

Abstract
Angiogenesis is a prerequisite for solid tumor growth. Glioblastoma multiforme, the most common malignant brain tumor, is characterized by extensive vascular proliferation. We previously showed that transgenic mice expressing a GFAP-v-src fusion gene in astrocytes develop low-grade astrocytomas that progressively evolve into hypervascularized glioblastomas. Here, we examined whether tumor progression triggers angiogenetic signals. We found abundant transcription of vascular endothelial growth factor (VEGF) in neoplastic astrocytes at surprisingly early stages of tumorigenesis. VEGF and v-src expression patterns were not identical, suggesting that VEGF activation was not only dependent on v-src. Late-stage gliomas showed perinecrotic VEGF up-regulation similarly to human glioblastoma. Expression patterns of the endothelial angiogenic receptors flt-1, flk-1, tie-1, and tie-2 were similar to those described in human gliomas, but flt-1 was expressed also in neoplastic astrocytes, suggesting an autocrine role in tumor growth. In crossbreeding experiments, hemizygous ablation of the tumor suppressor genes Rb and p53 had no significant effect on the expression of VEGF, flt-1, flk-1, tie-1, and tie-2. Therefore, expression of angiogenic signals is an early event during progression of GFAP-v-src tumors and precedes hypervascularization. Given the close similarities in the progression pattern between GFAP-v-src and human gliomas, the present results suggest that these mice may provide a useful tool for antiangiogenic therapy research.
Angiogenesis is defined as the sprouting of capillaries from existing blood vessels. With the exception of the female reproductive cycles, angiogenesis takes place in adult organisms mainly in pathological situations such as wound healing and tumor growth. 1 It has been suggested that tumor progression and metastasis require persistent new blood vessel growth and that manipulating the “angiogenic switch” is an important feature that a tumor must acquire for successful growth. 2 Some important evidence for this hypothesis was derived from transgenic mouse models of cancer. For example, mice expressing the SV 40 large T antigen in pancreatic β cells exhibit a stage-dependent onset of pathological angiogenesis during tumor development. 3,4 Although all β cells express the transgene, only 50% of the islets switch to a hyperproliferative stage (CIS). Angiogenesis is turned on in a small fraction of hyperplastic islets and facilitates progression to large solid tumor formation. 5 Similar observations were made in various other transgenic models for cancer. 6-8
Vessel density in invasive human cancers is clearly a significant prognostic indicator, in both breast 9 and prostate 10 cancers. Prognosis deteriorates with increasing density of blood vessels. In human gliomas, the presence of microvascular hyperproliferations is suggested to be the most important single predictor of survival. 11 Low-grade gliomas (WHO grade II), 12 which consist of highly differentiated neoplastic astrocytes and sparse blood vessel growth, have a mean survival time of several years, whereas higher-grade gliomas (WHO grade III) with highly proliferative cells have a prognosis of 2 to 5 years. 11 This is in stark contrast to glioblastoma multiforme (WHO grade IV), which is defined by the presence of extensive microvascular proliferations and necroses and has a mean survival time of less than 1 year. 13 Because pathological angiogenesis is such an ominous sign in human gliomas, glioblastoma multiforme may be an ideal candidate for antiangiogenic therapy. 14
A large body of direct evidence incriminates vascular endothelial growth factor (VEGF) as a central mediator of tumor-induced angiogenesis in gliomas in vivo. For example, tumor growth is inhibited by neutralizing antibodies against VEGF 15 by retrovirus-mediated gene transfer of a dominant-negative VEGF receptor (flk-1) mutant that inhibits its signaling pathway 16 and by expression of antisense VEGF RNA in tumor cells. 17 In human brain tumors, VEGF expression significantly correlates with the density of counted vessels, and thus represents a useful marker for glioma angiogenesis. 18
We have generated a transgenic mouse model for astrocytoma by expressing the viral v-src tyrosine kinase under control of the glial fibrillary acidic protein (GFAP) regulatory elements in astrocytes. 19 Abnormal astrogliosis and dysplastic changes were observed frequently, and within the first year of life 20% of the mice developed overt astrocytoma in the brain and in the spinal cord (Maddalena A and Aguzzi A, unpublished results). We have defined dysplasia in this context as abnormal accumulation of nondividing astrocytes without frank tumorigenesis. This preneoplastic stadium is obvious in all GFAP-v-src mice older than 2 weeks. While early lesions were histologically consistent with low-grade astrocytoma, at later stages most tumors were highly mitotic and frankly malignant. Late-stage tumors showed, in addition to cellular polymorphism and high proliferative activity, numerous highly vascularized areas with cells palisading around necroses, thereby mimicking the morphological characteristics of human glioblastoma multiforme. The stochastic character of tumorigenesis in GFAP-v-src mice is compatible with a multistep model carcinogenesis to which additional genetic lesions concur with v-src.
The characteristics described above suggest that GFAP-v-src transgenic mice may represent a useful model for testing antiangiogenic therapies. We therefore investigated the expression patterns of molecules involved in angiogenesis at various time points during malignant progression in astrocytomas derived from these mice. In situ hybridization was used to assess the transcription of VEGF, its receptors flt-1 and flk-1, and the endothelial-specific transmembrane receptor tyrosine kinases tie-1 and tie-2 (which act as receptors for angiopoietin-1) in early neoplastic lesions as well as in compact, frankly malignant tumors. Because inactivation of cell cycle regulatory genes seem to be an early genetic event in the development and progression of gliomas 20 we intercrossed GFAP-v-src transgenic mice with mice hemizygous for the tumor suppressor genes p53 and Rb and investigated the angiogenetic parameters described above in double mutant mice. This allowed us to ask whether p53 and Rb may have an effect on the angiogenic switch in our model of gliomagenesis.
Materials and Methods
Animals and Tissue Specimens
Neoplastic and non-neoplastic brain tissues from GFAP-v-src transgenic mice, GFAP-v-src × p53+/−21, and GFAP-v-src × Rb+/−22 double transgenic mice were analyzed. 19 Animals were sacrificed by CO2 overdose, brains were removed, fixed overnight in 4% (w/v) paraformaldehyde in phosphate-buffered saline, and embedded in paraffin. Two-μm sections were mounted on silanized slides and stained with hematoxylin and eosin (H&E). For each genotype, we chose samples from three low-grade and three high-grade lesions according to the following features: low-grade lesions had only very few mitotic figures, nuclear atypia, displayed diffuse infiltration of the surrounding brain parenchyme, and could not easily be distinguished from the surrounding tissue thus reflecting characteristics of human low-grade astrocytoma (WHO grade II). In addition, those early lesions, which had a small diameter (<1.5 mm), already showed cellular pleomorphism and small foci of endothelial proliferation. Malignant high-grade lesions consisted of compact tumor masses (diameter >2 mm) with higher mitotic activity, nuclear atypia, and cellular pleomorphism according to human anaplastic astrocytoma (WHO grade III). Some of them additionally had areas with extensive vascular proliferations and/or necroses, thus exhibiting characteristics of human glioblastoma multiforme (WHO grade IV). 23 High-grade lesions consistently showed higher cellular density than low-grade lesions.
In Situ Hybridization
Sense and antisense RNA probes were transcribed in vitro with T3 and T7 RNA polymerase from linearized pBluescript vectors carrying cDNAs for v-src (700 bp), GFAP 24 (1200 bp), VEGF 25 (600 bp), flk-1 26 (2600 bp), flt-1 27 (2000 bp), tie-1 (600 bp), and tie-2 (1200 bp) 28 in the presence of digoxigenin-11-dUTP (Boehringer Mannheim). As described, 400 to 800 ng of labeled transcripts (0.6 to 2.6 kb) were hybridized to tissue sections at 65°C. 29 tie-2 probes were hybridized at 60°C. Digoxigenin was detected with alkaline phosphatase-labeled anti-digoxigenin Fab fragments and 4-nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate (Boehringer Mannheim). Coverslips were mounted with glycerol gelatin.
Results
We investigated the transcription pattern of VEGF, flk-1, flt-1, tie-1, and tie-2 by in situ hybridization in 20 different tissue samples. The morphological features of the neoplastic lesions were analyzed and correlated to the in situ hybridization analysis (Table 1) ▶ .
Table 1.
Histological Characterization of the Tissue Samples Generated in GFAP-v-src, GFAP-v-src × p53+/− and GFAP-v-src × Rb+/− Transgenic Mice by H&E Staining and GFAP in Situ Hybridization
| Sample | Malignancy | Genotype | Age (weeks) | Mitotic figures† | Growth pattern | Neovascularization‡ | Necroses§ | GFAP MRNA |
|---|---|---|---|---|---|---|---|---|
| 1 | — | Wild-type | 6 | − | − | − | − | + |
| 2 | LG | Src | 4 | − | D | + | − | ++ |
| 3 | LG | Src | 4 | − | D | + | − | ++ |
| 4 | LG | Src | 4 | − | D | + | − | ++ |
| 5 | LG-HG | Src | 14 | + | D-C | + | − | ++ |
| 6 | LG | Src × Rb | 20 | − | D | + | − | ++ |
| 7 | LG | Src × Rb | 20 | − | D | + | − | ++ |
| 8 | LG | Src × Rb | 4 | − | D | + | − | ++ |
| 9 | LG | Src × p53 | 4 | − | D | + | − | ++ |
| 10 | LG | Src × p53 | 12 | ++ | D | + | − | ++ |
| 11 | LG-HG | Src × p53 | 4 | + | D-C | + | − | ++ |
| 12 | HG | Src | 11 | + | C | ++ | − | +++ |
| 13 | HG | Src | 10 | +++ | C | +++ | +++ | +++ |
| 14* | HG | Src | 20 | +++ | C | +++ | ++ | ++ |
| 15 | HG | Src × Rb | 30 | ++ | C | + | + | +++ |
| 16 | HG | Src × Rb | 9 | +++ | C | ++ | − | +++ |
| 17 | HG | Src × Rb | 16 | +++ | C | +++ | ++ | +++ |
| 18 | HG | Src × p53 | 36 | + | C | + | ++ | ++ |
| 19 | HG | Src × p53 | 18 | + | C | ++ | + | ++ |
| 20 | HG | Src × p53 | 46 | +++ | C | ++ | − | +++ |
src, Rb, and p53 mice had a hemizygous genotype for the respective gene. All samples were from different individual mice.
20 different tissue samples from nonmalignant wild-type brain, low-grade (LG) and high-grade (HG) gliomas were analyzed. One subcutaneous tumor was localized in the neck (*). Neoplastic lesions were either growing as compact masses (C), diffusely infiltrating (D), or presented a combination of both growth patterns (D-C).
† −, Normal mitotic activity; +, slightly elevated mitotic activity; ++, moderate elevated mitotic activity; +++, numerous mitotic figures.
‡ +, very few and small foci of neovascularization; ++, several larger foci of neovascularization; +++, areas with extensive microvascular proliferation.
§ −, no necrotic areas; +, 1 to 4 necrotic areas; ++, 5 to 10 necrotic areas; +++, over 11 necrotic areas (diameter >0.25 mm). For GFAP mRNA evaluation see Table 2 ▶ .
We discovered incipient abnormal endothelial proliferations in all of the low-grade lesions. However, these were very small and were present only in few foci per lesion (Figure 1a) ▶ . In contrast, most of the high-grade tumors had large areas with extensive microvascular proliferation (Figure 1b) ▶ . To unequivocally localize neoplastic cells and also to provide an internal quality control for mRNA, we performed additional in situ hybridizations for GFAP mRNA on consecutive sections. We found that all neoplastic lesions expressed abundant amounts of GFAP irrespective of their degree of anaplasia (Figure 2, c and d) ▶ .
Figure 1.

Vascular pattern in low-grade (a) and high-grade (b) gliomas of GFAP-v-src transgenic mice. Low-grade lesion were well vascularized (arrows) but showed very little or no endothelial proliferations. In contrast, high-grade lesions exhibited distinct areas with extensive microvascular proliferation (arrows). Original magnification, ×158.
Figure 2.
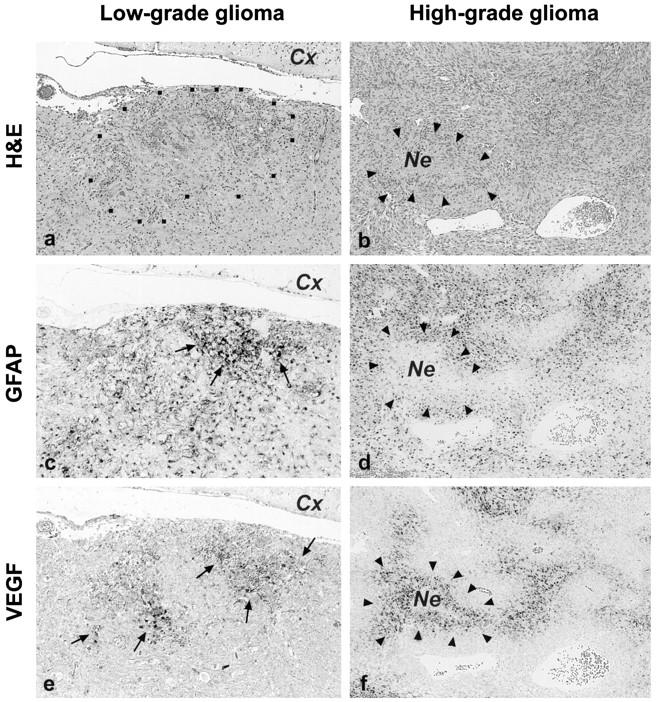
Low-grade (a, c, and e) and high-grade (b, d, and f) neoplastic lesions of GFAP-v-src transgenic mice: H&E stains (a and b) and in situ hybridization for GFAP (c and d) and for VEGF (e and f). Low-grade tumors consisted of small (<1.5 mm) nodular neoplastic lesions that diffusely infiltrated the surrounding brain parenchyme (a, region enclosed by dots). Neoplastic astrocytes showed strong GFAP mRNA expression when compared with the surrounding tissue (c, arrows) . In the cerebral cortex adjacent to the tumor (c; Cx), only a few reactive astrocytes were GFAP-positive, whereas neurons and other neuroectodermal cells failed to express GFAP (thus confirming the specificity of the GFAP in situ hybridization reaction). High-grade lesions were large tumors, some of them showed necrotic areas (Ne) surrounded by palisading cells (arrowheads). In situ hybridization for VEGF on consecutive sections (e and f) showed extensive transcription of the VEGF gene in clusters of neoplastic astrocytes of low-grade lesions (e, arrows). In contrast, neither brain tissue surrounding the neoplasms (peripheral areas of e), nor normal mouse brain control tissues (not shown) showed VEGF mRNA. High-grade tumors expressed the highest levels of VEGF mRNA in palisading cells surrounding necrotic foci (f, arrowheads), in areas of low vascular density, and at the periphery of the tumor (not shown). Original magnification, ×75.
VEGF, flt-1, and flk-1 Expression
In all low-grade lesions, we found extensive expression of VEGF in neoplastic astrocytes, whereas neither brain tissue surrounding the neoplasms, nor control tissues from normal mouse brains (data not shown), showed any detectable VEGF mRNA (Figure 2e) ▶ . In some areas of early neoplastic lesions, the expression pattern of VEGF and v-src were almost identical (Figure 3, a and b) ▶ , whereas in others, VEGF and v-src expression appeared to be independent from each other. We found both areas in which VEGF was transcribed in the absence of v-src (Figure 3, c and d) ▶ , and vice versa (Figure 3, e and f) ▶ .
Figure 3.
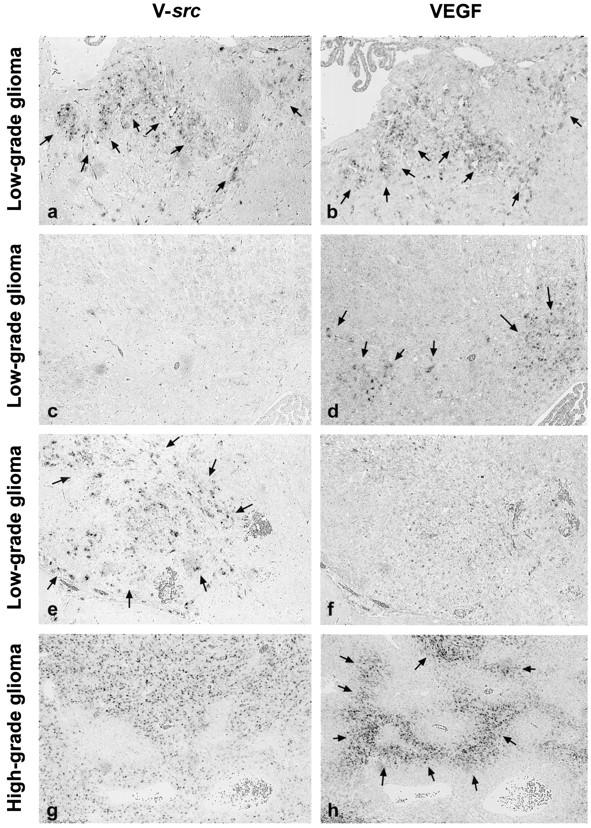
Comparative in situ hybridization analysis of the expression patterns of v-src (a, c, e, and g) and of VEGF (b, d, f, and h) on consecutive sections. In low-grade lesions, we found certain areas with superimposable spatial expression of v-src and VEGF (a and b, arrows). However, other areas displayed expression of only VEGF or only v-src (c-f, arrows). In high-grade tumors, v-src transcription showed a homogeneous expression pattern throughout the tumor without any specific relationship to its histoarchitecture (g). In contrast, VEGF up-regulation was most prominent in astrocytes palisading around necrotic areas (h, arrows; see Figure 2 ▶ ), indicating that the regulatory mechanisms of VEGF transcription active in human gliomas were conserved in GFAP-v-src tumors. Original magnification, ×75.
In high-grade neoplasms, extensive up-regulation of VEGF was evident in tumor cells palisading around necroses (Figure 2f) ▶ at areas with low vascular density and also at the periphery of each tumor (data not shown). In contrast to VEGF, v-src transcription showed a homogeneous expression pattern throughout the tumor without any specific relationship to its topography (Figure 3 ▶ , g and h).
We found that flt-1 (the receptor of VEGF, also named VEGFR-1) was expressed in a dot-like pattern in normal endothelial cells of mouse brains, especially in cells lining small vessels and capillaries (Figure 4a) ▶ . The same pattern was seen in non-neoplastic brain parenchyme of transgenic mice (data not shown). In some low-grade lesions, we found additional up-regulation of flt-1 in endothelial cells of newly formed, sprouting capillaries (Figure 4e) ▶ with no significant relationship to the areas of VEGF expression. Most of the highly malignant tumors showed strong staining for flt-1, especially in small vessels and capillaries and also at the periphery of the tumor, whereas larger vessels were only weakly stained (Figure 4i) ▶ . In one sample of a glioblastoma-like tumor there was additional expression of flt-1, flk-1, tie-1, and tie-2 in endothelial cells in the non-neoplastic tissue adjacent to the tumor (not shown).
Figure 4.
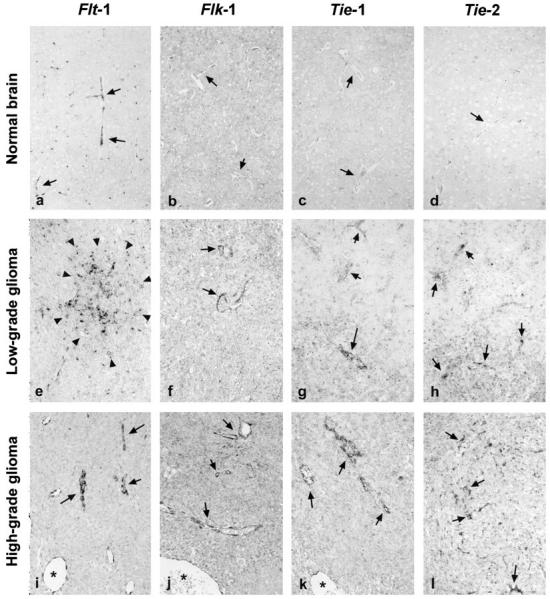
In situ hybridization with antisense probes for the VEGF receptors flt-1 and flk-1, the orphan/angiopoietin receptors tie-1 and tie-2 in normal mouse brain (a-d), and low-grade (e-h) and high-grade (i-l) neoplastic lesions. In normal mouse brains, flt-1 was expressed in a punctuate pattern exclusively endothelial cells (see arrows in Figure 3a ▶ ), whereas no flk-1, tie-1, or tie-2 mRNA could be detected (b-d, arrows). A similar pattern of expression was detected in non-neoplastic brain parenchyme of GFAP-v-src transgenic mice (not shown). A number of low-grade lesions expressed sizeable amounts of flk-1 and, to a lesser extent, of tie-1 and tie-2. In addition, we observed further up-regulation of flt-1 in endothelial cells of newly formed, sprouting vessels (e; see Figure 5 ▶ ). Moreover, we found flt-1 mRNA also in neoplastic astrocytes of most low-grade lesions (e, arrowheads; see Figure 5 ▶ ). In high-grade tumors, most small vessels and capillaries showed strong staining for flt-1 and flk-1 and to a lesser extent for tie-1 and tie-2 (i-l, arrows), whereas larger vessels were only weakly stained (see asterisks in Figure 3, i, j, and k ▶ ). Original magnification, ×158.
Surprisingly, we found expression of flt-1 also in neoplastic astrocytes in a proportion of low-grade lesions (Figure 5, a and b) ▶ . This finding was consistently reproduced in six of nine tumors analyzed.
Figure 5.

Details of the tumor shown in Figure 4e ▶ illustrating flt-1 mRNA expression in endothelial cells of sprouting vessels (arrows) as well as, to a lesser extent, in neoplastic astrocytes of low-grade lesions (arrowheads). Original magnification, ×500.
In contrast to flt-1, we found no or only very little expression of flk-1 (also named VEGFR-2) in normal brain tissue as well as in non-neoplastic tissue of transgenic mice (Figure 4b) ▶ . Some low-grade lesions clearly expressed flk-1 on endothelial cells of small and larger vessels but, in contrast to flt-1, failed to express on anaplastic astrocytes (Figure 4f) ▶ . In highly malignant lesions, the expression pattern of flk-1 in endothelial cells was similar to flt-1 but with even higher affinity to small vessels (Figure 4j) ▶ . There was no obvious topographical relationship of the presence of flk-1 mRNA to expression of VEGF (not shown).
tie-1 and tie-2 Expression
tie-1 and tie-2, both receptor tyrosine kinases known to be involved in embryonal angiogenesis 28 showed no expression in adult brain tissue (Figure 4, c and d) ▶ . Also, only few low-grade lesions exhibited expression of tie-1 or tie-2 on endothelial cells (Figure 4, g and h) ▶ . We found strong expression of both receptor mRNAs only in glioblastoma multiforme-like tumors (Figure 4, k and l) ▶ . As in the case of flt-1 and flk-1, endothelial cells of small vessels displayed a stronger staining for tie-1 and tie-2 than the larger ones. In general, we detected a stronger signal in endothelial cells for tie-1 than for tie-2.
Effect of p53+/− and Rb+/− on the Expression Pattern of Angiogenic Molecules
We were interested in potential effects of loss of the tumor suppressor genes, Rb and p53, on angiogenesis. We found that the neoplastic lesions arising in GFAP-v-src × p53+/− and in GFAP-v-src × Rb+/− double transgenic mice were morphological identical to those of GFAP-v-src mice (Maddalena A, Hainfellner J, and Aguzzi A, unpublished observations). In situ hybridization analysis of these neoplastic lesions revealed no significant alteration in the expression pattern of VEGF, flt-1, flk-1, tie-1, and tie-2 (Table 2) ▶ . Neither the localization nor the quantity of mRNA during malignant progression differed from those of GFAP-v-src single transgenic mice.
Table 2.
Distribution of the Expression of VEGF, flt-1, flk-1, tie-1, and tie-2 mRNA in Normal Brain, Low-Grade Lesions and High-Grade Tumors of GFAP-v-src Transgenic Mice, and in GFAP-v-src × p53+/− and GFAP-v-src × Rb+/− Double Transgenic Mice
| Sample | VEGF | flt-1 | flk-1 | tie-1 | tie-2 |
|---|---|---|---|---|---|
| 1 | − | + | − | − | − |
| 2 | ++ | ++* | + | + | + |
| 3 | ++ | ++* | + | − | − |
| 4 | ++ | + | + | − | − |
| 5 | ++ | ++* | + | + | − |
| 6 | + | n.d. | − | n.d. | n.d. |
| 7 | ++ | +* | ++ | − | − |
| 8 | ++ | + | − | − | − |
| 9 | n.d. | ++ | + | n.d. | + |
| 10 | ++ | +* | + | − | + |
| 11 | ++ | +* | + | − | − |
| 12 | ++ | +++ | ++ | + | − |
| 13 | +++ | +++ | +++ | ++ | + |
| 14 | ++ | +++ | +++ | ++ | ++ |
| 15 | +++ | ++ | n.d. | + | − |
| 16 | ++ | n.d. | ++ | ++ | + |
| 17 | +++ | + | + | + | + |
| 18 | ++ | +++ | ++ | + | − |
| 19 | +++ | ++ | +++ | − | + |
| 20 | ++ | + | + | + | + |
v-src and VEGF were expressed in neoplastic astrocytes, whereas flt-1, flk-1, tie-1, and tie-2 were expressed only in endothelial cells. Surprisingly, we found that some low-grade gliomas expressed flt-1 also in neoplastic astrocytes (*).
Semiquantitative evaluation of expression: −, no expression detectable; +, weak expression, at the threshold of detectability; ++, clearly detectable expression; +++, strong expression; n.d., not determined.
Discussion
Pathological angiogenesis is a crucial but poorly understood aspect of astrocytoma biology. Detailed knowledge of the interactions between angiogenic factors and their receptors, and particularly of their timing, during malignant progression may lead to improved imaging and therapy of human gliomas. Indeed, several studies have suggested a significant role for VEGF in tumor angiogenesis. 15,17,30
Before the various forms of antiangiogenic therapy can be attempted in humans, it is most desirable to assess their potential in a reliable animal model. The GFAP-v-src transgenic mice provide such a model because tumorigenesis in these mice recapitulates the sequence of events found in human “career gliomas”, ie, the progression of small pre-neoplastic foci to low-grade, slowly growing neoplastic lesions and finally to higher grade, rapidly growing glioblastoma-like tumors. 31
We have therefore analyzed the expression patterns of VEGF and its receptor tyrosine kinases flt-1 and flk-1 as well as the tie-1 and tie-2 receptor molecules in early neoplastic lesions and late end-stage gliomas in this transgenic mouse model for glioma. Our results strongly suggest that the angiogenic switch, defined as the abrupt up-regulation of molecules (growth factors and their receptors) capable of inducing abnormal vessel growth, is a very early event during malignant progression in astrocytoma produced by GFAP-v-src transgenic mice. In all early neoplastic lesions we found clusters of dysplastic astrocytes expressing VEGF mRNA.
In an earlier study, we had speculated that VEGF expression may be directly induced by v-src at this stage, 19 based on previous reports showing transactivation of the VEGF promoter by v-src in a dose-dependent manner in U87 glioblastoma cells. 32 However, although the expression pattern of VEGF and v-src mRNA was superimposable in some areas of low-grade lesions, other areas showed no correlation between the expression of these two genes. This may indicate that other mechanisms, in addition to direct transcriptional induction of VEGF by v-src, are likely to be involved in early VEGF expression. This phenomenon was even more evident in high-grade gliomas, in which VEGF mRNA was strongly up-regulated in tumor cells directly adjacent to necrotic foci, in the periphery of the tumors, and in areas of low vessel density. In contrast, v-src showed a uniform expression pattern throughout the tumors.
These findings are in agreement with previous reports showing enhanced VEGF mRNA transcription in pseudopalisading cells of human gliomas, ie, in those tumor cells that line areas of necrosis, as well as in areas of tumor infiltration into surrounding tissue. 14,33,34 Because VEGF levels are dramatically increased within a few hours of exposing different cell cultures to hypoxia, 35 it has been proposed that VEGF induction is driven by insufficient perfusion. Hypoxia-induced transcriptional activation is mediated by direct binding of hypoxia-inducible factor, to regulatory cis-acting enhancer elements located in the 5′-flanking region of the VEGF gene 36,37 and by increased RNA stability. 38 In addition to hypoxia, there may be other factors inducing VEGF up-regulation in high malignant tumors: for example, oncogenic Ras and hypoxia have been shown to synergistically up-regulate VEGF via hypoxia-inducible factor-1. 39 Other mechanisms of VEGF induction via glucose deprivation 40 and mutations in the VHL tumor suppressor gene 41 have been proposed. Because they are easily amenable to genetic and other manipulations, we hope that the GFAP-v-src transgenic mice will be of help in clarifying the role of these pathways in vivo.
Both VEGF receptor tyrosine kinases, flt-1 and flk-1, were expressed on endothelial cells in most of the low-grade lesions. In general flt-1 mRNA, which was found also on normal brain vasculature, was expressed to a greater extent in low-grade tumors than flk-1, again in strict analogy to the situation in human gliomas. 33,34 In highly malignant tumors, strong hybridization signals were observed in capillaries and small tumor vessels, and in one sample also in endothelial cells of tissues directly adjacent to the tumor. These findings suggest that VEGF acts specifically on those vessels that are primed to grow into the tumor from peripheral, unaffected tissue and are in agreement with observations made in human gliomas. 34
The factors that induce the expression of VEGF receptors in tumor endothelium are as yet unknown. It has been suggested that VEGF itself may induce expression of its receptors by means of a positive feed-back mechanism. 42 However, we found no significant correlation between areas of VEGF induction and the expression pattern of flt-1 and of flk-1, suggesting that other factors may be involved in flt-1/flk-1 induction. According to experiments with cultured cells, flt-1 is up-regulated by hypoxia in endothelial cells, and up-regulation is not prevented by addition of neutralizing antibodies against VEGF. 43
Apart from endothelial cells, we found flt-1 mRNA in neoplastic astrocytes, which, to the best of our knowledge, was not described before. Although in normal healthy organisms in which expression of flt-1 in vivo appears to be restricted to endothelial cells, there are reports demonstrating flt-1 expression in stromal cells of human hemangioblastoma 44 and in monocytes. 45 One could speculate that flt-1 is also up-regulated by hypoxia in dysplastic astrocytes and that stimulation by VEGF leads to tissue factor production and chemotaxis like in monocytes and endothelial cells. Tissue factor itself can also regulate VEGF expression and seems to be important for tumor angiogenesis as tumors expressing tissue factor antisense mRNA exhibit reduced angiogenesis. 46
The tie receptors comprise a novel family of endothelial-specific transmembrane receptor tyrosine kinases. 47 tie-1 and tie-2 are expressed in early stage of angiogenesis of the mouse, but the onset of their expression occurs somewhat later than that of flt-1 and flk-1. tie-1, whose ligand still is unknown, is required for the structural integrity of the endothelium, 48 whereas tie-2 and the recently identified ligands angiopoietin-1 and angiopoietin-2 are involved in vascular remodeling. 49,50 We found very little tie-1 and tie-2 mRNA in a few low-grade lesions, suggesting that the tie receptors may play a minor role in early angiogenesis. In high-grade tumors, a larger amount of tie-1 and tie-2 mRNA was identified, and again their expression was particularly pronounced in small vessels. A similar pattern of tie-1 expression was reported in human gliomas, 34 whereas tie-2 expression was not detectable in transplanted astrocytoma cell lines. 30
Alterations of the retinoblastoma (Rb) and p53 tumor suppressor genes have been reported in human gliomas. 20 It was proposed that progression of astrocytoma to glioblastoma involves clonal expansion of tumor cells carrying mutations of the p53 tumor suppressor gene. 51 Moreover, mutant p53 up-regulates VEGF in NIH 3T3 cells 52 and wild-type p53 protein enhances angiogenesis inhibitors, eg, TSP-1. 53,54 However, we did not find any significant changes in the transcription patterns of VEGF and any of the angiogenic receptor molecules in GFAP-v-src × p53+/− and in GFAP-v-src × Rb+/− double transgenic mice.
In summary, our results indicate that the angiogenic switch takes place at an early stage during the malignant progression of astrocytomas observed in GFAP-v-src transgenic mice. In addition, the expression pattern of VEGF, flt-1, flk-1, tie-1, and tie-2 is similar to what has been described in human gliomas. Therefore, we have reasons to hope that GFAP-v-src transgenic mice will provide a useful tool for deepening our insight into the molecular pathophysiology of glioma angiogenesis and will perhaps even claim a use in antiangiogenic therapy research.
Acknowledgments
We thank Marianne König and Beatrice Pfister for technical assistance, Dr. Joachim Weis for critical comments, and Norbert Wey for artwork.
Footnotes
Address reprint requests to Adriano Aguzzi, Institute of Neuropathology, Department of Pathology, Universitätsspital Zürich, Schmelzbergstrasse 12, CH-8091 Zürich, Switzerland. E-mail: adriano@pathol.unizh.ch.
Supported by grants of the Swiss National Foundation 31–40827.94 and of the Swiss Cancer League/Cancer League of the Kanton of Aarau (FOR673) and the Migros Foundation to A. Aguzzi.
Johannes Hainfellner’s current address is Neurologisches Institut, Neues allgemeines Krankenhaus 04 J, Währinger Gürtel 18–20, A-1090 Wien, Österreich.
Jakob Weissenberger’s current address is Universität Bern, Pathologisches Institut, Murtenstrasse 31, CH-3010 Bern, Switzerland.
References
- 1.Folkman J: Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995, 1:27-31 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogeneic switch during tumorigenesis. Cell 1996, 86:353-364 [DOI] [PubMed] [Google Scholar]
- 3.Christofori G, Naik P, Hanahan D: A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 1994, 369:414-418 [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D: Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 1985, 315:115-122 [DOI] [PubMed] [Google Scholar]
- 5.Folkman J, Watson K, Ingber D, Hanahan D: Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339:58-61 [DOI] [PubMed] [Google Scholar]
- 6.Lacey M, Alpert S, Hanahan D: Bovine papillomavirus genome elicits skin tumours in transgenic mice. Nature 1986, 322:609-612 [DOI] [PubMed] [Google Scholar]
- 7.Sippola Thiele M, Hanahan D, Howley PM: Cell-heritable stages of tumor progression in transgenic mice harboring the bovine papillomavirus type 1 genome. Mol Cell Biol 1989, 9:925-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arbeit JM, Munger K, Howley PM, Hanahan D: Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J Virol 1994, 68:4358-4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weidner N, Semple JP, Welch WR, Folkman J: Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N Engl J Med 1991, 324:1-8 [DOI] [PubMed] [Google Scholar]
- 10.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J: Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol 1993, 143:401-409 [PMC free article] [PubMed] [Google Scholar]
- 11.Daumas Duport C, Scheithauer B, O’Fallon J, Kelly P: Grading of astrocytomas: a simple and reproducible method. Cancer 1988, 62:2152-2165 [DOI] [PubMed] [Google Scholar]
- 12.Kleihues P, Burger PC, Scheithauer BW: Histological Typing of Tumours of the Central Nervous System. World Health Organization International Histological Classification of Tumours. 1993, Springer Verlag, Berlin Heidelberg
- 13.Plate KH, Breier G, Risau W: Molecular mechanisms of developmental and tumor angiogenesis. Brain Pathol 1994, 4:207-218 [DOI] [PubMed] [Google Scholar]
- 14.Plate KH, Breier G, Weich HA, Risau W: Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 1992, 359:845-848 [DOI] [PubMed] [Google Scholar]
- 15.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N: Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362:841-844 [DOI] [PubMed] [Google Scholar]
- 16.Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A: Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 1994, 367:576-579 [DOI] [PubMed] [Google Scholar]
- 17.Saleh M, Stacker SA, Wilks AF: Inhibition of growth of C6 glioma cells in vivo by expression of antisense vascular endothelial growth factor sequence. Cancer Res 1996, 56:393-401 [PubMed] [Google Scholar]
- 18.Takano S, Yoshii Y, Kondo S, Suzuki H, Maruno T, Shirai S, Nose T: Concentration of vascular endothelial growth factor in the serum and tumor tissue of brain tumor patients. Cancer Res 1996, 56:2185-2190 [PubMed] [Google Scholar]
- 19.Weissenberger J, Steinbach JP, Malin G, Spada S, Rulicke T, Aguzzi A: Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene 1997, 14:2005-2013 [DOI] [PubMed] [Google Scholar]
- 20.Tsuzuki T, Tsunoda S, Sakaki T, Konishi N, Hiasa Y, Nakamura M: Alterations of retinoblastoma, p53, p16(CDKN2), and p15 genes in human astrocytomas. Cancer 1996, 78:287-293 [DOI] [PubMed] [Google Scholar]
- 21.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A: Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356:215-221 [DOI] [PubMed] [Google Scholar]
- 22.Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H: Requirement for a functional Rb-1 gene in murine development (see comments). Nature 1992, 359:328-330 [DOI] [PubMed] [Google Scholar]
- 23.Kleihues P, Burger PC, Scheithauer BW: The new WHO classification of brain tumors. Brain Pathol 1993, 3:255-268 [DOI] [PubMed] [Google Scholar]
- 24.Lewis SA, Balcarek JM, Krek V, Shelanski M, Cowan NJ: Sequence of a cDNA clone encoding mouse glial fibrillary acidic protein: structural conservation of intermediate filaments. Proc Natl Acad Sci USA 1984, 81:2743-2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breier G, Albrecht U, Sterrer S, Risau W: Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development 1992, 114:521-532 [DOI] [PubMed] [Google Scholar]
- 26.Millauer B, Wizigmann Voos S, Schnurch H, Martinez R, Moller NP, Risau W, Ullrich A: High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72:835-846 [DOI] [PubMed] [Google Scholar]
- 27.Breier G, Clauss M, Risau W: Coordinate expression of vascular endothelial growth factor receptor-1 (flt-1) and its ligand suggests a paracrine regulation of murine vascular development. Dev Dyn 1995, 204:228-239 [DOI] [PubMed] [Google Scholar]
- 28.Sato TN, Qin Y, Kozak CA, Audus KL: tie-1 and tie-2 define another class of putative receptor tyrosine kinase genes expressed in early embryonic vascular system. Proc Natl Acad Sci USA 1993, 90:9355-9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marino S, Kretschmer C, Brandner S, Cavard C, Zider A, Briand P, Isenmann S, Wagner EF, Aguzzi A: Activation of HIV transcription by human foamy virus in transgenic mice. Lab Invest 1995, 73:103-110 [PubMed] [Google Scholar]
- 30.Millauer B, Longhi MP, Plate KH, Shawver LK, Risau W, Ullrich A, Strawn LM: Dominant-negative inhibition of Flk-1 suppresses the growth of many tumor types in vivo. Cancer Res 1996, 56:1615-1620 [PubMed] [Google Scholar]
- 31.Kleihues P, Soylemezoglu F, Schauble B, Scheithauer BW, Burger PC: Histopathology, classification, and grading of gliomas. Glia 1995, 15:211-221 [DOI] [PubMed] [Google Scholar]
- 32.Mukhopadhyay D, Tsiokas L, Sukhatme VP: Wild-type p53 and v-Src exert opposing influences on human vascular endothelial growth factor gene expression. Cancer Res 1995, 55:6161-6165 [PubMed] [Google Scholar]
- 33.Plate KH, Breier G, Weich HA, Mennel HD, Risau W: Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer 1994, 59:520-529 [DOI] [PubMed] [Google Scholar]
- 34.Hatva E, Kaipainen A, Mentula P, Jaaskelainen J, Paetau A, Haltia M, Alitalo K: Expression of endothelial cell-specific receptor tyrosine kinases and growth factors in human brain tumors. Am J Pathol 1995, 146:368-378 [PMC free article] [PubMed] [Google Scholar]
- 35.Shweiki D, Itin A, Soffer D, Keshet E: Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359:843-845 [DOI] [PubMed] [Google Scholar]
- 36.Damert A, Ikeda E, Risau W: Activator-protein-1 binding potentiates the hypoxia-induciblefactor-1-mediated hypoxia-induced transcriptional activation of vascular-endothelial growth factor expression in C6 glioma cells. Biochem J 1997, 327:419-423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL: Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 1996, 16:4604-4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikeda E, Achen MG, Breier G, Risau W: Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J Biol Chem 1995, 270:19761-19766 [DOI] [PubMed] [Google Scholar]
- 39.Mazure NM, Chen EY, Yeh P, Laderoute KR, Giaccia AJ: Oncogenic transformation and hypoxia synergistically act to modulate vascular endothelial growth factor expression. Cancer Res 1996, 56:3436-3440 [PubMed] [Google Scholar]
- 40.Shweiki D, Neeman M, Itin A, Keshet E: Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: implications for tumor angiogenesis. Proc Natl Acad Sci USA 1995, 92:768-772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wizigmann Voos S, Breier G, Risau W, Plate KH: Up-regulation of vascular endothelial growth factor and its receptors in von Hippel-Lindau disease-associated and sporadic hemangioblastomas. Cancer Res 1995, 55:1358-1364 [PubMed] [Google Scholar]
- 42.Breier G, Damert A, Plate KH, Risau W: Angiogenesis in embryos and ischemic diseases. Thromb Haemost 1997, 78:678-683 [PubMed] [Google Scholar]
- 43.Brogi E, Schatteman G, Wu T, Kim EA, Varticovski L, Keyt B, Isner JM: Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J Clin Invest 1996, 97:469-476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hatva E, Bohling T, Jaaskelainen J, Persico MG, Haltia M, Alitalo K: Vascular growth factors and receptors in capillary hemangioblastomas and hemangiopericytomas. Am J Pathol 1996, 148:763-775 [PMC free article] [PubMed] [Google Scholar]
- 45.Clauss M, Weich H, Breier G, Knies U, Rockl W, Waltenberger J, Risau W: The vascular endothelial growth factor receptor Flt-1 mediates biological activities: implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J Biol Chem 1996, 271:17629-17634 [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Deng Y, Luther T, Muller M, Ziegler R, Waldherr R, Stern DM, Nawroth PP: Tissue factor controls the balance of angiogenic and antiangiogenic properties of tumor cells in mice. J Clin Invest 1994, 94:1320-1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanahan D: Signaling vascular morphogenesis and maintenance. Science 1997, 277:48-50 [DOI] [PubMed] [Google Scholar]
- 48.Sato TN, Tozawa Y, Deutsch U, Wolburg Buchholz K, Fujiwara Y, Gendron Maguire M, Gridley T, Wolburg H, Risau W, Qin Y: Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376:70-74 [DOI] [PubMed] [Google Scholar]
- 49.Folkman J: Addressing tumor blood vessels. Nat Biotechnol 1997, 15:510. [DOI] [PubMed] [Google Scholar]
- 50.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD: Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277:55-60 [DOI] [PubMed] [Google Scholar]
- 51.Sidransky D, Mikkelsen T, Schwechheimer K, Rosenblum ML, Cavanee W, Vogelstein B: Clonal expansion of p53 mutant cells is associated with brain tumour progression. Nature 1992, 355:846-847 [DOI] [PubMed] [Google Scholar]
- 52.Kieser A, Weich HA, Brandner G, Marme D, Kolch W: Mutant p53 potentiates protein kinase C induction of vascular endothelial growth factor expression. Oncogene 1994, 9:963-969 [PubMed] [Google Scholar]
- 53.Dameron KM, Volpert OV, Tainsky MA, Bouck N: Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 1994, 265:1582-1584 [DOI] [PubMed] [Google Scholar]
- 54.Van Meir EG, Polverini PJ, Chazin VR, Su Huang HJ, de Tribolet N, Cavenee WK: Release of an inhibitor of angiogenesis upon induction of wild type p53 expression in glioblastoma cells. Nat Genet 1994, 8:171-176 [DOI] [PubMed] [Google Scholar]