

Abstract
Retinal astrocytes are located in the nerve fiber layer and along retinal blood vessels and have been hypothesized to participate in the induction and maintenance of the blood-retinal barrier. Platelet-derived growth factor-A (PDGF-A) is normally produced by retinal ganglion cells and is involved in astrocyte recruitment and proliferation. We used gain-of-function transgenic mice that express PDGF-A in photoreceptors to explore the roles of PDGF-A and astrocytes in the retina. Transgene-positive mice developed glial infiltration of the inner retina and had significantly less oxygen-induced retinal vascular closure and no neovascularization compared with littermate controls, which had prominent vascular closure and neovascularization. The increased survival of endothelial cells in transgenic mice in the face of oxygen-induced down-regulation of vascular endothelial growth factor was accompanied by an increase in astrocyte-derived fibroblast growth factor-2. Therefore, PDGF-A increases retinal astrocytes, which promote the survival of endothelial cells as well as their expression of barrier characteristics.
Several lines of evidence suggest that platelet-derived growth factors (PDGFs) function as wound repair paracrine signaling agents. PDGFs are released from aggregated platelets at wound sites and act as chemoattractants and mitogens for cell types that participate in wound repair at many sites in the body, including glial cells, smooth muscle cells, pericytes, and fibroblasts (for review, see Heldin and Westermark 1 ). Furthermore, exogenous PDGFs promote wound repair, and neutralizing antibodies for PDGFs impair wound healing. 2 However, PDGFs are produced by many cell types and also participate in functions other than wound repair. For instance, PDGFs have been implicated in development, 3-6 and expression of PDGFs in neurons suggests possible neurotrophic and/or gliotrophic effects. 7,8
The PDGF family is made up of dimers of the products from two genes, PDGF-A and PDGF-B, resulting in three family members, PDGF-AA, PDGF-AB, and PDGF-BB. There are two PDGF receptor (PDGFR) gene products, PDGFRα and PDGFRβ, that also form dimers (for review, see Hart and Bowen-Pope 9 ). PDGFRα binds both PDGF-A and -B, whereas PDGFRβ binds only PDGF-B. Differential expression of the receptors results in differences in activity of the isoforms.
The retina provides a great model system to study the effects of growth factors and/or cellular interactions in the nervous system. PDGFs have been implicated in the differentiation of O2-A progenitor cells in the optic nerve into astrocytes that migrate into the retina and become localized in the nerve fiber layer and around blood vessels. 10-13 Increased expression of PDGFs in retinal pigmented epithelial (RPE) cells after disruption of the RPE monolayer may be a mechanism by which defects are repaired, but may also contribute to scarring that underlies proliferative vitreoretinopathy, the major cause of failure of retinal reattachment surgery and disciform scarring, the end-stage of macular degeneration with choroidal neovascularization (NV). 14 PDGFs produced by endothelial cells in retinal NV in diabetic retinopathy or retinopathy of prematurity may also contribute to glial and RPE cell recruitment into epiretinal scar tissue that can lead to retinal detachment. 15,16 Therefore, neutralization of PDGFs may be a good strategy for preventing and treating these scarring processes. But to design the most specific treatments, it is necessary to know whether scarring is mediated by PDGF-A, PDGF-B, or both. It is also necessary to know what other effects PDGF-A and -B have in the retina, because these other effects are likely to be perturbed by neutralization of PDGFs. To investigate the effects of PDGF-A in the retina, we coupled the rhodopsin promoter to the coding region of the PDGF-A gene and generated transgenic mice that express PDGF-A in photoreceptors.
Materials and Methods
Generation of Transgenic Mice
A full-length complementary DNA (cDNA) for human PDGF-A was cloned into a plasmid containing the 2.2-kb HindIII/NaeI fragment from the bovine rhodopsin promoter. 17 The plasmid also contained an intron and a polyadenylic acid addition site derived from the mouse protamine gene and a eukaryotic consensus ribosomal binding site. After transformation, a clone with correct orientation was selected. DNA was double CsCl purified and cut with EcoRI to provide a 3546-bp fusion gene (Figure 1) ▶ . The fusion gene was purified, and transgenic mice were generated by established techniques as previously described. 17
Figure 1.

Schematic map of the rhodopsin/PDGF-A fusion gene. P1 and P2, oligonucleotide primers used to screen genomic DNA for presence of the transgene; P3 and P4, primers used for RT-PCR; + 1, transcription start site.
Mice were screened for the presence of the transgene by either Southern blot analysis or polymerase chain reaction (PCR) of tail DNA. 17 Tail pieces were digested overnight at 55°C in 50 mmol/L Tris (pH 7.5), 100 mmol/L ethylenediaminetetraacetic acid, 400 mmol/L NaCl, 0.5% sodium dodecyl sulfate (SDS) containing 0.6 μg/μl proteinase K. PCR was done at 58°C, with primers that amplify 580 bp of transgene-specific sequence P1 (5′-GTCCAGCCGGAGCCCCGTG-3′) and P2 (5′-TGGCACTTGACACTGCTCGTGTTG-3′; Figure 1 ▶ ).
Retinal Reverse Transcriptase-PCR
At appropriate time points, mice were sacrificed, eyes were removed, and retinas were dissected. Retinal RNA was isolated using the guanidine isothiocyanate method as described by Chomczynski and Sacchi. 18 Reverse transcription was carried out with ∼0.5 μg of total RNA, reverse transcriptase (RT; SuperScript II, Life Technologies, Inc., Gaithersburg, MD), and 5.0 μmol/L oligodeoxythymidylate primer. Aliquots of the cDNAs were used for PCR amplification with primers for the hPDGF-A/mP1 fusion gene that amplify across an intron-exon border, P3 (5′-AACACGAGCAGTGTCAAGTGCCAG-3′) and P4 (5′-GATGTGGCGAGATGCTCTTGAAGTCTGGTA-3′; Figure 1 ▶ ). The expected PCR products for the hPDGF-A/mP1 fusion gene fragment from genomic DNA and messenger RNA (mRNA) are 632 bp and 538 bp, respectively. Titrations were performed to ensure that PCR reactions were carried out in the linear range of amplification. Mouse S16 ribosomal protein primers (5′-CACTGCAAACGGGGAAATGG-3′ and 5′-TGAGATGGACTGTCGGATGG-3′) were used to provide an internal control for the amount of template in the PCR reactions.
Northern Blot Analysis
RNA blot hybridization analysis was done as previously described, 14 using 10 to 15 μg of total retinal RNA. The cDNA probe was the 1.1-kb EcoRI-HindIII fragment of hPDGF-A labeled with 32P by hexanucleotide random priming. The hybridization temperature was 65°C, and the membrane was washed twice for 60 minutes at room temperature in 2× standard saline citrate, 0.1% SDS, followed by a 15-minute wash at 58°C in 1× standard saline citrate, 0.1% SDS and a final 15-minute wash at 65°C in 0.5× standard saline citrate, 0.1% SDS.
Immunohistochemistry for PDGF
Transgene-positive and littermate control mice were sacrificed at various time points; their eyes were removed, fixed in 4% paraformaldehyde, and embedded in paraffin. Sections (10 μm each) were cut and immunohistochemically stained as previously described 14 with a 1:100 dilution of rabbit anti-hPDGF antibody (Genzyme, Cambridge, MA). Specificity of staining was assessed by substitution of nonimmune serum for primary antibody and by preabsorption of primary antibody with antigenic peptide.
Evaluation of the Retinal Phenotype of Transgenic Mice
At various time points, mice were sacrificed, and eyes were snap-frozen or briefly fixed in 4% paraformaldehyde and embedded in optical cutting temperature embedding media (OCT; Miles Diagnostics, Elkhart, IN) or paraffin. Frozen or paraffin sections were stained with hematoxylin and eosin (H&E), histochemically stained with biotinylated griffonia simplicifolia lectin B4 (GSA; Vector Laboratories, Burlingame, CA), 19 or immunohistochemically stained with a 1:100 dilution of a rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP; a gift of L. F. Eng, Palo Alto, CA) 20 or a 1:100 dilution of a rabbit polyclonal antibody to human cellular retinaldehyde binding protein (a gift from J. Saari, Seattle, WA), 21 a 1:50 dilution of a monoclonal anti-hFGF2 antibody (a gift from T. Reilly, Wilmington, DE) 19,22 or a 1:20 dilution of a monoclonal antibody directed against proliferating cell nuclear antigen (a gift from P. Hall, Aberdeen, Scotland). 14,23
Murine Model of Oxygen-Induced Ischemic Retinopathy
Ischemic retinopathy was produced in transgene-positive and littermate control mice by a method described by Smith et al. 24 Seven-day-old mice and their mothers were placed in an airtight incubator and exposed to an atmosphere of 75 ± 3% oxygen for 5 days. Incubator temperature was maintained at 23 ± 2°C, and oxygen was measured every 8 hours with an oxygen analyzer. After 5 days, the mice were removed from the incubator and placed in room air for 5 days. After 5 days, the mice were killed, and their eyes were rapidly removed and frozen in OCT or fixed in 10% formalin and embedded in paraffin. To perform quantitative assessments, 10-μm serial sections were cut through the entire extent of each eye. The entire eye was sampled by staining sections roughly 50 to 60 μm apart, which provided 13 sections per eye for analysis. GSA-stained sections were examined with an Axioskop microscope (Zeiss, Thornwood, NY), and images were digitized using a 3 charge-coupled device color video camera (IK-TU40A, Toshiba, Tokyo, Japan) and a frame grabber. Image-Pro Plus software (Media Cybernetics, Silver Spring, MD) was used to delineate lectin-stained cells on the surface of the retina, and their area was measured. The mean of the 13 measurements from each eye was used as a single experimental value.
Compound Transgenic Mice with Increased Expression of Both PDGF-A and Vascular Endothelial Growth Factor in the Retina
The production and screening of transgenic mice with overexpression of vascular endothelial growth factor (VEGF) in photoreceptors (rho/VEGF-transgenic mice) have been previously described. 25 Mice from the V-6 line that are heterozygous for the rhodopsin/VEGF transgene develop NV that originates from the deep capillary bed of the retina and grows into the subretinal space, where it gradually spreads and enlarges. V-6 rho/VEGF mice were mated with rho/PDGF-A2 or rho/PDGF-A3 mice and, at P21, the offspring were perfused with fluorescein-labeled dextran, and retinal NV was measured on retinal flat mounts as previously described. 26 Briefly, mice were anesthetized, the descending aorta was clamped, the right atrium was cut, and the mice were perfused through the left ventricle with 1 ml of phosphate-buffered saline containing 50 mg/ml of fluorescein-labeled dextran (2 × 10 6 average molecular weight; Sigma Chemical Co., St. Louis, MO). The eyes were removed and fixed for 1 hour in 10% buffered formalin phosphate. The cornea and lens were removed, and then the entire retina was carefully dissected from the eyecup, radially cut from the edge of the retina to the equator in all four quadrants, and flat-mounted in Aquamount with photoreceptors facing upward. Flat mounts were examined by fluorescence microscopy and photographed with Ectachrome 400 film (Kodak, Rochester, NY), and slides were scanned with a QuickScan 35 scanner (Minolta, Osaka, Japan). Image files were imported into Adobe PhotoShop 4.0, labeled, and printed with a Fujix pictography 3000 printer (Fuji Photo Film Co., Tokyo, Japan). For quantitative assessments, retinal flat-mounts were examined by fluorescence microscopy at ×400 magnification, which provides a narrow depth of field so that, when focusing on NV on the outer edge of the retina, the remainder of the retinal vessels are out of focus, allowing easy delineation of the NV. The outer edge of the retina, which corresponds to the subretinal space in vivo, is easily identified, and, therefore, there is standardization of focal plane from slide to slide. Images were digitized using a 3 charge-coupled device color video camera (IK-TU40A; Toshiba, Tokyo, Japan) and a frame grabber. Image-Pro Plus 3.0 software (Media Cybernetics, Silver Spring, MD) was used to delineate each of the lesions and calculate the number in each retina, the area of each lesion, and the total area of NV per retina. Measurements were repeated three times for each retina, and the mean was used for one experimental value; there was little variability among triplicate measurements.
Results
Generation of Transgenic Mice
Six independent lines that incorporated the rhodopsin promoter/PDGF-A fusion gene were obtained (designated rho/PDGF-A1–6). The founders were backcrossed with C57BL/6J mice to establish transgenic lines; rho/PDGF-A6 did not reproduce and therefore only 5 lines were established. Mice that were heterozygous at the transgene locus were used in all analyses.
Expression of PDGF-A mRNA in the Retinas of Transgenic Mice
RT-PCR using total retinal RNA as template and primers specific for transgene mRNA showed good expression in lines rho/PDGF-A1, -A2, and -A3, moderate expression in line 5, and weak expression in line 4 (Figure 2A) ▶ . The time course of transgene mRNA expression in the retina was assessed in lines 2 and 3 and showed that in both lines mRNA was first detected at P3 and increased to a steady-state level between P10 and P14 (Figure 2B) ▶ .
Figure 2.

Assessment of transgene mRNA levels in retinal RNA by RT-PCR. Total RNA was extracted from the retinas of rho/PDGF-A mice, and RT-PCR was done for PDGF-A transgene mRNA and S16 ribosomal protein mRNA. The size of the band amplified with transgene primers was compatible with the 538 bp predicted for amplification from mRNA and was absent (as was the band for S16) when RT was excluded from the reaction. A: In adult mice, a strong signal (lines 1–3), an intermediate signal (line 5), and a weak signal for (line 4) were seen. B: A weak signal was seen at P3 and increased to a steady-state level between P10 and P14 (lines 2 and 3).
The Phenotype of rho/PDGF-A Mice Is Subtle and Correlates with Expression of PDGF-A
Only very rough quantitative comparisons can be made with immunohistochemical staining, but it is safe to say that retinas of adult mice from lines 1 and 3 showed a strong signal for PDGF-A, whereas retinas from line 4 mice showed no signal above background (Figure 3A) ▶ . Lines 2 and 5 showed staining above background, but less than that seen in lines 1 and 3. The pattern of staining varied slightly among the lines, but in general there was diffuse staining throughout the entire retina, suggesting that PDGF-A is secreted by photoreceptors and spreads throughout the inner retina. In lines 1 and 3, there was a distinct band of more intense staining in the region of photoreceptor terminals (arrows), suggesting that in these lines, secretion may occur primarily from the synaptic terminals (Figures 3 and 4) ▶ ▶ .
Figure 3.

Immunohistochemical staining for PDGF-A, and morphological and proliferative changes in rho/PDGF-A mice. A: Retinal frozen sections from adult mice from each of the rho/PDGF-A lines were immunohistochemically stained using an affinity-purified anti-hPDGF antibody. Lines 1 and 3: Prominent staining with a distinct band in the outer plexiform layer in the region of the photoreceptor terminals. Lines 2 and 5: Intermediate staining. Line 4: No staining above background. B: The retinal phenotype is subtle and correlates with expression of PDGF-A. Lines 1–3: Good expression of PDGF-A, as shown by RT-PCR and immunohistochemistry, and consistently ectopic lightly staining cells in the inner nuclear layer of the peripheral retina (arrows). The posterior retina showed fewer changes and is not illustrated. Lines 4 and 5: Lowest expression of PDGF-A as shown by RT-PCR and immunohistochemistry. No identifiable changes are shown, even in the peripheral retina. C: Immunohistochemical staining for proliferating cell nuclear antigen shows more labeled nuclei on the surface of the retina and in the region of ectopic cells in the retinas of transgene-positive mice.
Figure 4.
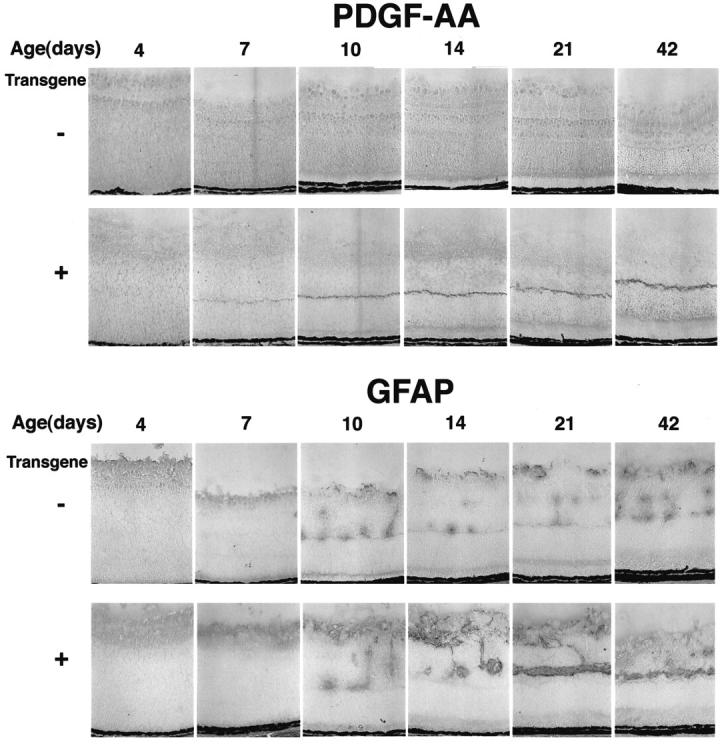
Time course of immunohistochemical staining for PDGF-AA and GFAP. Transgene-positive and -negative mice from the rho/PDGF-A3 line were sacrificed on P4, P7, P10, P14, P21, and P42. The eyes were frozen, sectioned, and stained for PDGF-AA or GFAP as described in Materials and Methods. Expression of PDGF-A is seen in the photoreceptors of transgene-positive mice starting on P7 and shows prominent staining, particularly in photoreceptor terminals, at all subsequent time points. At P4 and P7 in both transgene-positive and -negative mice, there is staining for GFAP in the nerve fiber layer, the normal location for astrocytes. At P10 and later time points, there is GFAP staining in the nerve fiber layer and in columns that extend into the inner nuclear layer, which likely represents perivascular astrocytes. The staining in both locations appears greater in transgene-positive mice and at P21 and P42, and there is a continuous horizontal streak of GFAP-positive cells that is not present in littermate controls.
rho/PDGF-A Mice Develop Retinal Gliosis
Light microscopy of H&E-stained retinal sections of adult rho/PDGF-A mice showed some irregularity and disruption of the inner nuclear layer, which were caused by ectopic cells with lightly staining cytoplasm in the periphery of the retina in lines 1–3 (Figure 3B) ▶ . Lines 4 and 5, which have the lowest expression of PDGF-A, showed no identifiable abnormalities. Staining for proliferating cell nuclear antigen demonstrated a few positive nuclei on the surface of the retina in transgene-negative mice, whereas transgene-positive mice showed increased labeled nuclei in the inner retina (Figure 3C) ▶ . All of the rho/PDGF-A lines showed increased GFAP staining in the inner nuclear layer, except line 4 (not shown). Figure 4 ▶ shows the time course of PDGF-A protein and GFAP expression in line 3. PDGF-A protein was first detectable in photoreceptors of transgenic mice on P7, at which time transgenic mice appeared to have increased GFAP staining in the nerve fiber layer compared with wild-type mice. At later time points there was increased staining for PDGF-A and a progressive increase in GFAP-reactive cells in the region corresponding to the ectopic cells in the inner nuclear layer seen in H&E-stained sections. Staining for GFAP was also done in line 2, and similar changes were seen (not shown).
Normally, astrocytes are the only cells in the retina that express GFAP, but after various insults such as retinal detachment, light damage, or genetic photoreceptor degeneration, Muller glia become GFAP-positive. 27,28 Staining for cellular retinaldehyde-binding protein (a marker for Muller cells 29 ) showed no differences between rho/PDGF-A2 or -A3 transgene-positive mice and wild-type mice (not shown).
Astrocytes normally reside in the nerve fiber layer and surround retinal blood vessels, 30 and they have been demonstrated to participate in the development of retinal blood vessels 31 and to affect their behavior, particularly for induction of barrier characteristics. 32 Staining with GSA, an endothelial cell-selective marker, demonstrated that the accumulation of GFAP-positive cells in the inner nuclear layer of rho/PDGF-A transgene-positive mice was accompanied by an accumulation of endothelial cells (Figure 5) ▶ , demonstrating that the development of the deep capillary beds was affected. The difference between transgene-positive and -negative mice was most prominent at P14 and became less noticeable at later time points.
Figure 5.

rho/PDGF-A transgenic mice have altered development of retinal vessels. Retinas from P14 and P21 rho/PDGF-A transgene-positive and littermate control mice were histochemically stained for GSA, a selective marker for endothelial cells. At P14 there was a horizontal band of endothelial cells in the inner nuclear layer, corresponding to the GFAP-positive band seen in Figure 4 ▶ . The band is less striking at P21, despite the continued prominence of the GFAP-positive band at P21.
rho/PDGF-A Transgenic Mice Are Protected from NV in Oxygen-Induced Ischemic Retinopathy
NV on the surface of the retina recruits cells to form epiretinal membranes that exert traction on the retina, often resulting in traction retinal detachments and severe loss of vision. Glial and RPE cells are major components of vascularized epiretinal membranes in diabetic retinopathy and other ischemic retinopathies. We hypothesized that endothelial cell-derived PDGFs are likely to stimulate the migration and proliferation of astrocytes and RPE cells and contribute to their recruitment and growth in neovascular membranes. Therefore, we anticipated that, in the oxygen-induced ischemic retinopathy model, the excess PDGF-A and excess astrocytes in rho/PDGF-A mice would result in increased NV compared with wild-type mice. Transgene-negative mice with ischemic retinopathy developed extensive NV on the surface of the retina, but, surprisingly, transgene-positive mice did not develop any NV (Figure 6, A and B) ▶ . A total of 10 transgene-positive mice with ischemic retinopathy were examined with GSA lectin staining of serial sections, and none had any NV (Figure 6C) ▶ .
Figure 6.

rho/PDGF-A transgenic mice do not develop hyperoxia-induced retinal NV. At P7, transgene-positive and littermate control mice were placed in 75% oxygen for 5 days followed by room air for 5 days and then sacrificed. Retinal frozen sections were stained with GSA. A: A transgene-negative mouse shows an area of retinal vascular nonperfusion (delimited by arrows) and large clumps of NV on the surface of the retina (arrowheads). B: A transgene-positive mouse shows no areas of vascular nonperfusion and no NV. C: Image analysis was performed to measure the area of NV, as described in Materials and Methods, on retinas from 10 transgene-negative and 10 transgene-positive mice. There was a dramatic difference with substantial NV in transgene-negative mice and no NV identified in any of the transgene-positive mice. *P < 0.0001 by two-tailed t-test.
PDGF-A Does Not Directly Inhibit NV
One possible explanation for the absence of NV in rho/PDGF-A mice with ischemic retinopathy is that the excess PDGF-A directly inhibits NV. Although this possibility was considered unlikely, it was tested by crossing rho/PDGF-A mice with rho/VEGF mice. rho/VEGF mice express VEGF in photoreceptors and develop intraretinal and subretinal NV. 25 Double transgenic mice that expressed both VEGF and PDGF-A in photoreceptors had more neovascular lesions per retina than mice that expressed only VEGF, but the area of individual lesions was significantly smaller, and therefore the total area of NV per retina was not significantly different (Figure 7) ▶ . These data indicate that PDGF-A does not directly inhibit retinal NV.
Figure 7.

Transgenic mice that express both PDGF-A and VEGF do not have less NV than mice that express only VEGF. rho/PDGF-A+/− mice were crossed with rho/VEGF+/+ mice. At P21, the pups were perfused with fluorescein-labeled dextran, and retinal whole mounts were examined by fluorescence microscopy. Mice were genotyped after quantitation of NV. rho/PDGF-A transgene-negative mice (A: ×100; B: ×400) had numerous areas of NV surrounded by retinal pigmented epithelial cells typical of rho/VEGF mice. rho/PDGF-A transgene-positive mice (C: ×100; D: ×400) also showed numerous areas of NV that appeared smaller with less participation of retinal pigmented epithelial cells. E: The number of neovascular lesions, the area per lesion, and the total area of NV per retina were measured by image analysis. There was no significant difference in the number of lesions or total area of NV per retina, but the average area of lesions was significantly less in mice that expressed both PDGF-A and VEGF, compared with mice that expressed only VEGF (*P = 0.0364 by two-tailed t-test).
rho/PDGF-A Mice Develop Less Hyperoxia-Induced Retinal Capillary Dropout
rho/PDGF-A and littermate control mice were placed in 75% oxygen at P7 and removed to room air at P12, which is the standard protocol for oxygen-induced ischemic retinopathy. At sequential time points after the onset of hyperoxia, mice were perfused with fluorescein-labeled dextran, and retinal flat mounts were prepared. At each time point, there was less capillary nonperfusion in the retinas of rho/PDGF-A mice compared with wild-type mice (Figure 8) ▶ . Measurements by image analysis confirmed that rho/PDGF-A mice developed significantly less nonperfused retina and had complete revascularization at P17, compared with P21 in wild-type mice (Figure 8B) ▶ .
Figure 8.
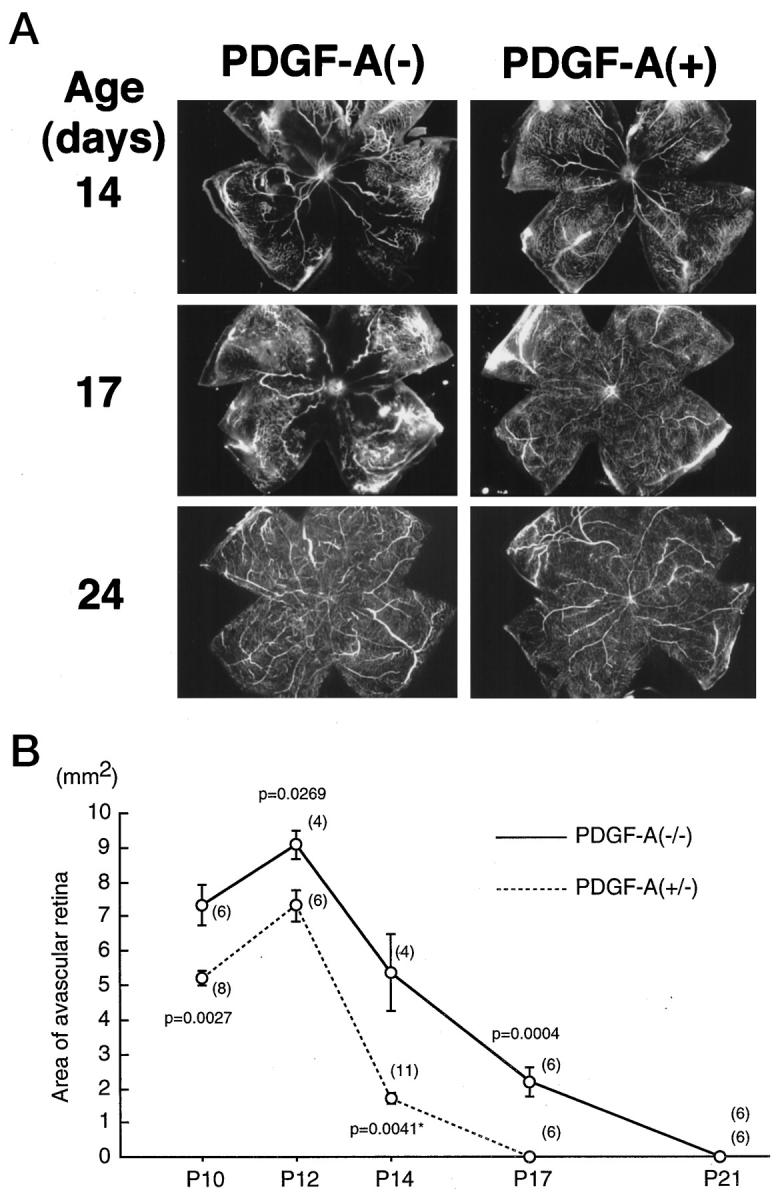
rho/PDGF-A transgene-positive (PDGF-A+) mice with ischemic retinopathy have less retinal vascular nonperfusion than littermate controls (PDGF-A−). Rho/PDGF-A+/− mice were crossed with wild-type mice, and resultant litters were placed in 75% oxygen at P7 and removed to room air at P12. At P10, P12, P14, P17, P21, and P24, mice were perfused with fluorescein-labeled dextran, and retinal whole mounts were prepared. The retinas were examined by fluorescence microscopy, and the area of vascular nonperfusion per retina was measured by image analysis. The mice were genotyped at the time of sacrifice, but the examiner was masked for genotype. A: At P14, PDGF-A− mice showed large areas of vascular nonperfusion, which are typical for wild-type mice after 5 days of hyperoxia, but PDGF-A+ mice showed only a small area of nonperfusion. At P17, PDGF-A+ mice had complete revascularization of the retina, but PDGF-A− mice still have a large area of nonperfusion. At P24, both types of mice were completely revascularized. B: The mean (± SEM) area of avascular retina was plotted for each time point for PDGF-A− and PDGF-A+ mice. The number of retinas used to calculate the mean for each time point (shown in parenthesis adjacent to each point) was determined by the litter sizes and chance breakdown of genotypes. At each time point, PDGF-A+ mice had significantly less avascular retina by two-tailed t-test than PDGF-A− mice.
VEGF Is Not Responsible for Retinal Endothelial Cell Rescue in rho/PDGF-A Mice
Hyperoxia results in capillary dropout in neonatal mice, because it causes decreased expression of VEGF, and endothelial cells of newly developed retinal vessels are dependent on VEGF for survival. 33 Exogenous VEGF rescues endothelial cells in hyperoxic retinas and astrocytes produce VEGF. 33 We examined whether the decrease in hyperoxia-induced retinal capillary dropout in rho/PDGF-A mice is caused by increased production of VEGF by ectopic astrocytes, which overcomes the down-regulation by hyperoxia. Northern blots demonstrated increased VEGF mRNA in the retinas of P7 transgene-positive mice compared with transgene-negative mice as predicted, but after either 4 hours or 3 days of hyperoxia, VEGF mRNA levels were decreased to essentially undetectable levels in both transgene-positive and -negative mice (Figure 9) ▶ . Within hours of removing mice from 75% oxygen to the relative hypoxia of room air, there was a marked increase in VEGF mRNA in the retinas of littermate controls, with only a small increase in VEGF mRNA in the retinas of transgene-positive mice. At later time points, there continued to be much greater VEGF mRNA in the retinas of control mice compared with transgenic mice. These data suggest that, despite increased VEGF expression by ectopic astrocytes in the retinas of rho/PDGF-A mice under normoxic conditions, hyperoxia effectively eliminates VEGF expression in both transgenic and wild-type mice. Therefore, VEGF is not responsible for retinal endothelial cell rescue from hyperoxic damage in rho/PDGF-A mice. The increased expression of VEGF mRNA after removal from 75% oxygen to room air in the retinas of littermate controls compared with rho/PDGF-A mice is consistent with the demonstration that rho/PDGF-A mice have less retinal capillary dropout and therefore less retinal ischemia.
Figure 9.

Retinal expression of mRNA for endothelial survival factors in neonatal rho/PDGF-A transgene-positive and -negative mice during and after hyperoxia. rho/PDGF-A+/− mice were crossed with wild-type mice, and resultant litters were placed in 75% oxygen at P7 and removed to room air at P12. Mice were sacrificed at P7, before placement in hyperoxia or 4 hours after placement in hyperoxia, P10 (after 3 days of hyperoxia), P12 (after 5 days of hyperoxia and 1 hour of hypoxia), P14 (2 days after removal from hyperoxia to the relative hypoxia of room air), and P17 (after 5 days of hypoxia when there is severe NV). Total retinal RNA was isolated and 10 μg were used for each lane for Northern blots. The blots were hybridized with a VEGF cDNA probe and, after exposure, were stripped and hybridized with a probe for IGF-I. Blots were successively stripped and rehybridized with probes for FGF2, PDGF-A, and 18S. At P12 and P14, during the period when there was an endothelial rescue effect in transgene-positive mice as judged by the decreased vascular nonperfusion, FGF2 mRNA expression was increased in transgene-positive mice compared with transgene-negative mice, although this was not true for the other survival factors.
Temporal and Spatial Correlation of FGF2 Expression and Improved Endothelial Cell Survival in rho/PDGF-A Mice
Two factors other than VEGF that have survival-promoting activity for endothelial cells are insulin-like growth factor-I (IGF-I) and fibroblast growth factor 2 (FGF2). Northern blots with probes for IGF-I showed that, after 5 days of hyperoxia followed by 1 hour in room air, IGF-I mRNA was markedly increased, but there was no difference between transgene-positive and -negative mice that would help to explain the rescue of endothelial cells in transgenic mice. At P14 and P17 there was somewhat greater IGF-I mRNA in the retinas of transgene-positive mice, that could have contributed to the more rapid revascularization. The differences for FGF2 mRNA were more striking, with a small increase in transgene-positive mouse retinas at P12 and a large difference at P14 (Figure 9) ▶ . This experiment was repeated with mice from lines 2 and 3 and the differential increase in FGF2 mRNA at P12 and P14 in transgenic mice compared with wild-type littermates was a consistent finding. Immunohistochemistry demonstrated increased staining for FGF2 in the inner retina in the region of the ectopic glial cells of transgenic mice (Figure 10) ▶ . The first time point examined was P10, which is 3 days after the onset of hyperoxia, suggesting that hyperoxia somehow induces the production of FGF2. These data are consistent with astrocyte-derived FGF2 acting as a survival factor for retinal endothelial cells of hyperoxic/hypoxic retinas of rho/PDGF-A mice.
Figure 10.
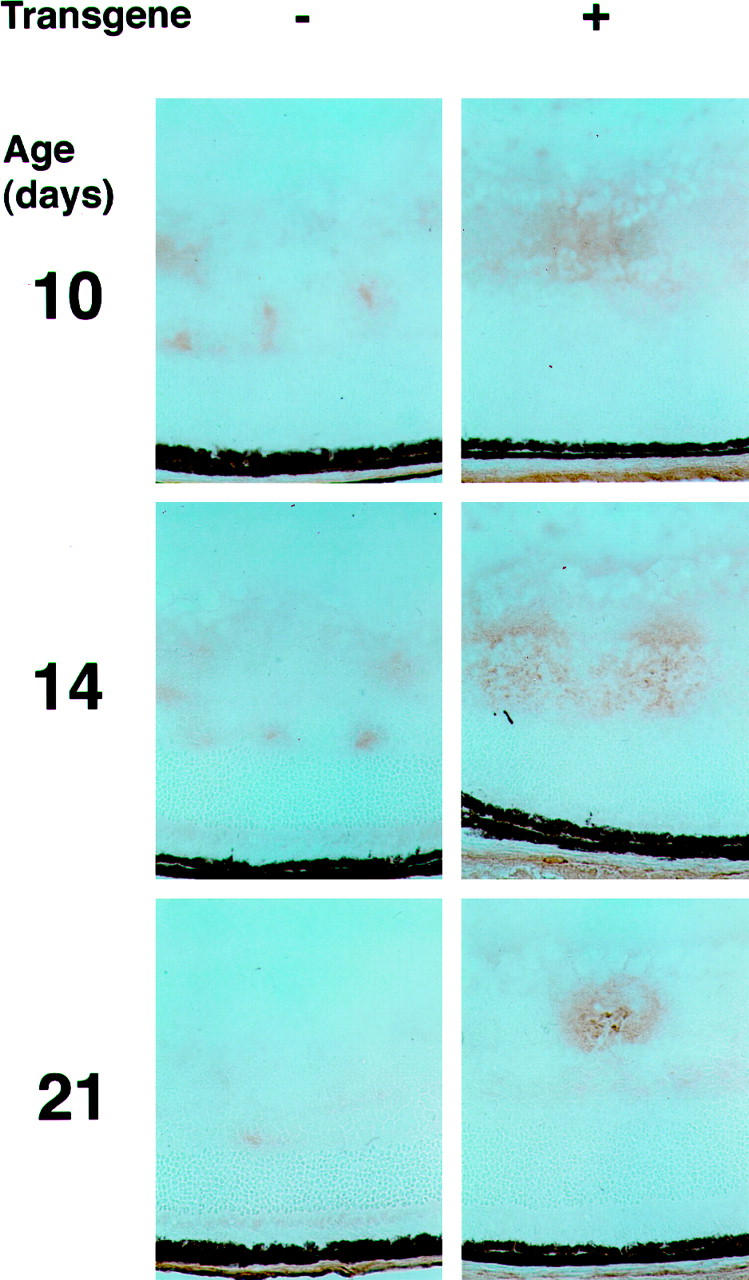
Immunohistochemistry for FGF2 in the retinas of P10, P12, and P21 rho/PDGF-A transgene-positive and -negative mice with ischemic retinopathy. rho/PDGF-A+/− mice crossed with wild-type mice and resultant litters were placed in 75% oxygen at P7 and removed to room air at P12. Mice were sacrificed at P10, after 3 days of hyperoxia; P14, 2 days after removal from hyperoxia to the relative hypoxia of room air; and P21, 9 days after onset of the hypoxic period. Eyes were rapidly removed, and frozen sections were immunohistochemically stained for FGF2. At each time point, there was increased staining for FGF2 in transgene-positive mice compared with that in transgene-negative mice. The FGF2 staining occurred in the inner part of the retina, where both the ectopic astrocytes and retinal blood vessels were located.
Discussion
In this study, we have demonstrated that increased expression of PDGF-A in photoreceptors causes migration and proliferation of GFAP-positive astrocytes, so that they are increased in the inner nuclear and nerve fiber layers of the retina. This results in some irregularity of the inner nuclear layer, particularly in the peripheral portion of the retina in some mice, and some alteration in the development of the deep retinal capillary beds, but the phenotype is quite subtle. However, in the setting of oxygen-induced ischemic retinopathy, rho/PDGF-A mice show a dramatic difference from littermate wild-type mice, because they do not develop any retinal NV. This is not because PDGF-A inhibits NV, because double transgenic mice with increased expression of both PDGF-A and VEGF do not have less NV than transgenic mice with increased expression of only VEGF. Instead, it is because rho/PDGF-A mice develop less hyperoxia-induced retinal capillary dropout and reestablish a complete retinal vasculature more quickly. As a result, the increase in VEGF expression on removal from hyperoxia to room air is much less in rho/PDGF-A mice compared with wild-type mice and is below the threshold needed for NV.
An important question is, why are rho/PDGF-A mice more resistant to hyperoxia-induced capillary dropout? The most likely explanation is that the additional astrocytes produce a factor(s) that promotes survival of vascular endothelial cells. Astrocytes are known to produce VEGF and exogenous VEGF rescues endothelial cells from hyperoxia-induced apoptosis. 33 However, although rho/PDGF-A mice show prominent constitutive expression of VEGF, possibly due to increased numbers of astrocytes, hyperoxia causes decreased expression of VEGF in the retinas of rho/PDGF-A-transgenic mice to essentially undetectable levels, just as occurs in wild-type mice. Therefore, it is unlikely that VEGF is responsible for the decreased susceptibility to hyperoxia in rho/PDGF-A mice.
Two other survival factors for endothelial cells are IGF-I and FGF2. Northern blots showed that IGF-I was increased during the hypoxic period in both rho/PDGF-A and wild-type mice. There was no difference in IGF-I mRNA levels at P12, when there was a substantial difference in capillary nonperfusion, so IGF-I is unlikely to be responsible for the rescue of endothelial cells in rho/PDGF-A mice. However, it is interesting that IGF-I mRNA is increased in hypoxic retina, since it has been demonstrated to contribute to ischemic retinopathy 34 and recently a hypoxia-inducible factor-1 response element has been demonstrated in the IGF-I promoter. 35 At P14 and P17 there was slightly increased levels of IGF-I mRNA in transgenic mice, but the most dramatic difference during the critical period when enhanced survival of endothelial cells became manifested in transgenic mice was in FGF2 expression. This was confirmed to occur in mice from both lines 2 and 3, and immunohistochemistry showed that the increased staining for FGF2 occurred throughout the inner retina in the same sort of pattern of increased staining for GFAP. Increased production of FGF2 in ectopic astrocytes provides a good explanation for the decreased susceptibility to hyperoxia-induced capillary dropout in rho/PDGF-A mice. Astrocytes normally associate with retinal blood vessels and produce substances that promote barrier characteristics. 32 Our findings in rho/PDGF-A mice suggest that another function of astrocytes in the retina is to promote endothelial cell survival and that FGF2 may be one mediator of this effect.
Previous studies have investigated the effect of increased expression of PDGF-A in neurons of the inner retina or the lens. Fruttiger and associates 36 used the neuron-specific enolase (NSE) promoter to drive expression of the short isoform of PDGF-A, beginning before the day of birth in retinal ganglion cells and some cells of the inner and outer nuclear layer of transgenic mice. These mice showed numerous ectopic astrocytes that extended throughout the entire depth of the retina, but the radial spread of astrocytes from the optic nerve to the periphery was decreased. NV occurred in association with the extensive astrocyte network in all retinal layers of posterior retina and did not occur in the periphery of the retina, where there were no astrocytes. Reneker and Overbeek 37 used the αA-crystallin promoter to drive expression of PDGF-A, beginning around embryonic day 12 in the lenses of transgenic mice. These mice developed a several-layer-thick carpet of astrocytes on the surface of the retina. NSE/PDGF-A 36 and αA-crystallin/PDGF-A 37 mice each have a larger number of additional astrocytes and a more dramatic phenotype than rho/PDGF-A mice. Astrocytes may be responsive to PDGF-A only between P0 and P14, 37,38 and therefore glial cell proliferation may proceed for 2 weeks in NSE/PDGF-A and αA-crystallin/PDGF-A mice, compared with 1 week in rho/PDGF-A mice, which would explain why the former two types of transgenic mice show greater numbers of additional astrocytes. The difference in location of ectopic astrocytes in αA-crystallin/PDGF-A mice compared with rho/PDGF-A mice is probably explained by the location of PDGF-A expression. The production of PDGF-A in the lens would provide a gradient favoring migration of astrocytes to the retinal surface, whereas photoreceptor-derived PDGF-A would provide a gradient favoring migration into the retina from the retinal surface.
Unlike αA-crystallin/PDGF-A or rho/PDGF-A mice, NSE/PDGF-A mice show extensive NV in association with astrocyte proliferation. Because astrocytes have been demonstrated to produce VEGF, it was postulated that the NV was caused by excess astrocyte-derived VEGF from the large number of ectopic astrocytes, although this was not demonstrated. However, if this were the case, one would expect prominent NV in αA-crystallin/PDGF-A mice, and, although there were a few GFAP-negative cells, which were postulated to be blood vessel cells, there was no prominent NV. In addition to large numbers of ectopic astrocytes, NSE/PDGF-A mice had areas of nonperfused peripheral retina, because retinal blood vessel development did not extend beyond the ectopic astrocytes located posteriorly. Nonperfused retina is a source of VEGF, and this, combined with astrocyte-derived VEGF, may be sufficient to cause NV, although the amount of VEGF produced by ectopic astrocytes alone in αA-crystallin/PDGF-A or rho/PDGF-A mice is insufficient to cause NV.
Taken together, the findings in rho/PDGF-A, αA-crystallin/PDGF-A, and NSE/PDGF-A mice suggest that differences in the temporal and/or spatial pattern of expression of PDGF-A in the eye can result in large phenotypic differences in the retina. An inducible promoter system combined with multiple retina-specific promoters would provide a means for detailed analysis of the effects of alterations in timing and location of PDGF-A expression. Such an analysis will be needed to determine whether altered expression of PDGF-A in the retinas of adult animals causes changes like those seen in proliferative retinopathies. But even without such a detailed analysis, it appears safe to conclude that alteration of PDGF-A expression in the eye during development alters the number and location of astrocytes and secondarily affects the development and survival of blood vessels.
Footnotes
Address reprint requests to Peter A. Campochiaro, M.D., Maumenee 719, The Johns Hopkins University School of Medicine, 600 N. Wolfe Street, Baltimore, MD 21287-9277. E-mail: pcampo@jhmi.edu.
Supported by Public Health Service grants EY05951, EY12609, EY10017, EY09769, and core grant P30EY1765 from the National Eye Institute; a Juvenile Diabetes Foundation fellowship grant (to N. O.); a Lew R. Wasserman Merit Award (to P. A. C.); a career development award (to D. J. Z.); an unrestricted grant from Research to Prevent Blindness, Inc.; the Rebecca P. Moon, Charles M. Moon, Jr., and Dr. P. Thomas Manchester Research Fund; a grant from Mrs. Harry J. Duffey; a grant from Dr. and Mrs. William Lake; a grant from Project Insight; and a grant from the Association for Retinopathy of Prematurity and Related Diseases. P. A. C. is the George S. and Dolores Dore Eccles Professor of Ophthalmology and Neuroscience.
Current address for H. Yamada and E. Yamada: The Department of Ophthalmology, Kansai Medical University, Osaka, Japan.
References
- 1.Heldin CH, Westermark B: Platelet-derived growth factor: mechanism of action and possible in vivo function. Cell Reg 1990, 8:555-566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierce GF, Mustoe TA, Altrock BW, Deuel TF, Thomason A: Role of platelet-derived growth factor in wound healing. J Cell Biochem 1991, 45:319-326 [DOI] [PubMed] [Google Scholar]
- 3.Mercola M, Wang C, Kelley J, Brownlee C, Jackson-Grusby L, Stiles C, Bowen-Pope D: Selective expression of PDGF and its receptor during early mouse embryogenesis. Dev Biol 1990, 138:114-122 [DOI] [PubMed] [Google Scholar]
- 4.Leveen P, Pekny M, Gerbre-Medhin S, Swolin B, Larsson E, Betsholtz C: Mice deficient for PDGF-B show renal, cardiovascular, and hematological abnormalities. Genes Dev 1994, 8:1875-1887 [DOI] [PubMed] [Google Scholar]
- 5.Soriano P: Abnormal kidney development and hematological disorders in PDGF β-receptor mutant mice. Genes Dev 1994, 8:1888-1896 [DOI] [PubMed] [Google Scholar]
- 6.Schatteman GC, Motley ST, Effmann EL, Bowen-Pope DF: Platelet-derived growth factor receptor α subunit deleted patch mouse exhibits severe cardiovascular dysmorphogenesis. Teratology 1995, 51:351-366 [DOI] [PubMed] [Google Scholar]
- 7.Smits A, Kato M, Westermark B, Nister M, Heldin CH, Funa K: Neurotrophic activity of platelet-derived growth factor (PDGF): rat neuronal cells possess functional PDGF β-type receptors and respond to PDGF. Proc Natl Acad Sci USA 1991, 88:8159-8163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mudhar HS, Pollock RA, Wang C, Stiles CD, Richardson WD: PDGF and its receptors in the developing rodent retina and optic nerve. Development 1993, 118:539-552 [DOI] [PubMed] [Google Scholar]
- 9.Hart CE, Bowen-Pope DF: Platelet-derived growth factor receptor: current views of the two subunit model. J Invest Dermatol 1990, 94:535-575 [DOI] [PubMed] [Google Scholar]
- 10.Watanabe T, Raff MC: Retinal astrocytes are immigrants from the optic nerve. Nature 1988, 332:834-837 [DOI] [PubMed] [Google Scholar]
- 11.Noble M, Murray K, Stroobant P, Waterfield MD, Riddle P: Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte-type-2 astrocyte progenitor cell. Nature 1988, 333:560-562 [DOI] [PubMed] [Google Scholar]
- 12.Pringle N, Collarini EJ, Mosley MJ, Heldin C-H, Westermark B, Richardson WD: PDGF A chain homodimers drive proliferation of bipotential (O-2 A) glial progenitor cells in the developing rat optic nerve. EMBO J 1989, 8:1049-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barres BA, Raff MC: Control of oligodendrocyte number in the developing rat optic nerve. Neuron 1994, 12:935-942 [DOI] [PubMed] [Google Scholar]
- 14.Campochiaro PA, Hackett SF, Vinores SA, Freund J, Csaky C, La Rochelle W, Henderer J, Johnson M, Rodriguez IR, Friedman Z, Derevjanik N, Dooner J: Platelet-derived growth factor is an autocrine growth stimulator in retinal pigmented epithelial cells. J Cell Sci 1994, 107:2459-2469 [DOI] [PubMed] [Google Scholar]
- 15.Robbins SG, Mixon KN, Wilson DJ, Hart CE, Robertson JD, Westra I, Planck SR, Rosenbaum JT: Platelet-derived growth factor ligands and receptors immunolocalized in proliferative retinal diseases. Invest Ophthalmol Vis Sci 1994, 35:3649-3663 [PubMed] [Google Scholar]
- 16.Vinores SA, Henderer JD, Mahlow J, Chiu C, Derevjanik NL, LaRochelle W, Csaky C, Campochiaro PA: Isoforms of platelet-derived growth factor and its receptors in epiretinal membranes: immunolocalization to retinal pigmented epithelial cells. Exp Eye Res 1995, 60:607-619 [DOI] [PubMed] [Google Scholar]
- 17.Zack DJ, Bennett J, Wang Y, Davenport C, Klaunberg B, Gearhart J, Nathans J: Unusual topography of bovine rhodopsin promoter-lac Z fusion gene expression in transgenic mouse retinas. Neuron 1991, 6:187-199 [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 19.Ozaki H, Okamoto N, Ortega S, Chang M, Ozaki K, Sadda S, Vinores MA, Derevjanik N, Zack DJ, Basilico C, Campochiaro PA: Basic fibroblast growth factor in neither necessary nor sufficient for the development of retinal neovascularization. Am J Pathol 1998, 153:757-765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vinores SA, Campochiaro PA, McGehee R, Orman W, Hackett SF, Hjelmeland LM: Ultrastructural and immunocytochemical changes in retinal pigment epithelium, retinal glia, and fibroblasts in vitreous culture. Invest Ophthalmol Vis Sci 1990, 31:2529-2545 [PubMed] [Google Scholar]
- 21.Vinores SA, Derevjanik NL, Mahlow J, Hackett SF, Haller JA, deJuan E, Frankfurter A, Campochiaro PA: Class III β-tubulin in human retinal pigment epithelial cells in culture, and in epiretinal membranes. Exp Eye Res 1993, 60:385-400 [DOI] [PubMed] [Google Scholar]
- 22.Reilly TM, Taylor DS, Herblin WF, Thoolen MJ, Chiu AT, Watson DW, Timmermans PBMWM: Monoclonal antibodies directed against basic fibroblast growth factor which inhibit its biological activity in vitro and in vivo. Biochem Biophys Res Comm 1989, 164:736-743 [DOI] [PubMed] [Google Scholar]
- 23.Hall PA, Levinson DA, Woods AL, Yu CCW, Kellock DB, Watkins JA, Barnes DM, Gillet CE, Camplejohn R, Dover R, Waseem NH, Lane DP: Proliferating cell nuclear antigen (PCNA) immunolocalization in paraffin section: an index of cell proliferation with evidence of deregulated expression in some neoplasms. J Pathol 1990, 162:285-294 [DOI] [PubMed] [Google Scholar]
- 24.Smith LEH, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA: Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 1994, 35:101-111 [PubMed] [Google Scholar]
- 25.Okamoto N, Tobe T, Hackett SF, Ozaki H, Vinores MA, LaRochelle W, Zack DJ, Campochiaro PA: Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol 1997, 151:281-291 [PMC free article] [PubMed] [Google Scholar]
- 26.Tobe T, Okamoto N, Vinores MA, Derevjanik NL, Vinores SA, Zack DJ, Campochiaro PA: Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest Ophthalmol Vis Sci 1998, 39:180-188 [PubMed] [Google Scholar]
- 27.Erickson PA, Fisher SK, Guerin CJ, Anderson DH, Kaska DD: Glial fibrillary acidic protein in Muller cells after retinal detachment. Exp Eye Res 1987, 44:37-48 [DOI] [PubMed] [Google Scholar]
- 28.Eisenfeld AJ, Bunt-Milam AH, Sarthy PV: Muller cell expression of glial fibrillary acidic protein after genetic and experimental photoreceptor degeneration in the rat retina. Invest Ophthalmol Vis Sci 1984, 25:1321-1328 [PubMed] [Google Scholar]
- 29.Bunt-Milam AH, Saari JC: Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. J Cell Biol 1983, 97:703-712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stone J, Dreher Z: Relationship between astrocytes, ganglion cells and vasculature of the retina. J Comp Neurol 1987, 255:35-49 [DOI] [PubMed] [Google Scholar]
- 31.Stone J, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T, Keshet E: Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 1995, 15:4738-4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janzer RC, Raff MC: Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325:253-257 [DOI] [PubMed] [Google Scholar]
- 33.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 34.Smith LEH, Kopchick JJ, Chen W, Knapp J, Kinose F, Daley D, Foley E, G. SR, Schaeffer JM: Essential role of growth hormone in ischemia-induced retinal neovascularization. Science 1997, 276:1706–1709 [DOI] [PubMed]
- 35.Lukiw WJ, Duhault J, Koenig-Berard E, Gordon WC, DeTurco ER, Bazan NG: Transcription factor HIF-1- and AP2-DNA binding orchestrate angiogenic gene transcription programs during retinal neovascularization. Invest Ophthalmol Vis Sci 1999, 40:S166 [Google Scholar]
- 36.Fruttiger M, Calver AR, Kruger WH, Mudhar HS, Michalovich D, Takakura N, Nishikawa SI, Richardson WD: PDGF mediates a neuron-astrocyte interaction in developing retina. Neuron 1996, 17:1117-1131 [DOI] [PubMed] [Google Scholar]
- 37.Reneker LW, Overbeek PA: Lens-specific expression of PDGF-A in transgenic mice results in retinal astrocytic hamartomas. Invest Ophthalmol Vis Sci 1996, 37:2455-2466 [PubMed] [Google Scholar]
- 38.Hart IK, Richardson WD, Heldin C-H, Westermark B, Raff MC: PDGF receptors on cells of the oligodendrocyte-type-2 astrocyte (O-2A) cell lineage. Development 1989, 105:594-603 [DOI] [PubMed] [Google Scholar]