

Abstract
Functional annotation of proteins is a fundamental problem in the post-genomic era. The recent availability of protein interaction networks for many model species has spurred on the development of computational methods for interpreting such data in order to elucidate protein function. In this review, we describe the current computational approaches for the task, including direct methods, which propagate functional information through the network, and module-assisted methods, which infer functional modules within the network and use those for the annotation task. Although a broad variety of interesting approaches has been developed, further progress in the field will depend on systematic evaluation of the methods and their dissemination in the biological community.
Keywords: data integration, function prediction, protein interaction networks, protein modules
Introduction
The past decade has seen a revolution in sequencing technologies, resulting in hundreds of sequenced genomes. A fundamental challenge of the post-genomic era is the interpretation of this wealth of data to elucidate protein function. To date, even for the most well-studied organisms such as yeast, about one-fourth of the proteins remain uncharacterized (Figure 1).
Figure 1.

Extent of annotation of proteins in model species. For each species, the charts give the fractions and numbers of annotated and unannotated proteins, according to the three ontologies of the GO annotation. The numbers are based on the Entrez Gene and the WormBase databases as of September 2006.
Classical computational approaches to gene annotation collect for each protein a set of features characterizing it, and apply machine-learning algorithms to infer annotation rules based on those features (Pavlidis et al, 2001). The newly available large-scale networks of molecular interactions within the cell have made it possible to go beyond these one-dimensional approaches, and study protein function in the context of a network. In particular, novel high-throughput technologies for protein–protein interaction (PPI) measurements (Aebersold and Mann, 2003; Fields, 2005) have created large-scale data on protein interaction across human and most model species. These data are commonly represented as networks, with nodes representing proteins and edges representing the detected PPIs.
In this review, we survey the growing body of works on functional annotation of proteins via their network of interactions (summarized in Table I). We distinguish two types of approaches (Figure 2): direct annotation schemes, which infer the function of a protein based on its connections in the network, and module-assisted schemes, which first identify modules of related proteins and then annotate each module based on the known functions of its members. Naturally, the presented methods and the emphasis on particular ones reflect the opinions of the authors.
Table 1.
A summary of functional annotation methods
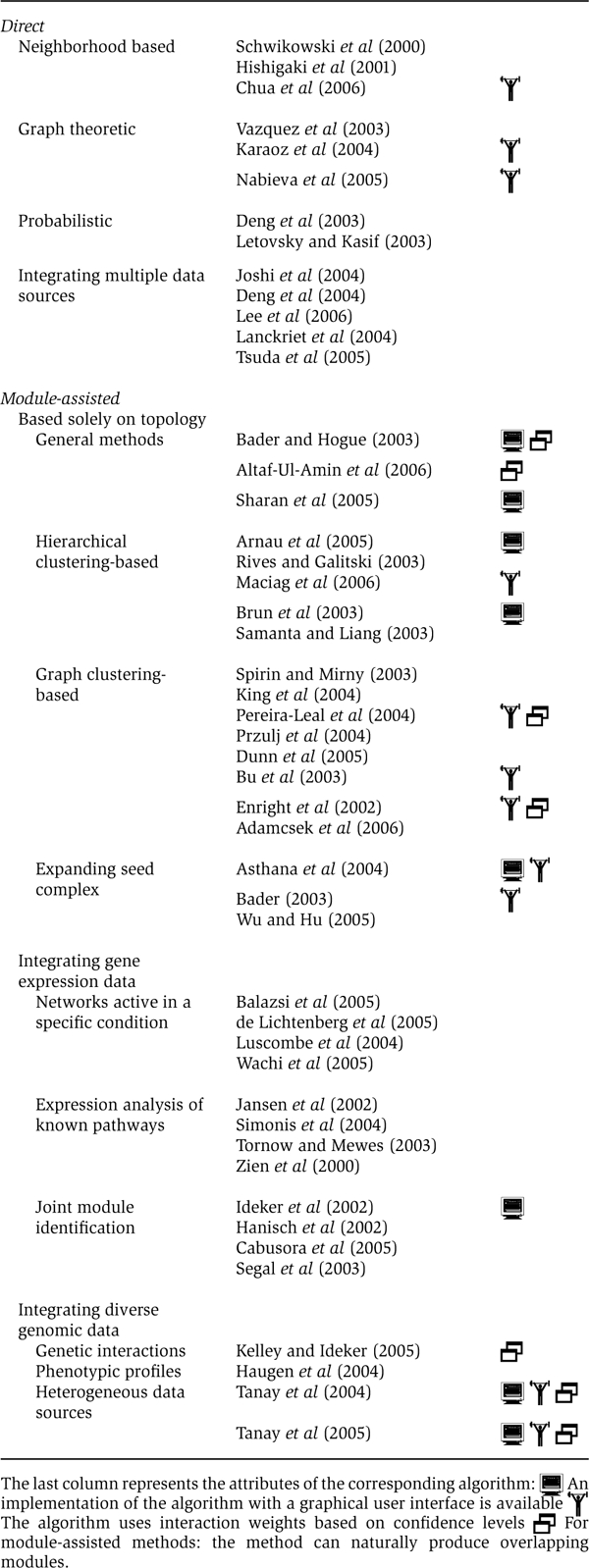 |
Figure 2.

Direct versus module-assisted approaches for functional annotation. The scheme shows a network in which the functions of some proteins are known (top), where each function is indicated by a different color. Unannotated proteins are in white. In the direct methods (left), these proteins are assigned a color that is unusually prevalent among their neighbors. The direction of the edges indicates the influence of the annotated proteins on the unannotated ones. In the module-assisted methods (right), modules are first identified based on their density. Then, within each module, unannotated proteins are assigned a function that is unusually prevalent in the module. In both methods, proteins may be assigned with several functions.
Direct methods
The common principle underlying all direct methods for functional annotation is that proteins that lie closer to one another in the PPI network are more likely to have similar function. As can be seen in Figure 3, there is an evident correlation between network distance and functional distance, that is, the closer the two proteins are in the network the more similar are their functional annotations. The methods described below differ in the way they capture and exploit this correlation. In the following, we denote the PPI network as a graph G=(V,E) (see Box 1 for graph-theoretic definitions).
Figure 3.

Correlation between protein functional distance and network distance. X-axis: distance in the network. Y-axis: average functional similarity of protein pairs that lie at the specified distance. The functional similarity of two proteins is measured using the semantic similarity of their GO categories (Lord et al, 2003).
Graph-theoretic concepts.
A graph is a pair G=(V,E), where V is a set of vertices (or nodes) and E is a set of edges connecting pairs of vertices. In PPI networks, the vertices represent proteins and the edges represent interactions.
The distance between two vertices in a graph is the number of edges on a shortest path between them.
The diameter of a graph is the maximum distance between any two of its vertices.
The neighborhood of a vertex is the set of vertices connected to it.
The n-neighborhood of a vertex is the set of vertices whose distance from it is at most n.
A clique in a graph is a fully connected subgraph, that is, a subgraph in which every two vertices are connected by an edge.
The degree of a vertex is the number of its neighbors.
A cut in a graph is a partition of the vertices into two non-overlapping sets. A multiway cut is a partition of the vertices into several disjoint sets. The value of the cut is the number of edges going between different sets.
Network flow: Imagine a graph as a network of interconnected pipes. Suppose water gets into one or more vertices (sources) from the outside, and can exit the network at certain other vertices (sinks). Then, it will spread in the pipes and reach other nodes, until it exits at sinks. The capacities of the edges (i.e., how much the pipe can carry per unit time) and the input at the sources determine the amount of flow along every edge (i.e., how much each pipe actually carries) and the amount exiting at each sink. In the context of a PPI network, by considering proteins that have a certain function as sources and simulating flow in the network, the amount of flow at edges and sinks can be used to annotate additional proteins.
The density of a graph is the fraction of edges it actually has out of all possible vertex pairs. Hence, the density of G=(V,E) is 2∣E∣/(∣V∣(∣V∣−1)), and a clique graph has the maximum possible density, that is, 1.
The clustering coefficient of a vertex is the density of its neighborhood (Watts and Strogatz, 1998).
A graph is called a k-core if the minimal degree in it is k.
Core clustering coefficient: For a parameter k, the core clustering coefficient of a vertex is the density of the largest k-core of its immediate neighborhood (Bader and Hogue, 2003).
An adjacency matrix of a graph G=(V,E) is a matrix A∣V∣×∣V∣={aij} where aij=1 if and only if vi and vj are neighbors. As PPI networks usually do not contain loops, in our context aii=0.
A graph is called bipartite if its vertices can be partitioned into two disjoint sets such that no edge connects two vertices of the same set.
Neighborhood counting
The simplest and most direct method for function prediction determines the function of a protein based on the known function of proteins lying in its immediate neighborhood. Schwikowski et al (2000) predict for a given protein up to three functions that are most common among its neighbors. Although simple and effective, the obvious caveats of this approach are that associations are not assigned any significance values and the full topology of the network is not taken into account in the annotation process.
Hishigaki et al (2001) try to tackle the first problem by computing χ2-like scores for function assignment. In detail, they examine the n-neighborhood of a protein (Box 1). For a protein p, each function f is assigned a score (nf-ef)2/ef, where nf is the number of proteins in the n-neighborhood of p that have the function f and ef is the expectation of this number based on the frequency of f among the network's proteins. A shortcoming of this approach is that within the n-neighborhood, proteins at different distances from p are treated in the same way. Chua et al (2006) try to tackle the second problem by investigating the relation between network distance and functional similarity. They focus on the 1- and 2-neighborhoods of a protein, and devise a functional similarity score that gives different weights to proteins according to their distances from the target protein.
Graph theoretic methods
As the PPI network is a graph, it is natural to apply graph algorithms for its functional analysis. Two main approaches have been suggested in this context: cut-based approaches and a flow-based algorithm. In contrast to the local neighborhood counting methods, these approaches are global and take into account the full topology of the network.
Vazquez et al (2003) aim at assigning a function σv to each unannotated protein v so as to maximize the number of edges that connect proteins (unannotated or previously annotated) assigned with the same function. Precisely, they try to maximize
where E′ is the set of edges incident on two unannotated proteins, δ is a function that equals 1 if x=y and 0 otherwise, and hv(f) denotes the number of neighbors of v previously annotated with function f. The first term in the optimization criterion concerns unannotated proteins, whereas the second term accounts for interactions between unannotated and previously annotated proteins. This optimization problem, which generalizes the computationally hard problem of minimum multiway cut (Box 1) (Dahlhaus et al, 1994), is heuristically solved using simulated annealing.
Karaoz et al (2004) use a similar approach but handle one function at a time. Each annotated protein v receives a state sv that equals +1 if v has the function in question and equals −1 otherwise. Next, an assignment of −1 and +1 states to unannotated proteins is sought so as to maximize 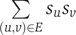 . The advantage of this formulation is that a partition of the vertices into two sets only is sought. To find a good partition, Karaoz et al (2004) apply a local search procedure in which for every vertex in turn (until convergence), the state of the vertex is changed according to the majority of the states of its neighbors. This procedure guarantees a solution with value at least half of the optimum.
. The advantage of this formulation is that a partition of the vertices into two sets only is sought. To find a good partition, Karaoz et al (2004) apply a local search procedure in which for every vertex in turn (until convergence), the state of the vertex is changed according to the majority of the states of its neighbors. This procedure guarantees a solution with value at least half of the optimum.
A related method suggested by Nabieva et al (2005) again formulates the annotation problem as a minimum multiway cut problem, where the goal is to assign a unique function to all unannotated proteins so as to minimize the cost of edges connecting proteins with different assignments. They propose an integer programming reformulation of this problem, which allows them to solve the problem in practice (on the yeast PPI network) by applying a commercial solver (CPLEX).
Finally, Nabieva et al (2005) also suggest a flow-based (Box 1) approach for the annotation problem. As they note that the maximum-cut-based approaches described above take into account global properties of the network but do not reward local proximity, they propose a novel method that aims at considering both local and global effects. They handle one function at a time. The basic idea is to treat each protein annotated with the function as the source of a ‘functional flow'. After simulating the spread over time of this functional flow through the network, each unannotated protein is assigned a score for having the function based on the amount of flow it received during the simulation.
Markov random field
A number of probabilistic approaches to the annotation problem have been suggested, all relying on a Markovian assumption: the function of a protein is independent of all other proteins given the functions of its immediate neighbors. This assumption gives rise to a Markov random field (MRF) model (Box 2), initially proposed by Deng et al (2003). Deng et al (2003) devise an MRF model in which the probability that a protein v is assigned with a certain function that occurs with frequency f is a logistic function of  , where α and β are model parameters and N(v,1) and N(v,0) are the numbers of neighbors of v that are assigned or not assigned with the function, respectively. The parameters α and β of the model are first estimated using a quasi-likelihood method, and then Gibbs sampling is used for inferring the functions of unannotated proteins. Interestingly, as pointed out by Deng et al (2004), setting α to 0 and β to 1 yields the exact optimization criterion used by Karaoz et al (2004).
, where α and β are model parameters and N(v,1) and N(v,0) are the numbers of neighbors of v that are assigned or not assigned with the function, respectively. The parameters α and β of the model are first estimated using a quasi-likelihood method, and then Gibbs sampling is used for inferring the functions of unannotated proteins. Interestingly, as pointed out by Deng et al (2004), setting α to 0 and β to 1 yields the exact optimization criterion used by Karaoz et al (2004).
Function prediction using the MRF method.
The Markov random field (MRF) model provides a probabilistic framework for simulating the mutual influence of random variables via a neighborhood system. Given a network of influence, the state of any random variable is assumed to be independent of all other random variable states given those of its immediate neighbors. In the function prediction setting, each random variable corresponds to a protein, and its states correspond to certain functional annotations. The joint distribution of the random variables can be shown to factorize over the cliques (Box 1) of the network (Besag, 1974). That is, the probability of a certain assignment of discrete states x=(x1,…,xN) is
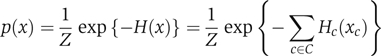 |
where N is the total number of variables, Z is a normalizing constant, C is the set of all cliques in the network, Hc is a potential function associated with clique c and xc is the assignment of states to the members of c.
Inference in this general model is computationally hard, hence it is common to assign 0 potentials to all cliques of size greater than 2, and further homogenize the model by associating the same potential function with all cliques of the same size. For such a homogeneous second-order MRF, we have
Deng et al (2003) treat one function at a time. To obtain a second-order MRF model, they assume that the probability of a 0/1 annotation over the entire network is proportional to exp(αN01+βN11+N00), where α,β are parameters for weighting the contributions of the different terms and Nij is the number of interacting pairs with assignment i,j (unordered). Combining the a priori probability of an assignment with N1 1 s, which depends on the frequency f of the function and is proportional to (f/(1 − f))N1, they obtain a homogeneous second-order MRF for which
Hence, the probability that protein v is assigned with the function given the annotations of its neighbors N(v) is
where N(v,i) is the number of neighbors of v that are assigned with i∈{0,1} and logit is the logistic function logit(x)=1/(1+e−x). Deng et al (2003) estimate the two parameters of the model using a quasi-likelihood method and apply Gibbs sampling to infer the unknown functional annotations.
Letovsky and Kasif (2003) also use an MRF model. Their main assumption is that the number of neighbors of a protein that are annotated with a given term is binomially distributed, where the distribution's parameter depends on whether the protein has that function or not. They employ loopy belief propagation (Murphy et al, 1999) to perform inference in their model.
Integrating multiple information sources
Several authors have integrated data from multiple sources for the annotation task. The approaches differ in the way the sources are combined. The simplest approach treats each data source independently. Such an approach for function prediction was first introduced by Marcotte et al (1999) in a different context. Joshi et al (2004) integrated PPIs, genetic interactions and coexpression interactions. For each interaction type they estimated the a priori probability of functional association given an interaction of this type. By treating all interactions (within- and between-types) as independent of one another, they combined them into a single reliability score for the functional association of a protein given the annotations of its neighbors.
A second approach for combining multiple data types constructs a joint probabilistic model or a prediction function of the different types. Deng et al (2004) generalized their earlier MRF approach to allow using multiple networks in the annotation process, and applied the method to annotate yeast proteins using both PPIs and genetic interactions (Deng et al, 2004; Lee et al, 2006). Lanckriet et al (2004) and later Tsuda et al (2005) represent each data type using a matrix of kernel similarity values. These matrices are then combined by learning optimal relative weights for the different kernels.
Module-assisted methods
The prevalence of the modularity paradigm in molecular cell biology has led to an extensive use of modules in prediction of molecular functions. By functional module one typically means a group of cellular components and their interactions that can be attributed to a specific biological function (Hartwell et al, 1999). Instead of predicting functions for individual genes, a module-assisted approach attempts to first identify coherent groups of genes and then assign functions to all the genes in each group. The module-assisted methods differ mainly in their module detection technique. Once a module is obtained, simple methods are usually used for function prediction within the module. For example, every function shared by the majority of the module's genes is assigned to all the genes in the module. Alternatively, a hypergeometric enrichment P-value is computed for every function:
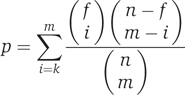 |
where n is the number of nodes in the PPI network, f is the number of genes in the network annotated with the function and m is the module size. The functions enriched within the module (i.e. obtaining P-value below some threshold) are then predicted for all the genes in the module.
Module finding algorithms can be divided into methods using solely network topology information and methods that utilize addition data sources, such as gene expression measurements or deletion phenotypes. The algorithms vary in their ability to detect overlapping modules and in the use of interaction reliabilities. The exact definition of a functional module also varies. The purpose of some algorithms is the detection of molecular complexes, but others can also detect sparser structures, such as signaling pathways (Steffen et al, 2002; Yeang et al, 2005; Scott et al, 2006). Here, we focus on the former, as the latter methods are currently not aimed at function prediction.
Detecting functional modules from network topology
Several works focused on the detection of functional modules based solely on protein interaction data. Most of the works described below decompose the PPI network into subnetworks based on some topological properties.
The molecular complex detection algorithm (MCODE) (Bader and Hogue, 2003) consists of three stages: vertex weighting, complex prediction and an optional post-processing step. The weighting of nodes is based on the core clustering coefficient (Box 1). Bader and Hogue (2003) propose the use of this coefficient instead of the standard clustering coefficient, as it increases the weights of heavily interconnected graph regions while giving small weights to the less connected vertices, which are abundant in the scale-free protein interaction networks. Once the weights are computed, the algorithm traverses the weighted graph in a greedy fashion to isolate densely connected regions. The post-processing step filters or adds proteins based on connectivity criteria. MCODE has been used in several recent publications describing mapping of large-scale interaction networks (LaCount et al, 2005; Rual et al, 2005). It is available as a plug-in for the Cytoscape network visualization software (Shannon et al, 2003).
Altaf-Ul-Amin et al (2006) use a similar approach: they define a cluster property of a node n with respect to a cluster C as the number of edges between n and the nodes of C divided by that number averaged over the nodes of C. Starting from single nodes, clusters are gradually grown as long as the cluster property of the added nodes and the density of the cluster both exceed a certain threshold.
Sharan et al (2005) proposed the NetworkBlast algorithm for detecting protein modules in protein interaction networks. Each candidate set of proteins is assigned a likelihood ratio score that measures its fit to a protein complex model versus the chance that its connections arise at random (Box 3). A greedy network search algorithm is subsequently used for the detection of high-scoring modules. The method can be generalized to identify modules that are conserved over several networks.
Detecting dense subgraphs using maximum likelihood scoring.
Maximum likelihood-based scoring has been used for detecting molecular complexes in NetworkBlast (Sharan et al, 2005) and for integration of heterogeneous data through biclustering in SAMBA (Tanay et al, 2002, 2004, 2005). For a given subnetwork H, this technique compares the probabilities of two alternatives: (i) H has dense structure (the subnetwork model) and (ii) H is random (null model).
When assigning a score to a complex, in the subnetwork model, every possible interaction in the subnetwork exists with some high probability β, independently of other protein pairs. In the null model, every two proteins u,v are connected with probability pu,v that depends on their degrees. pu,v can be estimated by generating a collection of random networks preserving the degree of every protein and calculating the fraction of networks in which an interaction between u and v exists. The log-likelihood ratio of the two alternatives for a set of proteins C with a set of interactions E(C) is thus
 |
In case edge reliabilities are available, it is possible to incorporate them into the subnetwork scoring model by assessing the probability of the available observations, given that the protein pair u,v interacts (P(Ou,v)∣Tu,v)) and does not interact (P(Ou,v)∣Fu,v)). The log-likelihood score then becomes
 |
In the SAMBA algorithm (Tanay et al, 2002), a bipartite graph represents the genomic data: nodes on side A represent genes and nodes on side B represent different properties (Figure 4). A likelihood ratio is used to score a potential bicluster (A,B) by summing over all the edges between A and B:
 |
Using these scores, optimization algorithms based on hashing (Tanay et al, 2002) and local search (Sharan et al, 2005) can be used to detect high-scoring subnetworks.
Hierarchical clustering-based methods
Several works described the use of hierarchical clustering for module detection. A key decision in the use of this approach is the selection of the similarity measure between protein pairs. An intuitive similarity metric is based on pairwise distances (Box 1) between proteins in the network. One of the problems in using it in a hierarchical clustering setting is that the distances between many protein pairs are identical (the ties in proximity problem) (Arnau et al, 2005). Rives and Galitski (2003) postulated that module members are likely to have similar shortest path distance profiles. They therefore used hierarchical clustering on the all-pair shortest path distances matrix and outlined modules by manual inspection. Although such an approach is impractical for genome-scale networks, it has been successfully applied to focused subnetworks, such as the network of interactions between nuclear proteins and a regulatory network of filamentation in Saccharomyces cerevisiae (Rives and Galitski, 2003). A more sophisticated version of this approach was recently applied to the gene expression machinery network (Maciag et al, 2006). The authors used a special weighted form of mutual clustering coefficient (Goldberg and Roth, 2003) for quantifying similarities between proteins, and a modified version of the k-means algorithm (Hartigan, 1975) was used for cluster detection.
Arnau et al (2005) used the shortest path length between proteins as a distance measure and attempted to overcome the ‘ties in proximity' problem by obtaining multiple, equally valid hierarchical clustering solutions with a random choice when ties are encountered. The fraction of the solutions in which the protein pair was clustered together was then used as a similarity measure for clustering using standard hierarchical algorithms. Additional similarity measures proposed for using in a hierarchical clustering setting include Czekanovski–Dice distance (Brun et al, 2003) and the statistical significance of the number of common interaction partners (Samanta and Liang, 2003).
Graph clustering methods
Numerous graph-clustering algorithms have been applied to the graph representing the binary interactions. Spirin and Mirny (2003) proposed two such algorithms. The first algorithm is based on superparamagnetic clustering (SPC) (Blatt et al, 1996) and the second is a Monte Carlo algorithm maximizing the density of the obtained clusters. Both algorithms require the size of the sought clusters as input. The SPC algorithm is specifically shown to perform well in detecting dense structures loosely connected to other areas of the network. In addition to detecting protein complexes, Spirin and Mirny (2003) also show that their method is capable of detecting sparsely connected functional modules, such as the MAPK signaling cascade.
Przulj et al (2004) used the highly connected subgraphs (HCS) algorithm (Hartuv and Shamir, 2000) to determine complexes in PPI data. A highly connected subgraph is defined as a subgraph with n nodes such that more than n/2 edges must be removed in order to disconnect it. It can be shown that this property ensures that the diameter of the subgraph (Box 1) is at most two, and that it is at least half as dense as a clique of the same size. The HCS algorithm finds a minimum cut (Box 1) in the graph and uses it to partition the graph. This process is repeated recursively until highly connected components are reached.
The restricted neighborhood search clustering (RNSC) algorithm proposed by King et al (2004) partitions the node set of the network into clusters based on a cost function that is used to evaluate the partitioning. The algorithm starts with a random cluster assignment and proceeds by reassigning nodes, so as to maximize the partition's score. In addition, a tabu list is maintained to avoid cycling back to previously explored partitions. Finally, the clusters are filtered based on their size, density and functional homogeneity.
The Markov clustering (MCL) algorithm (Enright et al, 2002) has been recently proposed for complex detection in PPI data (Krogan et al, 2006). The algorithm simulates flow on the PPI graph by constructing its adjacency matrix (Box 1) and computing its successive powers to increase the contrast between regions with high flow and regions with a low flow. This process can be shown to converge towards a partition of the graph into high-flow regions corresponding to protein complexes, separated by regions of no flow.
Additional graph clustering techniques applied include clique percolation (Adamcsek et al, 2006), graph flow simulation (Pereira-Leal et al, 2004), edge-betweenness clustering (Dunn et al, 2005) and spectral methods (Bu et al, 2003).
Expansion of complex seeds
In contrast to finding complexes de novo in the protein interaction network, several works attempted prediction of new members for partially known protein complexes. The Complexpander software (Asthana et al, 2004) receives a particular ‘core' set of proteins and produces a list of candidate proteins, ranked by the probability of membership in the complex. This approach approximates the probability that a given candidate protein is co-complexed with the core by the probability that a path consisting of stable protein interactions exists between the candidate and a member of the core. The algorithm assigns a weight to each pair of proteins, representing the probability that they interact directly and stably. These weights are then used to estimate the probability that two proteins are connected by a path, by generating a collection of random networks and counting how many of them contain a short path between the protein pair.
For a similar problem, an algorithm called SEEDY was proposed by Bader (2003). SEEDY constructs complexes by adding proteins to a given seed, as long as the reliability of the most reliable path from a candidate to the seed does not fall below a given threshold. The reliability of a path in SEEDY is defined as the product of the reliabilities of its edges.
Wu and Hu (2005) proposed an algorithm that detects ‘community structures' (Girvan and Newman, 2002) for a given protein seed, in a network without edge reliabilities. The algorithm first identifies a ‘core' by a heuristic search for a maximum clique containing the seed, and then expands it through a breadth-first-search graph traversal (Cormen et al, 1990) with additional vertices, such that it will meet a certain ‘community' criterion.
Integrated analysis of interactions and expression profiles
Several studies have integrated PPI data with diverse additional sources to infer modular structures. These modular structures can serve as potent function predictors, and also shed light on the interplay between the different sources of information.
Microarray technologies (Schena et al, 1995), which measure expression levels on a genome-wide scale, are currently the largest source of high-throughput genomic information. A natural question is the relation between transcription pattern similarity of a pair of genes and the existence of a protein interaction between their products. As shown in both simple and complex organisms (Ge et al, 2001; Hahn et al, 2005), genes of interacting proteins tend to share similar expression patterns. Following this observation, the use of both information sources together for analysis of functional modules has been an appealing concept adopted by many research groups.
One line of works proposed a two-step approach: extraction of a group of genes that are highly expressed in a certain condition, and then analysis of the topological properties of PPI networks induced by these genes (Luscombe et al, 2004; Balazsi et al, 2005; de Lichtenberg et al, 2005; Wachi et al, 2005). Other works used the reverse approach by analyzing the expression coherence of known pathways or complexes (Zien et al, 2000; Jansen et al, 2002; Tornow and Mewes, 2003; Simonis et al, 2004).
Several approaches have been proposed for identifying functional modules by simultaneous analysis of the network and the expression data. Ideker et al (2002) introduced a framework for identification of active subnetworks, that is, connected regions of the network that show significant changes in expression over a particular subset of the conditions. This method uses P-values calculated for every measurement in the expression data to derive a statistical score for every candidate subnetwork, and utilizes simulated annealing to search for high-scoring subnetworks. A similar methodology was recently employed by (Cabusora et al (2005), using shortest-paths algorithms for module finding.
The co-clustering methodology (Hanisch et al, 2002) uses a distance function that combines similarity of gene expression patterns and network topology. The network distance between two nodes is an edge-weighted version of their topological distance in the network. The expression distance is based on the Pearson correlation between the expression patterns. The two distances are combined into a similarity score using a logistic function, and hierarchical clustering is applied to the matrix of the combined distances.
Segal et al (2003) provided a probabilistic formulation, in which a module is a group of genes with high pairwise similarities and with a significant portion of the possible interactions. A probabilistic graphical model was used to extract a pre-specified number of modules from gene expression measurements combined with a protein interaction data set. This method thus produces groups of genes that are both coexpressed and exhibit a dense interaction pattern.
Integrated analysis of interactions and diverse genomic data
Kelley and Ideker (2005) described the first module-assisted integration of protein interaction data with genetic interaction data. Synthetic lethality and synthetic sickness are interactions between two nonessential genes whose combined deletion is lethal or produces a severe growth effect, respectively. Kelley and Ideker (2005) addressed the question of whether genetic interactions occur mostly within or between different pathways by identifying statistically significant modular structures in the combined network of physical (protein–protein and protein–DNA) and genetic interactions. By defining pathways as dense subnetworks in the physical interaction network, Kelley and Ideker (2005) show that genetic interactions occur between pathways much more often than within pathways. The modular structures led to better function prediction than using physical interactions alone.
Another source of rich genomic information that is becoming increasingly available is the collection of phenotypes of strains containing a deletion for a single nonessential gene and exposed to diverse conditions (Brown et al, 2006). Haugen et al (2004) measured deletion phenotypes along with expression profiles in the arsenic response of S. cerevisiae. The data were then integrated with a metabolic network and a physical network using the ActiveModules algorithm (Ideker et al, 2002). This type of integration was used to reveal proteins central to the arsenic response and to provide network-based explanations to the differences between the phenotypically sensitive pathways and the differentially expressed ones.
Tanay et al (2004) described an integrative framework allowing the integration of protein interaction data with gene expression, phenotypic sensitivity and transcription factor (TF) binding, using the SAMBA biclustering algorithm (Tanay et al, 2002). SAMBA models the genomic data as a bipartite graph (Box 1), where nodes on one side represent genes and that on the other side represent different properties derived from the genomic data (Figure 4). For protein interaction data, a property represents the presence of an interaction with a specific protein. A gene expression experiment is represented by several properties corresponding to different expression level ranges. A TF property represents the binding of the gene promoter by the TF. The bipartite graph is weighted using a maximum likelihood-based score (Box 3). Heavy subgraphs in the bipartite graph correspond to groups of genes that manifest a common behavior across a large set of heterogeneous experiments. This approach was experimentally shown to provide accurate function prediction, and was later extended to analyze a compendium of some 2000 distinct experiments in S. cerevisiae, yielding 1200 statistically significant modules (Tanay et al, 2005).
Figure 4.

Integration of multiple data sources using the SAMBA framework. In the SAMBA framework (Tanay et al, 2004), different gene characteristics are represented by properties (A). Quantitative characteristics, such as gene expression levels, are discretized first. The genes and the properties are represented by nodes in a bipartite graph (B), where edges connect genes with the properties they have. The SAMBA algorithm seeks modules consisting of a subset of genes and a subset of properties, such that these subsets are densely connected in the graph.
Performance comparison
The availability of such a wide range of methods calls for a comprehensive comparison among them. Below we summarize some of the key comparisons reported so far.
Direct methods
For direct methods, no systematic comparison has been reported so far, but some information on the performance of the different methods can be gleaned from comparisons made in the annotation studies reviewed above, one of which we describe below.
Several measures have been suggested to evaluate the quality of a direct annotation method (see e.g., Deng et al, 2003; Nabieva et al, 2005). All these measures are close variants of the one proposed by in Deng et al (2003). The latter is based on measuring the precision and recall of an annotation, computed in a leave-one-out setting (i.e., the known annotation of a single protein at a time is hidden and predicted using the network and the annotations of all other proteins), taking into account multiple annotations per protein. Specifically, let ni be the number of known functions for protein i, let mi be the number of predicted functions for the protein when hiding its true annotations and let ki the overlap between the two sets. The precision and recall of the predictions are defined as
 |
A discussion of other measures that are applicable if the Gene Ontology (GO) annotation is used and of weighted matches between known and predicted functions based on their positions in the GO hierarchy (Ashburner et al, 2000) appears in Deng et al (2004).
Chua et al (2006) compared several schemes, including neighborhood counting (Schwikowski et al, 2000), the χ2 method of Hishigaki et al (2001), the MRF method of Deng et al (2003) and the flow-based method of Nabieva et al (2005). (To avoid possible bias, we do not report here on the method of Chua et al (2006), which was also included in the comparison.) Each method was applied to the MIPS interaction set (Mewes et al, 2002) using the GO annotation, and was evaluated using the precision and recall measures. The MRF method outperformed the others by a significant margin, whereas the other three methods exhibited similar performance. These results were consistent across the three main MIPS categories: cellular role, biochemical function and subcellular localization. The advantage of MRF is probably owing to the use of a more sophisticated probabilistic model.
Module-assisted methods
One of the obstacles to systematic evaluation of the different module-assisted methods for functional annotation is the lack of agreed upon technique for function prediction within a module. However, if one considers a module as a functional unit whose member proteins have identical functions, then one could evaluate a module-assisted method by the fit between the produced modules and either the MIPS complexes catalog (Mewes et al, 2002) or GO categories (Ashburner et al, 2000), using measures similar to those described in the Direct methods section. A systematic quantitative evaluation of four module-assisted clustering algorithms has been presented recently by Brohee and van Helden (2006): RNSC, SPC, MCODE and MCL. As the authors of this study were not involved in the development of any of the algorithms, the chances of inadvertent evaluation bias are low. Brohee and van Helden (2006) used a test graph constructed by representing 220 known complexes as cliques in the graph and generated 41 altered graphs by random addition or removal of edges in different proportions. The parameters of each of the clustering algorithms were then tuned based on this data set using a statistic combining detection sensitivity and specificity. In addition, the four module-assisted techniques were applied to six PPI graphs formed based on high-throughput experiments, and their performance was evaluated by their success in recovering known complexes.
The authors found that the MCL algorithm is remarkably robust to graph alternations. MCL had the best performance on both simulated and real data sets, whereas RNSC was relatively less sensitive to suboptimal parameters. MCODE and SPC were shown to be clearly inferior under most conditions. The comparative analysis also highlighted intrinsic strengths and weaknesses of the algorithms. MCODE performed best on random data, by detecting the fewest false positives, owing to its ability to report complexes covering only part of the network, rather than partitioning all the nodes into complexes. SPC tended to generate ‘mega-complexes' of very large size, thus obtaining very high sensitivity but very low specificity. Notably, this comparison used unweighted networks, whereas the MCL and SPC algorithms can deal with weighted graphs and are likely to give better performances if weights are assigned to reflect the reliability of the interactions (Pereira-Leal et al, 2004).
Comparing direct and module-assisted methods
To the best of our knowledge, no systematic comparison of network-based function prediction, covering both direct and module-assisted methods, has been undertaken to date. In a simplistic comparison of two basic methods (Figure 5), we found that a simple neighbor-counting method has a higher specificity in predicting functions when compared to the more involved module-assisted MCODE algorithm. This may be explained by the focus of MCODE on processes imposing subgraphs with a dense interaction pattern. A more comprehensive analysis alongside the development of better prediction techniques will highlight these differences.
Figure 5.

Performance comparison of a direct method versus a module-assisted one. Two receiver operating characteristic (ROC) curves comparing the accuracy of a neighborhood-counting method (Schwikowski et al, 2000) and of the MCODE method (Bader and Hogue, 2003) in predicting GO Biological Process annotations using a PPI network obtained from BioGRID (Stark et al, 2006). A ROC curve is commonly used to assess prediction performance by plotting the true positive rate versus the false positive rate when varying the prediction threshold. In the neighborhood-counting variant used here, a protein is assigned with a function if the hypergeometric enrichment P-value for the function in the protein's direct neighborhood is below a certain threshold. MCODE clusters were obtained using the Cytoscape plug-in with the ‘node score cutoff' parameter set to 0.05 and the other parameters at their default values. Using MCODE, we predict a function for a protein if that function's P-value in the protein's module is below a certain threshold. Each ROC curve was obtained by varying the threshold. Only proteins assigned to at least one MCODE cluster were used in the analysis for both methods.
Discussion
Efforts for network-based function prediction have been going on for over 6 years now, since the introduction of molecular techniques capable of mapping protein interactions on a genome-wide scale. Despite the large number of techniques suggested for functional annotation using networks, systematic annotation is still mostly based on other data sources, such as sequence homology. Several goals, reviewed below, have to be accomplished in order for the network-based functional annotation tools to become widely used.
Despite the large number of different algorithms developed for both direct and module-assisted function prediction, the implementations of only a small fraction of them are publicly accessible. Only a handful, such as MCODE (Bader and Hogue, 2003), PRODISTIN (Baudot et al, 2006), CFinder (Adamcsek et al, 2006) and NetworkBlast (http://www.pathblast.org/), are currently supported with a graphical interface. Networks are highly visualizable, and as the human eye is better in pattern detection than any computer, a good graphical interface will help make such computational tools widely used by the biological community.
Although the field has advanced considerably in recent years on the methodological side, comprehensive comparisons of the plethora of available annotation methods, similar to that performed by Brohee and van Helden (2006), are greatly in need. Such systematic evaluation efforts were recently performed in other fields, for example, discovery of TF-binding sites (Tompa et al, 2005), biclustering of expression data (Prelic et al, 2006) and protein structure prediction (Kryshtafovych et al, 2005), which has a long and successful history of community evaluations. Owing to the fundamental differences between the different annotation types, such as biological process and molecular complexes, it is clear that different methods are best suited for different types of annotation. An important prerequisite for a comprehensive comparison is the definition of golden standards for functional annotation. In most of the studies described here, the MIPS complexes catalog and Gene Ontology were used as a benchmark for prediction success. However, both data sets are currently not comprehensive and some annotations are found in one but not in the other.
Although several methods described above use diverse functional genomic data sources, they are still greatly outnumbered by methods utilizing only the network topology. Owing to the increasing accessibility of microarray technology, gene expression measurements have become widely available for diverse conditions across species. As of August 2006, almost 95 000 and 45 000 hybridization samples were available in Gene Expression Omnibus and ArrayExpress databases, respectively. This huge body of data is currently poorly exploited by integrative annotation methods, as most of them focus on expression data derived in a single study. Following the success of several methods integrating expression data from multiple studies (Ihmels et al, 2002; Lee et al, 2004; Segal et al, 2005; Tanay et al, 2005), we expect that techniques based on large compendia of expression data and protein interaction networks will significantly increase the accuracy of functional annotation. Additional large-scale genomic data, such as deletion phenotypes (Brown et al, 2006), proteomic measurements (Kislinger et al, 2006) and protein cellular localization (Huh et al, 2003), can also be used in an integrative framework. Data of a high diversity and dimensionality have been integrated using biclustering (Tanay et al, 2005) and kernel-based methods (Lanckriet et al, 2004).
This review focused on methods aimed at genome-scale functional annotation using network data from a single species. Several additional studies developed methods for detection of functional modules in slightly different contexts. In particular, specific algorithms were developed for detection of molecular complexes from lists of proteins identified in biochemical purification experiments, rather than from binary interaction networks (Krause et al, 2003; Hollunder et al, 2005; Scholtens et al, 2005; Gavin et al, 2006). Another set of works attempted to identify evolutionarily conserved functional modules via the integration of networks from multiple organisms (Kelley et al, 2003; Sharan et al, 2005; Campillos et al, 2006; Flannick et al, 2006; Gandhi et al, 2006; see also the review in Sharan and Ideker, 2006).
Which methods should be used by a newcomer to the field? As mentioned above, the limited information about the comparative performance of the methods presented here makes it difficult to decide which method should be used in a specific setting. When using only PPI data, our initial and limited comparison does seem to indicate that direct methods are currently slightly superior to module-assisted ones, with MRF and MCL being the leading techniques for direct and module-assisted function prediction, respectively. New techniques should thus be compared to these methods to prove their superiority. If the goal is actual function prediction rather than methodological improvement, the use is mainly limited to methods that are implemented as a tool with a graphical user interface or available as a web server (Table I). As to methods integrating multiple data sources, no comparative assessment is currently available.
When using interaction networks, whether as a sole information source or in conjunction with other data sources, the current limitations of these data have to be recognized. The currently available protein interaction data are known to be both noisy (von Mering et al, 2002) and partial (Hart et al, 2006). In addition, as large-scale interaction mappings are conducted only in a single growth condition or in a single tissue type, interaction data currently lack any spatial or temporal information. Clearly, for some functional annotations, the relevant interactions may occur only under specific conditions in a specific time point. In addition, not every functional aspect of the protein is expected to be manifested in its interaction pattern. Some proteins, such as metabolic enzymes, are most functional on their own without the need for cooperation from other proteins.
Despite these caveats, analysis of interaction networks is a young, promising and very active research area. The utilization of such networks for function prediction is just one of a plethora of possible ways by which this rich source of information can be exploited. Although techniques for network-based function prediction have been continuously improving, there is still a lot of room for improvement, both in terms of the methodologies and in terms of their evaluation. We expect that improved, more accurate methods that are made readily accessible to the biological community will make interaction networks a prevalent instrument for functional annotation, among their many other important uses.
Acknowledgments
We thank Ken Chua for providing us with his comparison data. RS was supported by an Alon Fellowship. IU is a fellow of the Edmond J Safra Bioinformatics Program at Tel-Aviv University. This research was supported by a grant from the Israeli Science Foundation.
References
- Adamcsek B, Palla G, Farkas IJ, Derenyi I, Vicsek T (2006) CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics 22: 1021–1023 [DOI] [PubMed] [Google Scholar]
- Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- Altaf-Ul-Amin M, Shinbo Y, Mihara K, Kurokawa K, Kanaya S (2006) Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinformatics 7: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnau V, Mars S, Marin I (2005) Iterative cluster analysis of protein interaction data. Bioinformatics 21: 364–378 [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana S, King OD, Gibbons FD, Roth FP (2004) Predicting protein complex membership using probabilistic network reliability. Genome Res 14: 1170–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Hogue CW (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader JS (2003) Greedily building protein networks with confidence. Bioinformatics 19: 1869–1874 [DOI] [PubMed] [Google Scholar]
- Balazsi G, Barabasi AL, Oltvai ZN (2005) Topological units of environmental signal processing in the transcriptional regulatory network of Escherichia coli. Proc Natl Acad Sci USA 102: 7841–7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudot A, Martin D, Mouren P, Chevenet F, Guenoche A, Jacq B, Brun C (2006) PRODISTIN Web Site: a tool for the functional classification of proteins from interaction networks. Bioinformatics 22: 248–250 [DOI] [PubMed] [Google Scholar]
- Besag J (1974) Spatial interaction and the statistical analysis of lattice systems. J R Stat Soc Ser B 36: 192–236 [Google Scholar]
- Blatt M, Wiseman S, Domany E (1996) Superparamagnetic clustering of data. Phys Rev Lett 76: 3251–3254 [DOI] [PubMed] [Google Scholar]
- Brohee S, van Helden J (2006) Evaluation of clustering algorithms for protein–protein interaction networks. BMC Bioinformatics 7: 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Sherlock G, Myers CL, Burrows NM, Deng C, Wu HI, McCann KE, Troyanskaya OG, Brown JM (2006) Global analysis of gene function in yeast by quantitative phenotypic profiling. Mol Syst Biol 2: 2006.0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun C, Chevenet F, Martin D, Wojcik J, Guenoche A, Jacq B (2003) Functional classification of proteins for the prediction of cellular function from a protein–protein interaction network. Genome Biol 5: R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu D, Zhao Y, Cai L, Xue H, Zhu X, Lu H, Zhang J, Sun S, Ling L, Zhang N, Li G, Chen R (2003) Topological structure analysis of the protein–protein interaction network in budding yeast. Nucleic Acids Res 31: 2443–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabusora L, Sutton E, Fulmer A, Forst CV (2005) Differential network expression during drug and stress response. Bioinformatics 21: 2898–2905 [DOI] [PubMed] [Google Scholar]
- Campillos M, von Mering C, Jensen LJ, Bork P (2006) Identification and analysis of evolutionarily cohesive functional modules in protein networks. Genome Res 16: 374–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua HN, Sung WK, Wong L (2006) Exploiting indirect neighbours and topological weight to predict protein function from protein–protein interactions. Bioinformatics 22: 1623–1630 [DOI] [PubMed] [Google Scholar]
- Cormen TH, Leiserson CE, Rivest RL (1990) Introduction to Algorithms. Cambridge, MA, New York: MIT Press, McGraw-Hill [Google Scholar]
- CPLEX. http://www.ilog.com/products/cplex/
- Dahlhaus E, Johnson DS, Papadimitriou CH, Seymour PD, Yannakakis M (1994) The complexity of multiterminal cuts. SIAM J Comput 23: 864–894 [Google Scholar]
- de Lichtenberg U, Jensen LJ, Brunak S, Bork P (2005) Dynamic complex formation during the yeast cell cycle. Science 307: 724–727 [DOI] [PubMed] [Google Scholar]
- Deng M, Tu Z, Sun F, Chen T (2004) Mapping Gene Ontology to proteins based on protein–protein interaction data. Bioinformatics 20: 895–902 [DOI] [PubMed] [Google Scholar]
- Deng M, Zhang K, Mehta S, Chen T, Sun F (2003) Prediction of protein function using protein–protein interaction data. J Comput Biol 10: 947–960 [DOI] [PubMed] [Google Scholar]
- Dunn R, Dudbridge F, Sanderson CM (2005) The use of edge-betweenness clustering to investigate biological function in protein interaction networks. BMC Bioinformatics 6: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30: 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S (2005) High-throughput two-hybrid analysis. The promise and the peril. FEBS J 272: 5391–5399 [DOI] [PubMed] [Google Scholar]
- Flannick J, Novak A, Srinivasan BS, McAdams HH, Batzoglou S (2006) Graemlin: general and robust alignment of multiple large interaction networks. Genome Res 16: 1169–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi TK, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, Sharma S, Pinkert S, Nagaraju S, Periaswamy B, Mishra G, Nandakumar K, Shen B, Deshpande N, Nayak R, Sarker M, Boeke JD, Parmigiani G, Schultz J, Bader JS, Pandey A (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38: 285–293 [DOI] [PubMed] [Google Scholar]
- Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636 [DOI] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M (2001) Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet 29: 482–486 [DOI] [PubMed] [Google Scholar]
- Girvan M, Newman ME (2002) Community structure in social and biological networks. Proc Natl Acad Sci USA 99: 7821–7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg DS, Roth FP (2003) Assessing experimentally derived interactions in a small world. Proc Natl Acad Sci USA 100: 4372–4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn A, Rahnenfuhrer J, Talwar P, Lengauer T (2005) Confirmation of human protein interaction data by human expression data. BMC Bioinformatics 6: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch D, Zien A, Zimmer R, Lengauer T (2002) Co-clustering of biological networks and gene expression data. Bioinformatics 18 (Suppl 1): S145–S154 [DOI] [PubMed] [Google Scholar]
- Hart GT, Ramani AK, Marcotte EM (2006) How complete are current yeast and human protein-interaction networks? Genome Biol 7: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan JA (1975) Clustering Algorithms. New York: Wiley [Google Scholar]
- Hartuv E, Shamir R (2000) A clustering algorithm based on graph connectivity. Inform Process Lett 76: 175–181 [Google Scholar]
- Hartwell LH, Hopfield JJ, Leibler S, Murray AW (1999) From molecular to modular cell biology. Nature 402: C47–C52 [DOI] [PubMed] [Google Scholar]
- Haugen AC, Kelley R, Collins JB, Tucker CJ, Deng C, Afshari CA, Brown JM, Ideker T, Van Houten B (2004) Integrating phenotypic and expression profiles to map arsenic-response networks. Genome Biol 5: R95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishigaki H, Nakai K, Ono T, Tanigami A, Takagi T (2001) Assessment of prediction accuracy of protein function from protein–protein interaction data. Yeast 18: 523–531 [DOI] [PubMed] [Google Scholar]
- Hollunder J, Beyer A, Wilhelm T (2005) Identification and characterization of protein subcomplexes in yeast. Proteomics 5: 2082–2089 [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK (2003) Global analysis of protein localization in budding yeast. Nature 425: 686–691 [DOI] [PubMed] [Google Scholar]
- Ideker T, Ozier O, Schwikowski B, Siegel AF (2002) Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18 (Suppl 1): S233–S240 [DOI] [PubMed] [Google Scholar]
- Ihmels J, Friedlander G, Bergmann S, Sarig O, Ziv Y, Barkai N (2002) Revealing modular organization in the yeast transcriptional network. Nat Genet 31: 370–377 [DOI] [PubMed] [Google Scholar]
- Jansen R, Greenbaum D, Gerstein M (2002) Relating whole-genome expression data with protein–protein interactions. Genome Res 12: 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi T, Chen Y, Becker JM, Alexandrov N, Xu D (2004) Genome-scale gene function prediction using multiple sources of high-throughput data in yeast Saccharomyces cerevisiae. Omics 8: 322–333 [DOI] [PubMed] [Google Scholar]
- Karaoz U, Murali TM, Letovsky S, Zheng Y, Ding C, Cantor CR, Kasif S (2004) Whole-genome annotation by using evidence integration in functional-linkage networks. Proc Natl Acad Sci USA 101: 2888–2893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BP, Sharan R, Karp RM, Sittler T, Root DE, Stockwell BR, Ideker T (2003) Conserved pathways within bacteria and yeast as revealed by global protein network alignment. Proc Natl Acad Sci USA 100: 11394–11399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley R, Ideker T (2005) Systematic interpretation of genetic interactions using protein networks. Nat Biotechnol 23: 561–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AD, Przulj N, Jurisica I (2004) Protein complex prediction via cost-based clustering. Bioinformatics 20: 3013–3020 [DOI] [PubMed] [Google Scholar]
- Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, Rossant J, Hughes TR, Frey B, Emili A (2006) Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125: 173–186 [DOI] [PubMed] [Google Scholar]
- Krause R, von Mering C, Bork P (2003) A comprehensive set of protein complexes in yeast: mining large scale protein–protein interaction screens. Bioinformatics 19: 1901–1908 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643 [DOI] [PubMed] [Google Scholar]
- Kryshtafovych A, Venclovas C, Fidelis K, Moult J (2005) Progress over the first decade of CASP experiments. Proteins 61 (Suppl 7): 225–236 [DOI] [PubMed] [Google Scholar]
- LaCount DJ, Vignali M, Chettier R, Phansalkar A, Bell R, Hesselberth JR, Schoenfeld LW, Ota I, Sahasrabudhe S, Kurschner C, Fields S, Hughes RE (2005) A protein interaction network of the malaria parasite Plasmodium falciparum. Nature 438: 103–107 [DOI] [PubMed] [Google Scholar]
- Lanckriet GR, De Bie T, Cristianini N, Jordan MI, Noble WS (2004) A statistical framework for genomic data fusion. Bioinformatics 20: 2626–2635 [DOI] [PubMed] [Google Scholar]
- Lee H, Tu Z, Deng M, Sun F, Chen T (2006) Diffusion kernel-based logistic regression models for protein function prediction. Omics 10: 40–55 [DOI] [PubMed] [Google Scholar]
- Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P (2004) Coexpression analysis of human genes across many microarray data sets. Genome Res 14: 1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letovsky S, Kasif S (2003) Predicting protein function from protein/protein interaction data: a probabilistic approach. Bioinformatics 19 (Suppl 1): i197–i204 [DOI] [PubMed] [Google Scholar]
- Lord PW, Stevens RD, Brass A, Goble CA (2003) Investigating semantic similarity measures across the Gene Ontology: the relationship between sequence and annotation. Bioinformatics 19: 1275–1283 [DOI] [PubMed] [Google Scholar]
- Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, Gerstein M (2004) Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431: 308–312 [DOI] [PubMed] [Google Scholar]
- Maciag K, Altschuler SJ, Slack MD, Krogan NJ, Emili A, Greenblatt JF, Maniatis T, Wu LF (2006) Systems-level analyses identify extensive coupling among gene expression machines. Mol Syst Biol 2: 2006.0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D (1999) A combined algorithm for genome-wide prediction of protein function. Nature 402: 83–86 [DOI] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B (2002) MIPS: a database for genomes and protein sequences. Nucleic Acids Res 30: 31–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K, Weiss Y, Jordan M (1999) Loopy belief propagation for approximate inference: an empirical study. Proceedings of the 15th Conference on Uncertainty in Artificial Intelligence, p 479 [Google Scholar]
- Nabieva E, Jim K, Agarwal A, Chazelle B, Singh M (2005) Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics 21 (Suppl 1): i302–i310 [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Weston J, Cai J, Grundy WN (2001) Gene functional classification from heterogeneous data. Proceedings of the Fifth Annual International Conference on Computational Biology. Montreal, Quebec, Canada: ACM Press [Google Scholar]
- Pereira-Leal JB, Enright AJ, Ouzounis CA (2004) Detection of functional modules from protein interaction networks. Proteins 54: 49–57 [DOI] [PubMed] [Google Scholar]
- Prelic A, Bleuler S, Zimmermann P, Wille A, Buhlmann P, Gruissem W, Hennig L, Thiele L, Zitzler E (2006) A systematic comparison and evaluation of biclustering methods for gene expression data. Bioinformatics 22: 1122–1129 [DOI] [PubMed] [Google Scholar]
- Przulj N, Wigle DA, Jurisica I (2004) Functional topology in a network of protein interactions. Bioinformatics 20: 340–348 [DOI] [PubMed] [Google Scholar]
- Rives AW, Galitski T (2003) Modular organization of cellular networks. Proc Natl Acad Sci USA 100: 1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A (2005) Towards a proteome-scale map of the human protein–protein interaction network. Nature 437: 1173–1178 [DOI] [PubMed] [Google Scholar]
- Samanta MP, Liang S (2003) Predicting protein functions from redundancies in large-scale protein interaction networks. Proc Natl Acad Sci USA 100: 12579–12583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270: 467–470 [DOI] [PubMed] [Google Scholar]
- Scholtens D, Vidal M, Gentleman R (2005) Local modeling of global interactome networks. Bioinformatics 21: 3548–3557 [DOI] [PubMed] [Google Scholar]
- Schwikowski B, Uetz P, Fields S (2000) A network of protein–protein interactions in yeast. Nat Biotechnol 18: 1257–1261 [DOI] [PubMed] [Google Scholar]
- Scott J, Ideker T, Karp RM, Sharan R (2006) Efficient algorithms for detecting signaling pathways in protein interaction networks. J Comput Biol 13: 133–144 [DOI] [PubMed] [Google Scholar]
- Segal E, Friedman N, Kaminski N, Regev A, Koller D (2005) From signatures to models: understanding cancer using microarrays. Nat Genet 37 (Suppl): S38–S45 [DOI] [PubMed] [Google Scholar]
- Segal E, Wang H, Koller D (2003) Discovering molecular pathways from protein interaction and gene expression data. Bioinformatics 19 (Suppl 1): i264–i271 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan R, Ideker T (2006) Modeling cellular machinery through biological network comparison. Nat Biotechnol 24: 427–433 [DOI] [PubMed] [Google Scholar]
- Sharan R, Ideker T, Kelley B, Shamir R, Karp RM (2005) Identification of protein complexes by comparative analysis of yeast and bacterial protein interaction data. J Comput Biol 12: 835–846 [DOI] [PubMed] [Google Scholar]
- Simonis N, van Helden J, Cohen GN, Wodak SJ (2004) Transcriptional regulation of protein complexes in yeast. Genome Biol 5: R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spirin V, Mirny LA (2003) Protein complexes and functional modules in molecular networks. Proc Natl Acad Sci USA 100: 12123–12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen M, Petti A, Aach J, D'Haeseleer P, Church G (2002) Automated modelling of signal transduction networks. BMC Bioinformatics 3: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Sharan R, Shamir R (2002) Discovering statistically significant biclusters in gene expression data. Bioinformatics 18 (Suppl 1): S136–S144 [DOI] [PubMed] [Google Scholar]
- Tanay A, Sharan R, Kupiec M, Shamir R (2004) Revealing modularity and organization in the yeast molecular network by integrated analysis of highly heterogeneous genomewide data. Proc Natl Acad Sci USA 101: 2981–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Steinfeld I, Kupiec M, Shamir R (2005) Integrative analysis of genome-wide experiments in the context of a large high-throughput data compendium. Mol Syst Biol 1: 2005.0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E (2005) Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol 23: 137–144 [DOI] [PubMed] [Google Scholar]
- Tornow S, Mewes HW (2003) Functional modules by relating protein interaction networks and gene expression. Nucleic Acids Res 31: 6283–6289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda K, Shin H, Scholkopf B (2005) Fast protein classification with multiple networks. Bioinformatics 21 (Suppl 2): ii59–ii65 [DOI] [PubMed] [Google Scholar]
- Vazquez A, Flammini A, Maritan A, Vespignani A (2003) Global protein function prediction from protein–protein interaction networks. Nat Biotechnol 21: 697–700 [DOI] [PubMed] [Google Scholar]
- von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P (2002) Comparative assessment of large-scale data sets of protein–protein interactions. Nature 417: 399–403 [DOI] [PubMed] [Google Scholar]
- Wachi S, Yoneda K, Wu R (2005) Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics 21: 4205–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts DJ, Strogatz SH (1998) Collective dynamics of 'small-world' networks. Nature 393: 440–442 [DOI] [PubMed] [Google Scholar]
- Wu DD, Hu X (2005) An efficient approach to detect a protein community from a seed. 2005 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB 2005). La Jolla, CA, USA: IEEE pp. 135–141 [Google Scholar]
- Yeang CH, Mak HC, McCuine S, Workman C, Jaakkola T, Ideker T (2005) Validation and refinement of gene-regulatory pathways on a network of physical interactions. Genome Biol 6: R62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zien A, Kuffner R, Zimmer R, Lengauer T (2000) Analysis of gene expression data with pathway scores. Proc Int Conf Intell Syst Mol Biol 8: 407–417 [PubMed] [Google Scholar]