications. Arch Pathol Lab Med. In press. [DOI] [PubMed]
- 29.Okamoto E, Couse T, De Leon H, Vinten-Johansen J, Goodman RB, Scott NA, Wilcox JN. Perivascular inflammation after balloon angioplasty of porcine coronary arteries. Circulation 2001;104:2228–35. [DOI] [PubMed]
- 30.Hansson GK. Regulation of immune mechanisms in atherosclerosis. Ann N Y Acad Sci 2001;947:157–66. [PubMed]
- 31.Wick G, Knoflach M, Xu Q. Autoimmune and inflammatory mechanisms in atherosclerosis. Annu Rev Immunol 2004; 22:361–403. [DOI] [PubMed]
- 32.Houtkamp MA, de Boer OJ, van der Loos CM, van der Wal AC, Becker AE. Adventitial infiltrates associated with advanced atherosclerotic plaques: structural organization suggests generation of local humoral immune responses. J Pathol 2001;193:263–9. [DOI] [PubMed]
- 33.Bobryshev YV, Lord RS, Parsson H. Immunophenotypic analysis of the aortic aneurysm wall suggests that vascular dendritic cells are involved in immune responses. Cardiovasc Surg 1998;6:240–9. [DOI] [PubMed]
- 34.Hackam DG, Mamdani M, Li P, Redelmeier DA. Statins and sepsis in patients with cardiovascular disease: a population-based cohort analysis. Lancet 2006;367:413–8. [DOI] [PubMed]
- 35.Merx MW, Weber C. Statins: a preventive strike against sepsis in patients with cardiovascular disease? Lancet 2006;367: 372–3. [DOI] [PubMed]
- 36.Madjid M, Casscells SW. Of birds and men: cardiologists' role in influenza pandemics. Lancet 2004;364:1309. [DOI] [PubMed]
- 37.Madjid M, Miller C, Zarubaev VV, Marinich IG, Kiselev OI, Lobzin YV, et al. Influenza epidemics are associated with a surge in autopsy-confirmed coronary artery disease death: results from eight years of autopsies in 34,892 subjects. Eur Heart J. In press. [DOI] [PMC free article] [PubMed]
- 38.Buja LM, Willerson JT. Role of inflammation in coronary plaque disruption. Circulation 1994;89:503–5. [DOI] [PubMed]
- 39.Sharma S, Kumar A. Septic shock, multiple organ failure, and acute respiratory distress syndrome. Curr Opin Pulm Med 2003;9:199–209. [DOI] [PubMed]
- 40.Almog Y, Shefer A, Novack V, Maimon N, Barski L, Eizinger M, et al. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation 2004;110:880–5. [DOI] [PubMed]
- 41.Rutger Persson G, Ohlsson O, Pettersson T, Renvert S. Chronic periodontitis, a significant relationship with acute myocardial infarction. Eur Heart J 2003;24:2108–15. [DOI] [PubMed]