

Abstract
A synthesis of EPO 114–166 glycopeptide (1), presenting the O-linked glycophorin of erythropoietin, is described.
The naturally occurring glycoprotein, erythropoietin (EPO), is a 166-residue protein possessing four carbohydrate domains.1 In the context of a major initiative underway in our laboratory aimed at achieving the de novo total synthesis of homogeneous erythropoietin, we disclosed, in the preceding communication,2 the synthesis of the EPO 22–37 fragment displaying the mature N-linked dodecasaccharide domain. Herein, we describe the synthesis of the EPO 114–166 glycopeptide, presenting the glycophorin glycan at Ser126 (1).
A synthetic plan toward erythropoietin with a view toward optimal convergency was developed. The program would entail synthesis of the required peptide fragments, each of which would be equipped with a mature carbohydrate domain. These glycopeptides would then be merged by exploiting chemical ligation methods developed elsewhere,3 as well as in our laboratory.4 One of the target fragments, 1, containing the 53 amino acid residues from Ala114 to Arg166 with a glycophorin domain attached at Ser126, is depicted in Figure 1.
Figure 1.
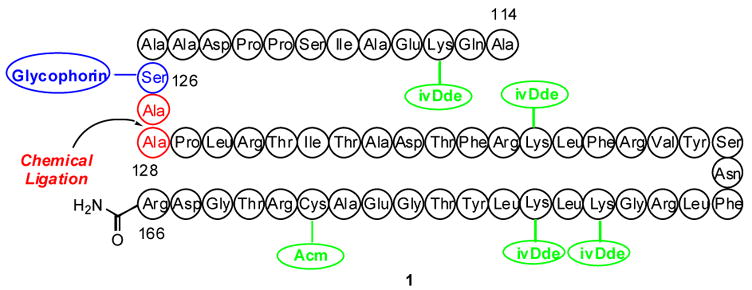
EPO 114–166 glycopeptide (1).
Several strategies might have been pursued toward the synthesis of 1. One obvious approach would involve utilizing the glycophorin-presenting serine as a cassette and preparing the entire fragment, from the C to N terminus, through iterative Fmoc-based solid phase peptide synthesis (SPPS). In the context of this complex target, however, the costs associated with the significant loss of the valuable serine glycosylamino acid (cf. 2) and the low overall yields typically associated with the preparation of such large peptide fragments in this manner rendered such a non-convergent strategy quite unattractive.
Rather, we favored an approach that would make use of glycopeptide ligation methods (including those developed in our laboratory),4b which can be employed to join together two peptide fragments, one of which bears an O-linked carbohydrate. Given the difficulties in achieving non-cysteine native chemical ligation with sterically hindered amino acids, we elected to disconnect 1 at the Ala127–Ala128 junction. We envisioned employing our newly developed cysteine-free chemical ligation techniques. In the event that this application proved to be non-feasible or impractical it would be possible to resort to a cysteine-based method, which would simp ly require a subsequent desulfurization to convert Cys to Ala.5
Our synthesis of the Ala114–Ala127 coupling fragment (7) would first require the preparation of ample quantities of the Ser-glycophorin glycosylamino acid (2). In light of the well-documented challenges associated with the selective appendage of O-linked carbohydrates to serine and threonine residues, we elected to employ the cassette approach, developed some years earlier in our laboratory in anticipation of just such a need.6 In the case at hand, the cassette method was successfully applied to reaching the glycophorin-presenting glycosylamino acid, wherein an Fmoc-masked serine benzyl ester would be O-linked to a galactosamine moiety at an early stage of the synthesis. This intermediate was ultimately advanced to the fully protected glycosylamino acid 2.6
In order to avoid compatibility issues associated with removal of the Fmoc group at a later stage in the synthesis, it was replaced with a Boc function, following the one-step protocol developed by Joullie (Scheme 1).7 Hydrogenation of the benzyl ester provided carboxylic acid 3. EDC and HOOBt-mediated amide coupling between 3 and 4 proceeded without epimerization.8 These results are in accord with those reported by Sakakibara, who observed that the use of HOOBt as an additive in peptide coupling reactions is superior to the more commonly employed HOBt reagent.9 Treatment of the resultant amide with 4M HCl in dioxane afforded the dipeptide amine HCl salt 5. The latter was successfully coupled with the fully protected polypeptide 6 (itself obtained through SPPS), in the presence of EDC and HOOBt in TFE and CHCl3, to afford the desired amide along with small amounts of TFE ester. The resultant compound was exposed to the action of 95% TFA and water to furnish glycopeptide 7, presenting the protected glycophorin domain.
Scheme 1.
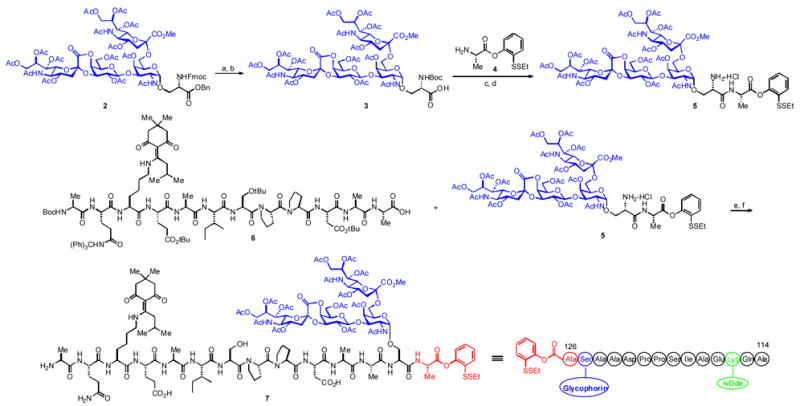
Synthesis of compound 7. a) Boc2O, KF, Et3N, DMF, b) H2, Pd/C, MeOH, 78% over two steps, c) EDC, HOOBt, DMF, CH2Cl2, 4, d) 4M HC l in dioxane, e) EDC, HOOBt, TFE, CHCl3, f) TFA, H2O, PhOH, TESH, 46% over four steps.
The next task was that of preparing the Ala128–Arg166 polypeptide coupling fragment, bearing a Tmb group on the N-terminus. Due care was taken to ensure that the Cys161 protecting group would be orthogonal to the functionality masking the Tmb thiol moiety. It was ultimately found that when the Cys161 thiol was masked as an S-acetamidomethyl group (Acm), the Tmb could be equipped with a 2,4-dinitrophenyl (DNP) group, which would be selectively removed upon exposure to sodium 2-mercapto-ethanesulfonate (MESNa) and K2CO3 in MeOH.10
Having devised what seemed to be a feasible protecting group strategy, we prepared the poylpeptide 8 using a peptide amide linker on a polystyrene resin (Scheme 2). Reductive amination between aldehyde 9 and peptide 8, following methodology which we had developed earlier,4b yielded 10, equipped with the Tmb auxiliary at Ala128. Treatment with MESNa exposed the required free thiophenol (see compound 11), which would couple with glycopeptide 7.
Scheme 2.
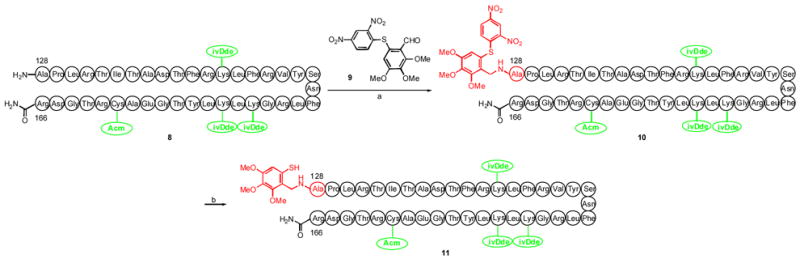
a) NaCNBH3, 9, MeOH, DMF, 63%, b) MESNa, K2CO3, MeOH, 88%.
In the event, the two peptides (7 and 11) were subjected to concurrent treatment with TCEP in DMF, fo llowed by sodium phosphate. We were expecting to exploit our recently established O→S migration, upon cleavage of the disulfide linkage in 7.11 It was presumed that the O→S intramolecular transacylation would be followed by an second trans acylation (intermolecular thioester exchange) with the liberated thiol linkage 11, generated from 10. The process advanced through another transacylation, (this time an intramolecular S→N migration), culminating in 12. Initially, two peaks corresponding to the desired molecular weight were observed early on by LC-MS. These were assumed to arise from the ligated adduct and the intermediate thioester through which the two peptide fragments were temporarily joined prior to the actual ligation event, (i.e. S→N migration). After 24 h at room temperature, only one peak remained. The product was isolated and confirmed to be the desired amide (12), as opposed to the thioester intermediate. Interestingly, the terminating intramolecular S→N migration was found to be reversible, as evidenced by the fact that the coupled thioester reappeared upon treatment with 95% trifluoroacetic acid.4b In order to block the undesired reverse N→S acyl migration pathway, the thio l group of the ligated product was selectively methylated to provide 13. We note that the successful methylation of intermediate 12 provides further evidence for the assignment of the latter as an amide bearing a free thio l functionality.
In order to demonstrate the compatibility of the glycan moiety with standard auxiliary removal conditions, the methylated glycopeptide 13 was subjected to 95% TFA and 5% TIPSH for 2 h (Scheme 3). Following removal of solvents and treatment with phosphate buffer, the target glycopeptide 1 was isolated intact, without loss of the glycophorin domain.
Scheme 3.
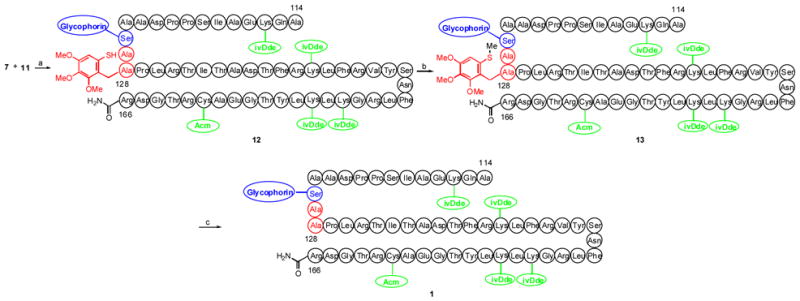
a) TCEP, DMF, Na2HPO4, 57%, b) methyl 4-nitrobenzenesulfonate, PBS, 7.7, 81%, c) TFA, TIPSH.
In summary, we have described herein the preparation of the Ala114–Arg166 fragment of EPO, bearing the requis ite glycophorin domain by strictly chemical synthesis. This disclosure, in concert with the preceding manuscript, provides encouragement that the EPO goal might be reached and offers a futuristic view of how one might assemble designed glycopeptides, and even glycoproteins, by synthesis through the powers of organic chemistry.
Acknowledgments
This work was supported by the NIH (CA28824 to SJD). We thank Dr. George Sukenick, Ms. Sylvi Rusli and Ms. Hui Fang of the Sloan-Kettering Institute’s NMR core facility for mass spectral and NMR spectroscopic analysis (SKI core grant no.: CA02848). Postdoctoral fellowship support is gratefully acknowledged by BW (New York State Department of Health, New York State Breast Cancer Research and Education Fund).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lai P, Everett R, Wang F, Arakawa T, Goldwasser E. J Biol Chem. 1986;261:3116–3121. [PubMed] [Google Scholar]
- 2.Wu B, Tan Z, Chen G, Chen J, Hua Z, Wan Q, Ranganathan K, Danishefsky SJ. Tetrahedron Lett. 2006;47 doi: 10.1016/j.tetlet.2006.09.045. Preceding Communication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Offer J, Boddy CNC, Dawson PE. J Am Chem Soc. 2002;124:4642–4646. doi: 10.1021/ja016731w. [DOI] [PubMed] [Google Scholar]; b) Offer J, Dawson PE. Org Lett. 2000;2:23–26. doi: 10.1021/ol991184t. [DOI] [PubMed] [Google Scholar]
- 4.a) Warren JD, Miller JC, Keding SJ, Danishefsky SJ. J Am Chem Soc. 2004;126:6576–6578. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]; b) Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ. Angew Chem Int Ed. 2006;45:4116–4125. doi: 10.1002/anie.200600538. [DOI] [PubMed] [Google Scholar]; c) Chen J, Warren JD, Wu B, Chen G, Wan Q, Danishefsky SJ. Tetrehedron Lett. 2006;47:1969–1972. [Google Scholar]; d) Wu B, Warren JD, Chen J, Chen G, Hua Z, Danishefsky SJ. Tetrehedron Lett. 2006;47:5219–5223. [Google Scholar]
- 5.Yan LZ, Dawson PE. J Am Chem Soc. 2001;123:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 6.Schwarz JB, Kuduk SD, Chen XT, Sames D, Glunz PW, Danishefsky SJ. J Am Chem Soc. 1999;121:2662–2673. [Google Scholar]
- 7.Li WR, Jiang J, Joullie MJ. Tetrehedron Lett. 1993;34:1413–1415. [Google Scholar]
- 8.a) Kuroda H, Chen YN, Kimura T, Sakakibara S. Int J Pept Protein Res. 1992;40:294. doi: 10.1111/j.1399-3011.1992.tb00304.x. [DOI] [PubMed] [Google Scholar]; b) Sakakibara S. Biopolymers. 1995;37:17. doi: 10.1002/bip.360370105. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Jiang X, Ye Y, Fan C, Romoff T, Goodman M. Org Lett. 1999;1:91–93. doi: 10.1021/ol990573k. [DOI] [PubMed] [Google Scholar]
- 10.Halcomb RL, Wittman MD, Olson SH, Danishefsky SJ, Golik J, Wong H, Vyas D. J Am Chem Soc. 1991;113:5080–5082. [Google Scholar]
- 11.Chen G, Warren JD, Chen J, Wu B, Wan Q, Danishefsky SJ. J Am Chem Soc. 2006;128:7460–7462. doi: 10.1021/ja061588y. [DOI] [PubMed] [Google Scholar]