

Abstract
We report here original data on the biological basis of prolonged neuromuscular paralysis caused by the toxic phospholipase A2 β-bungarotoxin. Electron microscopy and immunocytochemical labeling with anti-synaptophysin and anti-neurofilament have been used to show that the early onset of paralysis is associated with the depletion of synaptic vesicles from the motor nerve terminals of skeletal muscle and that this is followed by the destruction of the motor nerve terminal and the degeneration of the cytoskeleton of the intramuscular axons. The postjunctional architecture of the junctions were unaffected and the binding of fluorescein-isothiocyanate-conjugated α-bungarotoxin to achetylcholine receptor was not apparently affected by exposure to β-bungarotoxin. The re-innervation of the muscle fiber was associated by extensive pre- and post-terminal sprouting at 3 to 5 days but was stable by 7 days. Extensive collateral innervation of adjacent muscle fibers was a significant feature of the re-innervated neuromuscular junctions. These findings suggest that the prolonged and severe paralysis seen in victims of envenoming bites by kraits (elapid snakes of the genus Bungarus) and other related snakes of the family Elapidae is caused by the de-pletion of synaptic vesicles from motor nerve terminals and the degeneration of the motor nerve terminal and intramuscular axons.
The venoms of snakes are rich sources of phospholipases (PL)A2. Although the venom phospholipases probably have a primary digestive function, many of them are extremely toxic.
The most important toxic phospholipases of snake venoms are the neurotoxic/myotoxic phospholipases of the elapid snakes. These toxins are the dominant toxic fractions of the venoms of a number of snakes, including the kraits of Southeast Asia and the taipans of Australia, Papua-New Guinea, and Irian Jaya. 1,2
Envenoming bites by kraits and taipans are associated with a syndrome of neuromuscular paralysis that falls into three distinct phases. The first is a rapid-onset phase leading to profound paralysis within 30 to 60 minutes. The second is a stable phase of deep paralysis lasting for 2 to 3 days. The third is a recovery phase lasting 2 to 3 weeks. 3,4 The treatment of envenoming bites by kraits and taipans is extremely difficult. Severely envenomed patients respond poorly to antivenoms, anticholinesterases, and 2,4-diaminopyridines 3-7 and require prolonged periods of ventilatory support and intensive care. It is considered that the most likely toxic components causing long-lasting paralysis are the neurotoxic phospholipases A2.
The neurotoxic phospholipases are presynaptically active. 1,2 Their precise mechanism of action is not understood, 8-10 but consistent reports that the density of synaptic vesicles in nerve terminals is reduced when neuromuscular junctions are exposed to the presynaptically active toxins 11,12 and occasional reports of nerve terminal damage 13-15 suggested to us that damage to the motor innervation may be the primary cause of the prolonged paralysis caused by these toxins.
In this paper we report our observations on the neuropathology of peripheral neuromuscular systems in rats after the administration in vivo of sublethal doses of the principle toxin of the venom of kraits, the presynaptically active, toxic phospholipase A2 β-bungarotoxin (β-BTX). We show that the toxin causes the depletion of synaptic vesicles and the degeneration of the nerve terminal and intramuscular axons. As a result of our investigations we believe we have identified the primary pathological processes responsible for both the acute and chronic phases of neurotoxicity observed in humans envenomed by snakes whose venoms contain neurotoxic PLA2s.
Materials and Methods
Materials
β-BTX was obtained from Sigma Chemical Supplies (Poole, UK). Fluorescein isothiocyanate (FITC)-conjugated α-BTX was obtained from Molecular Probes (Eugene, OR). Mouse anti-synaptophysin monoclonal antibodies (SY 38) and rabbit anti-mouse rhodamine-labeled polyclonal antibodies were obtained from Dako (High Wycombe, UK). Mouse anti-neurofilament protein monoclonal antibodies (RT 97) were a gift from Dr. N. Leigh, Institute of Psychiatry, London, UK. The polyclonal antibody used to recognize junctional acetylcholinesterase (AChE) was a gift from Dr. J. Massoulie, Ecole Normale Supérieure, Paris, France. Vectashield aqueous mountant was supplied by Vector Laboratories (Burlingame, CA). Other reagents were obtained from regular commercial supplies and were routinely of the highest available quality.
Animals
Thirty female Wistar rats (90 to 100 g) and 11 female Balb/C mice (20 to 25 g) were obtained from an accredited breeder and were maintained in accordance with the requirements of the Animals (Scientific Procedure) Act of 1986 under the day-to-day supervision of a veterinarian.
Isolated Nerve Muscle Preparations
The hemidiaphragm with the phrenic nerve intact 16 was isolated from nine mice and suspended in an organ bath containing 25 ml of Liley’s physiological saline solution (12 mmol/L NaHCO3, 4 mmol/L KCl, 1 mmol/L KH2PO4, 138.8 mmol/L NaCl, 1 mmol/L MgCl2, 2 mmol/L CaCl2, 11 mmol/L glucose, pH 7.2) oxygenated with 95% O2/5% CO2 and maintained at 27°C. The mechanical response was indirectly elicited by supramaximal stimulation of the phrenic nerve (four 200-microsecond pulses at 30 Hz every 3 seconds) and was measured isometrically with a force transducer. Five preparations served as controls. The other four preparations were exposed to Mg2+ (approximately 8 mmol/L) so that the twitch response was reduced to 50% of the initial values. (Mg2+ competitively inhibits Ca2+-mediated transmitter release, reducing the safety factor of neurotransmission. This sensitizes the preparation to compounds that alter neurotransmitter release.) β-BTX was then added to the bathing medium to give a final concentration of 3 μg/ml (approximately), and the twitch tension was monitored as described above. At the point of failure of the mechanical response, the hemidiaphragm was removed and prepared for transmission electron microscopy. Control preparations were prepared for electron microscopy after 3 hours of continual stimulation in normal bathing fluid.
Nerve Muscle Preparation in Vivo
The soleus muscle of the female rat was used to study the neuropathology of the neuromuscular system after the inoculation of β-BTX. Briefly, 2 μg of β-BTX (0.2 ml of 10 μg/ml in 0.9% w/v NaCl) was injected into the dorsolateral aspect of one hind limb of 30 rats so as to bathe the underlying soleus muscle in toxin. At various time intervals (3 hours to 21 days after the injection of β-BTX), the muscle and its contralateral control were removed, pinned onto dental wax at 1.2 × the resting length, and prepared for electron microscopy or for the labeling of acetylcholine receptor (AChR), synaptophysin, or axonal neurofilament.
To study the acute effects of a lethal dose of β-BTX, 15 μg of the toxin in 0.15 ml of 0.9% w/v NaCl) was injected into the tail vein of two mice. When the respiration of the animals became critically impaired, they were killed by cervical crush. The diaphragms were then removed and prepared for transmission electron microscopy (TEM).
Transmission Electron Microscopy
Tissues for TEM were first fixed in Karnovsky’s fixative for 1 hour. 17 The end plates were labeled with cholinesterase 18 before secondary fixation in OsO4, dehydration, and embedding in araldite resin. The end-plate regions of the blocks of tissue were located, and ultrathin sections (50 to 70 nm) were prepared for examination.
Immunocytochemical Labeling
Isolated muscles were fixed in paraformaldehyde (0.5% w/v in 0.1 mol/L phosphate-buffered saline (PBS)) for 10 minutes. The muscle was then teased into three to four bundles of muscle fibers and returned for an additional 20-minute period of fixation. The muscle pieces were then rinsed in PBS and teased into smaller bundles of 10 to 12 fibers. These were permeabilized in absolute alcohol for 10 minutes at −18°C and again rinsed in PBS. The bundles were placed in 200 μl of incubation medium (3% w/v bovine serum albumin and 0.1 mol/L lysine in PBS) containing either mouse anti-synaptophysin monoclonal antibodies (100:1) or mouse anti-neurofilament antibody (100:1) and incubated at 4°C overnight. The following morning, the preparations were washed in PBS and incubated for 1 hour at 20°C in a medium containing FITC-α-BTX (to label AChR) and rhodamine-labeled rabbit anti-mouse polyclonal IgG. This secondary antibody had been preincubated with normal rat serum (2:1 by volume), the precipitate being spun down and discarded.
Preparations were once more washed in PBS, mounted in Vectashield on standard glass slides, and examined under a Bio-Rad MRC 600 confocal scanning laser microscope.
Specificity of Major Reagents
Specificity of FITC-α-BTX was checked by labeling frozen sections of soleus muscles with a polyclonal rabbit antibody to rat brain AChE and tetraethylrhodamine-conjugated swine anti-rabbit IgG. This enabled end plates to be unequivocally labeled. The sections were then incubated for 60 minutes in PBS with or without native α-BTX (30 μg/ml) or β-BTX (30 μg/ml). The sections were rinsed and incubated for 30 minutes with FITC-α-BTX. Control sections and sections incubated with native β-BTX were labeled with FITC-α-BTX at every AChE-identified end plate. Sections incubated with native α-BTX did not label with FITC-α-BTX.
The monoclonal antibody to synaptophysin (SY38) does not label Western blots. Nonspecific binding is minimized by overlaying sections with fetal calf serum or BSA. The product used in these studies labeled no part of the muscle fiber other than the neuromuscular junction and did not label denervated muscle fibers. The antibody cross-reacts with vesicles in a variety of other tissues (eg, adrenal medulla and islet cells) of no relevance to the study reported in this paper.
Specificity of anti-neurofilament antibody was originally demonstrated by Western blotting. The antibody recognizes both phosphorylated and nonphosphorylated forms of 160- and 200-kd neurofilaments. It does not recognize tubulin.
The secondary antibody (rhodamine-labeled rabbit anti-mouse polylonal IgG) was pre-absorbed with normal rat serum before use. There was no labeling with the secondary antibody in any preparation that had not been previously exposed to the primary antibody.
Collection of Data
To ensure that morphological data obtained during this study were truly representative, at least three muscles were used at each time point. For electron microscopy, blocks of tissue were randomly selected from each muscle and screened for the presence of end plates. Those blocks containing end plates (identified by the presence of positive staining for AChE) were used to prepare thin sections of tissue for electron microscopy. End plates were photographed only if the nerve terminal was clearly situated within a structure that possessed postsynaptic folds. If no folds were present it was assumed that the nerve terminal had been sectioned in a region just on the periphery of the neuromuscular junction. The tissue blocks were sectioned, and sections were examined blind. Between 10 and 40 randomly encountered end plates were photographed from each muscle.
Data on AChR, synaptophysin, and neurofilament distributions were similarly collected from muscle fiber bundles teased from at least three muscles. A typical muscle yielded approximately 20 to 30 useable bundles of 10 to 12 muscle fibers. Images were selected for analysis and photography only if the end plate, identified from FITC-α-BTX labeling, was en face. These data, and those concerning the classification of patterns of innervation, were collected blind and analyzed later by examination of stored images.
It should be acknowledged that the pathology we were recording was so significant that the term blind cannot be considered absolute.
Analysis of Data
A semiquantitative analysis of the synaptophysin labeling was conducted by visual comparison with the area of the synapse labeled by FITC-α-BTX and placing the examined terminals in one of three groups: >50%, complete; <50%, incomplete; <10%, absent.
The patterns of innervation visualized by the neurofilament labeling were categorized using a simplified version of the system of Tuffery. 19 The categories were as follows: absent, no evidence of any innervation; simple, a single axon innervating a single myofiber; preterminal or ultra-terminal sprouts, an axon innervating a myofiber but also bearing a sprout, either before the nerve terminal or from the terminal itself; collateral innervation, an axon innervating two adjacent myofibers; and multiple innervation, more than one axon innervating the same myofiber. The occurrence of these various patterns of innervation were assessed blind at each time point examined.
Where appropriate, data are expressed as the mean ± the SEM. Populations of data were compared using the unpaired Student’s t-test, with those with a probability P < 0.5 considered as significantly different.
Results
Experiments in Vitro
Muscle Twitches
Control preparations responded to stimulation of the phrenic nerve with a twitch response generating a mean force equivalent to 8.5 ± 0.8 g/g wet weight (n = 5). There was no significant reduction in the twitch tension over a period of 3 hours. Increasing [Mg2+]o to 8 mmol/L reduced the indirect twitch response by approximately 50%. The application of β-BTX (3 μg/ml) to partially blocked preparations resulted in a period of facilitation followed by a gradual, irreversible failure of neurotransmission. Maximal facilitation occurred 29.5 ± 4.6 minutes (n = 4) after the application of the toxin, the twitch tension reaching 134 ± 5% of control. The irreversible failure of neurotransmission occurred at 212 ± 19 minutes (n = 4).
Transmission Electron Microscopy
The nerve terminals of mouse diaphragm muscles maintained in normal bathing fluid were filled with synaptic vesicles and mitochondria. Fine Schwann cell processes covered the exposed part of the nerve terminal (Figure 1a) ▶ . The nerve terminals of muscles that had failed to respond to indirect excitation contained very few synaptic vesicles (Figure 1, b–e) ▶ , but they exhibited large numbers of omega profiles on the nerve terminal plasma membrane (Figure 1, b–e) ▶ , many of which were coated in electron-dense material thought to be clathrin. The terminals also contained large, coated vesicles (diameter, 72 ± 2 nm; range, 54 to 104 nm). Schwann cell processes were thickened. Although mitochondria were apparently normal in some terminals that had been exposed to β-BTX (Figure 1d) ▶ , many mitochondria had no discernible inner christae (Figure 1, b–e) ▶ , and some were devoid of any formal architecture (Figure 1e) ▶ .
Figure 1.
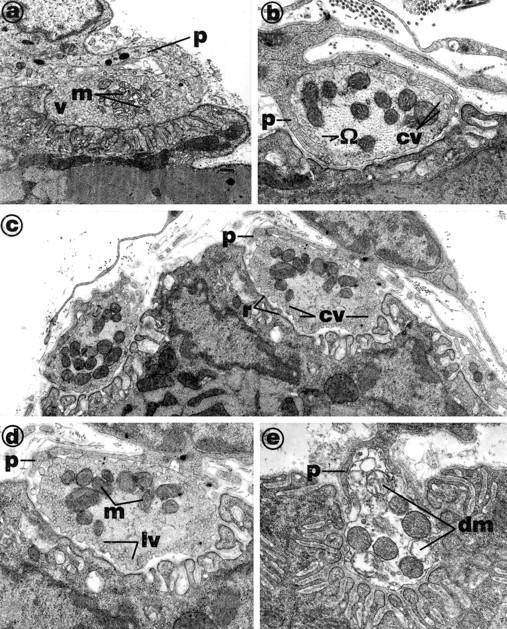
Electron micrographs of nerve terminals in mouse diaphragm muscles incubated in vitro in either normal bathing fluid for 3 hours (a) or in the presence of 3 μg/ml β-BTX (b to e) until neuromuscular transmission was blocked (∼200 minutes). The control terminals (a) were packed with synaptic vesicles (v) and mitochondria (m). The internal cristae of the mitochondria (m) were clearly defined. Schwann cell processes (p) were fine and well clear of the synaptic cleft. Nerve terminals from muscles exposed to β-BTX (b to d) contained few synaptic vesicles, but omega profiles (Ω) were common on nerve terminal membranes, and the membranes were ruffled (r; see c). Some terminals contained very large synaptic vesicles (lv; see d). In many sections, mitochondrial cristae were indistinct (see m in d). Schwann cell processes (p) often penetrated the synaptic cleft (see b and e). Some terminals were in advanced stages of degeneration (e) and contained many damaged mitochondria (dm).
Experiments in Vivo
Transmission Electron Microscopy
The inoculation of 15 μg of β-BTX intravenously into the tail vein of two mice caused respiratory distress within 30 minutes. The mice were killed at this stage, and their diaphragm muscles were prepared for electron microscopy. A total of 50 end plates were randomly photographed from sections of the muscles.
The density of synaptic vesicles was greatly reduced in all terminals; omega profiles, coated pits, and large coated vesicles were found in the terminals (Figure 2, a–e) ▶ . Many mitochondria exhibited a loss of internal cristae. Schwann cell processes were thickened and, in many cases, were apparently invading the synaptic cleft (Figure 2b) ▶ . Between 10% and 15% of terminals in these muscles were in an advanced state of degeneration; mitochondria were devoid of defining architecture and Schwann cell processes appeared to be engulfing the terminal (Figure 2e) ▶ . The plasma membrane of the nerve terminals, although ruffled and containing many omega profiles, was intact in every junction examined.
Figure 2.
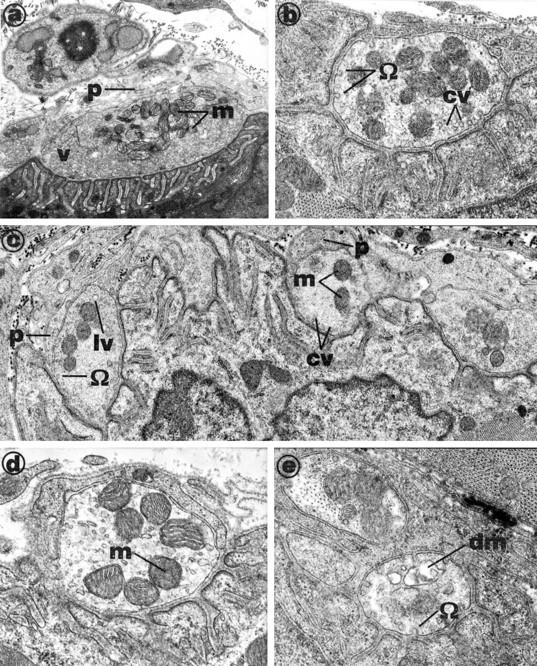
Electron micrographs of nerve terminals in mouse diaphragm muscles after the inoculation of a lethal dose of β-BTX (15 μg in 0.15 ml of NaCl, 0.9% w/v) administered via the tail vein. Animals were killed when respiration was significantly impaired and diaphragms removed. The control terminals, viewed in muscles of mice not inoculated with toxin (a) were packed with synaptic vesicles (v) and mitochondria (m). The internal cristae of the mitochondria (m) were clearly defined. Schwann cell processes (p) were fine and well clear of the synaptic cleft. Nerve terminals from muscles exposed in vivo to β-BTX (b to e) were mostly devoid of vesicles, although some contained small populations of large vesicles (lv), many of which were coated with an electron-dense material, possibly clathrin (cv; see b, c, and e). In complex end plates (see c), some terminals contained only large coated vesicles (cv) and others contained small clusters of apparently normal vesicles (v). Schwann cell processes (p) appeared to be invading the synaptic cleft (c, d, and e). Nerve terminal membranes exhibited numerous omega profiles (Ω), and many contained damaged mitochondria (dm in e). Note that c is an enlargement of the central terminal shown in b.
Immunofluorescence Studies of Synaptophysin and Neurofilament
Control preparations were labeled with FITC-αBTX and anti-synaptophysin antibody to determine the normal distribution of AChR and intraterminal synaptic vesicles. A total of 113 junctions was examined under the confocal microscope; 93% of junctions labeled with FITC-αBTX were also labeled with anti-synaptophysin antibody. The remaining 7% did not react with anti-synaptophysin antibody. In all cases where dual labeling was achieved, the distributions of the labels were exactly matched (Figure 3) ▶ .
Figure 3.

Combined AChr and synaptophysin labeling in soleus muscles was used to study the fate of vesicles in β-BTX-treated muscle fibers. In control fibers, the distribution of AChR was closely matched by the distribution of synaptophysin (A and D). A preparation studied 3 hours after exposure in vivo to β-BTX revealed the loss of synaptophysin labeling but the retention of AChR labeling (B and E). Synaptophysin reactivity did not return until 5 days after the inoculation of toxin. Combined neurofilament and AChR labeling in the muscles was used to study the status and pattern of innervation. Control preparations revealed the typical (one axon per end plate) pattern of innervation (C). A preparation made 6 hours after the inoculation with toxin reveals the breakdown of the axon cytoskeleton but the retention of AChR labeling (F). Reinnervation was associated with extensive collateral innervation (G and H) rarely seen in control preparations. This change in innervation pattern was first seen at 7 days and retained at least until 21 days after inoculation. Scale bars, 50 μm.
Muscle fiber bundles examined 3 hours after inoculation of β-BTX showed no obvious diminution in reactivity toward FITC-αBTX, but of 130 junctions examined, only 50% exhibited complete anti-synaptophysin antibody labeling. By 12 hours, anti-synaptophysin antibody labeling was absent from 90% of a total of 177 junctions studied (Figure 3) ▶ .
Similar bundles of fibers were used for combined labeling with FITC-α-BTX and anti-neurofilament antibody. Of 238 junctions studied on control muscle fibers, 91% of FITC-α-BTX-labeled fibers exhibited clear, mono-axonal innervation (Figure 3e) ▶ . The remaining 9% did not exhibit positive labeling with anti-neurofilament antibody. We assume these latter junctions were effectively denervated during the teasing process. The majority of fibers exhibited a simple pattern of innervation. Only 3% exhibited collateral innervation, and 1% exhibited multiple innervation (Table 1) ▶ .
Table 1.
Innervation Patterns on Muscle Fibers of Soleus Muscles of Female Rats
| Innervation pattern | % of fibers | ||||||
|---|---|---|---|---|---|---|---|
| Control | 3 hours | 6 hours | 12 hours | 5 days | 7 days | 14 days | |
| Simple | 87 | 40 | 9 | 1 | 35 | 60 | 68 |
| Preterminal sprouting | 0 | 0 | 0 | 0 | 4 | 7 | 3 |
| Ultraterminal sprouting | 0 | 0 | 0 | 0 | 27 | 4 | 0 |
| Collateral innervation | 3 | 0 | 0 | 0 | 5 | 8 | 19 |
| Multiple innervation | 1 | 0 | 0 | 0 | 2 | 5 | 1 |
| Total | 91 | 40 | 9 | 1 | 73 | 84 | 91 |
Muscles were examined 3 hours to 14 days after the inoculation of β-bungarotoxin (2 μg in 0.2 ml 0.9% w/v NaCl s.c. over the muscle).
The innervation of junctions was studied in 219 muscle fibers isolated 3 hours after exposure to β-BTX. Only 40% were innervated by an intact axon; 10% were unlabeled with anti-neurofilament antibody, and the remaining 50% were labeled, but the labeling of exonal neurofilament was fragmented (Figure 3b) ▶ . By 6 hours only 9% of the 92 junctions studied exhibited continuous labeling with anti-neurofilament antibody. Twelve hours after inoculation, no examples of continuous labeling with anti-neurofilament antibody were identified, although some examples of fragmented labeling were identified. By 24 hours no anti-neurofilament labeling of any kind was seen at any of 187 junctions studied.
Re-Innervation
There was clear evidence of significant levels of reinnervation by 5 days after the inoculation of β-BTX. Anti-synaptophysin antibody labeling was successful in 80% of the 169 FITC-α-BTX-labeled synapses studied, although the labeling was usually sparse. By 14 days, anti-synaptophysin antibody labeling could be identified at 95% of the 147 junctions studied, and in every case labeling patterns were indistinguishable from normal.
Similar results were obtained when axonal integrity was studied using labeling for axonal neurofilament. By 5 days after exposure to β-BTX, 73% of 148 junctions examined were innervated by axons exhibiting continuous labeling with anti-neurofilament antibody. Preterminal and ultraterminal sprouts were common, and there were numerous examples of collateral innervation. Many of the axons were of small diameter and appeared wispy. By 7 days, 84% of 166 junctions studied were innervated, and all axons appeared to be of normal caliber. There was a reduction in the degree of both pre- and ultraterminal sprouting, and a number of surviving sprouts terminated in strongly labeled retraction cones. By 21 days, 91% of junctions were innervated. Data on the degeneration and regeneration of intramuscular components of the motor innervation are summarized in Figure 4 ▶ . There were no free sprouts by this time, but collateral innervation was common, involving 19% of junctions studied (Figure 3) ▶ . The incidence of sprouting at various times after the inoculation of β-BTX is summarized in Table 1 ▶ .
Figure 4.

The chronology of loss of labeling of axonal neurofilament and its restoration in rat soleus muscles after the inoculation of a single dose of β-BTX. By 3 hours, only 40% of junctions were innervated by axons with an intact cytoskeleton; most axons exhibited a fragmented cytoskeleton. By 6 hours, labeling was continuous in only 9% of axons. No continuous labeling was seen at 12 hours, and labeling was non-existent at 24 hours. At 120 hours, all junctions were innervated by axons with an intact cytoskeleton, but axons were thin and wispy and many exhibited both pre- and post-terminal sprouts. Cytoskeleton labeling was normal by 14 days. Each point represents data from 92 to 238 junctions visualized in muscle fiber bundles obtained from three soleus muscles.
Discussion
Exposure to β-BTX, the presynaptically active neurotoxin isolated from the venom of kraits, caused an acute failure of neuromuscular transmission in mammalian nerve-muscle preparations in vitro and in vivo. Using a combination of electron microscopy and immunocytochemical staining for synaptophysin P.38, a transmembrane protein specific for synaptic vesicles, 20-22 we showed that the failure of transmission was associated with the depletion of synaptic vesicles from motor nerve terminals. There was no obvious damage to the skeletal muscle fibers; AChRs were not blocked, and nerve terminal plasma membranes were intact. Omega-shaped (Ω) profiles, often coated in an electron-dense material, were commonly seen on the plasma membranes of the terminals, and the terminals often contained a few large coated vesicles.
As coated Ω-shaped profiles and large coated vesicles are typically associated with high levels of transmitter release and the impaired turnover of vesicle membrane, our data suggest that the depletion of synaptic vesicles is the result of a combination of enhanced transmitter release and impaired retrieval and recycling of emptied vesicles.
It has long been acknowledged that β-BTX, and other presynaptically active neurotoxic phospholipases A2, cause, in vitro, an early enhancement of transmitter release before the failure of transmission (reviewed in Refs. 1 and 2 ). This is generally thought to reflect a response to the hydrolytic activity of the phospholipases, but the mechanism of action of the toxin at the molecular level is not known.
Significant damage to motor nerve terminals was seen after exposure to β-BTX in vitro and in vivo, and the longer exposure times in vivo revealed a progressive axonopathy that resulted in the destruction of the intramuscular axons of the soleus muscle by 24 hours. At this stage, no axons with an intact cytoskeleton (as visualized by anti-neurofilament antibodies) were seen in any of the three muscles examined. Such extensive damage has not been reported before, although several groups have shown occasional images of damaged nerve terminals or intramuscular axons. 13-15 We do not know how far back the process of degeneration extends or whether it shows any selectivity for motor or sensory systems, but preliminary evidence suggests that it is largely limited to the intramuscular compartment and that it is not selective.
The degenerative response was reversible; all identified junctions were innervated by 5 days, a rate of re-innervation that is similar to that reported by Grubb et al 23 following their studies with notexin.
The very extensive collateral sprouting and re-innervation seen in muscles exposed to β-BTX was not expected. Collateral sprouting is commonly seen when a muscle is partially denervated, and it arises when intact axons sprout to re-innervate the denervated muscle fibers. 24 Sprouting is encouraged by the activation of terminal Schwann cells on axonal remnants after axon damage. The Schwann cells form bridges between adjacent muscle fibers along which axonal sprouts from adjacent surviving axons can spread. 25 Whether a similar mechanism occurs after exposure to β-BTX is not clear. If the Schwann cells do survive, they might act as bridges along which sprouts of the most rapidly regenerating axons spread. The result would be the complete reorganization of the motor units. Whether this reorganization of motor units has any functional significance remains to be seen.
Clinical Correlations with These Data
The clinical experience of treating patients bitten by snakes such as the kraits and the taipans of Papua New Guinea (snakes whose venoms contain large amounts of presynaptically active phospholipases A2) is that the administration of antivenom, anticholinesterases, and diaminopyridines is largely ineffective in reversing or even alleviating the neuromuscular weakness. Victims require artificial ventilation and, in some cases, intensive care for prolonged periods. 3-7 It is now possible to offer a coherent and factually based explanation of the difficulties experienced by clinicians. Antivenom, if administered quickly after the bite, will neutralize any unbound toxins as they are released from the bite site, and anticholinesterases and diaminopyridines might be helpful during the recovery phase as neuromuscular contacts are restored, but no treatment currently available will prevent the destruction of the innervation once the process of degeneration has been initiated, nor will any available treatment enhance the speed with which the structural damage to the peripheral innervation is repaired or the rate at which the victims recover neuromuscular competency.
The extensive collateral re-innervation we have seen may reflect the simple regrouping of the motor unit. If, however, the collateral re-innervation reflects a loss of motor units and the enlargement of surviving motor units, the consequences might be serious, because larger-than-normal motor units are frequently unstable. Motor neurones supporting very large motor units die prematurely, leading to a delayed, but irreversible secondary phase of progressive neuromuscular weakness. 24
We suggest that this potentially important problem needs to be studied by mounting long-term studies on surviving victims of severe envenoming bites by kraits and taipans.
Footnotes
Address reprint requests to Dr. John B. Harris, School of Neurosciences and Psychiatry, Department of Neurobiology, Medical School, University of Newcastle upon Tyne, Newcastle upon Tyne NE2 4HH, UK. E-mail: j.b.harris@nc1.ac.uk.
Supported by the Wellcome Trust.
References
- 1.Harris JB: Phospholipases in snake venoms and their effects on nerve and muscle. Harvey AL eds. Snake Toxins. 1991, :pp 91-124 Pergamon Press, New York [DOI] [PubMed] [Google Scholar]
- 2.Hawgood B, Bon C: Snake venom presynaptic toxins. Tu AT eds. Handbook of Natural Toxins, Reptile Venoms and Toxins 1991, vol 5.:pp 3-52 Marcel Dekker, New York [Google Scholar]
- 3.Pearn JH: Survival after snake-bite with prolonged neurotoxic envenomation. Med J Aust 1971, 2:259-261 [DOI] [PubMed] [Google Scholar]
- 4.Connolly S, Trevett AJ, Nwokolo NC, Lalloo DG, Naraqi S, Mantle D, Schoffield IS, Fawcett PRW, Harris JB, Warrell DA: Neuromuscular effects of Papuan taipan venom. Ann Neurol 1995, 88:916-920 [DOI] [PubMed] [Google Scholar]
- 5.Trevett AJ, Lalloo DG, Nwokolo NC, Naraqi S, Kevan IH, Theakston RDG, Warrell DA: Failure of 3,4-diaminopyridine and edrophonium to produce significant clinical benefit in neurotoxicity following the bite of Papuan taipan (Oxyuranus scutellatus canni). Trans R Soc Trop Med Hyg 1995, 89:444-446 [DOI] [PubMed] [Google Scholar]
- 6.Trevett AJ, Lalloo DG, Nwokolo NC, Naraqi S, Kevan IH, Theakston RDG, Warrell DA: The efficacy of antivenom in the treatment of bites by the Papuan taipan (Oxyuranus scutellatus canni). Trans R Soc Trop Med Hyg 1995, 89:322-325 [DOI] [PubMed] [Google Scholar]
- 7.Trevett AJ, Lalloo DG, Nwokolo NC, Naraqi S, Kevan IH, Theakston RDG, Warrell DA: Electrophysiological findings in patients envenomed following the bite of a Papuan taipan (Oxyuranus scutellatus canni). Trans R Soc Trop Med Hyg 1995, 89:415-417 [DOI] [PubMed] [Google Scholar]
- 8.Mollier P, Brochier G, Morot Gaudry-Talarmain Y: The action of notexin from tiger snake venom (Notechis scutatus scutatus) on acetylcholine release and compartmentation in synaptosomes from electric organ of Torpedo marmorata. Toxicon 1990, 28:1039-1052 [DOI] [PubMed] [Google Scholar]
- 9.Ueno E, Rosenberg P: Inhibition of phosphorylation of rat synaptosomal proteins by snake venom phospholipase A2 neurotoxins (β-bungarotoxin, notexin) and enzymes (Naja naja atra, Naja nigricollis). Toxicon 1990, 28:1423-1437 [DOI] [PubMed] [Google Scholar]
- 10.Rowan EG, Harvey AL: Potassium channel blocking actions of β-bungarotoxin and related toxins on mouse and frog motor nerve terminals. Br J Pharmacol 1988, 94:839-847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cull-Candy SG, Fohlman J, Gustavsson D, Lüllmann-Rauch R, Thesleff S: The effects of taipoxin and notexin on the function and fine structure of the murine neuromuscular junction. Neuroscience 1976, 1:175-180 [DOI] [PubMed] [Google Scholar]
- 12.Strong PN, Heuser JE, Kelly RP: Selective enzymatic hydrolysis of nerve terminal phospholipids by β-bungarotoxin: biochemical and morphological studies. Hall Z Kelly R Fox CF eds. Cellular Neurobiology. 1977, :pp 227-249 Alan Liss, New York [PubMed] [Google Scholar]
- 13.Abe T, Limbrick AR, Miledi R: Acute muscle denervation induced by β-bungarotoxin. Proc R Soc B 1976, 194:545-553 [DOI] [PubMed] [Google Scholar]
- 14.Harris JB, Johnson MA, MacDonell CA: Muscle necrosis induced by some presynaptically active neurotoxins. Eaker D Wadström T eds. Natural Toxins. 1980, :pp 569-578 Pergamon Press, Oxford [Google Scholar]
- 15.Gopalakrishnakone P, Hawgood BJ: Morphological changes induced by crotoxin in murine nerve and neuromuscular junction. Toxicon 1984, 22:791-804 [DOI] [PubMed] [Google Scholar]
- 16.Büllbring E: Observations on the isolated phrenic nerve diaphragm preparation of the rat. Br J Pharmacol 1946, 1:38-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karnovsky MJ: A formaldehyde-glutaraldehyde fixative of high osmolarity for use in electron microscopy. J Cell Biol 1965, 27:1374 [Google Scholar]
- 18.Strum JM, Hall-Craggs ECB: A method of demonstrating motor endplates for light and electron microscopy. J Neurosci Methods 1982, 6:305-309 [DOI] [PubMed] [Google Scholar]
- 19.Tuffery AR: Growth and degeneration of motor end-plates in normal cat hind limb muscles. J Anat 1971, 110:221-247 [PMC free article] [PubMed] [Google Scholar]
- 20.Navone F, Jahn R, Di Gioia G, Stukenbrok H, Greengard P, De Camilli P: Protein P38: an integral membrane protein specific for small vesicles of neurons and neuroendocrine cells. J Cell Biol 1986, 103:2511-2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiedenmann B, Franke WW: Identification and localisation of synaptophysin, an integral membrane of glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 1985, 41:1017-1028 [DOI] [PubMed] [Google Scholar]
- 22.De Camilli P, Vitadello M, Canevini MP, Zanoric R, Jahn R, Gorio A: The synaptic vesicle proteins synapsin 1 and synaptophysin (protein P38) are concentrated both in efferent and afferent nerve endings of the skeletal muscle. J Neurosci 1988, 8:124-1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grubb BD, Harris JB, Schoffield I: Neuromuscular transmission at newly formed neuromuscular junctions in the regenerating soleus muscle of the rat. J Physiol 1991, 441:405-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafuse VR, Gordon T, Orozco R: Proportional enlargement of motor units after partial denervation of cat triceps surae muscles. J Neurophysiol 1992, 68:1261-1276 [DOI] [PubMed] [Google Scholar]
- 25.Son Y-J, Thompson WJ: Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron 1995, 14:133-141 [DOI] [PubMed] [Google Scholar]