

Abstract
The twin-to-twin transfusion syndrome (TTS) results from an unbalanced blood supply through placental anastomoses in monochorionic twins. It induces growth restriction, renal tubular dysgenesis, and oliguria in the donor and visceromegaly and polyuria in the recipient. A better understanding of its pathophysiology could contribute to improving the management of TTS, which still carries a high perinatal mortality in both twins. As well as several other candidates, the renin-angiotensin system might be involved in TTS. To evaluate its role in the pathogenesis of the syndrome, we studied the kidneys of 21 twin pairs who died from TTS at 19 to 30 weeks, compared with 39 individuals in a control group, using light microscopy, immunohistochemistry, and in situ hybridization. The overexpression of the renin protein and transcript with frequent evidence of renin synthesis by mesangial cells was observed in the donor kidneys, presumably as a consequence of chronic renal hypoperfusion. This upregulation of renin synthesis might be beneficial to restore euvolemia. In severe cases of TTS, however, angiotensin-II-induced vasoconstriction acts as an additional deleterious factor by further reducing the renal blood flow in donors. In recipients, renin expression was virtually absent, possibly because it was down-regulated by hypervolemia. However, in addition to congestion and hemorrhagic infarction, there were severe glomerular and arterial lesions resembling those observed in polycythemia- or hypertension-induced microangiopathy. We speculate that fetal hypertension in the recipient might be partly mediated by the transfer of circulating renin produced by the donor, through the placental vascular shunts.
The twin-to-twin transfusion syndrome (TTS) is a severe complication occurring in 5% of monochorionic twin pregnancies. This condition is thought to result from an unbalanced fetal blood supply through the placental vascular shunts, with the larger twin being the recipient and the smaller twin the donor. At an early stage, the disease is characterized by polyuria and polyhydramnios in the recipient fetus, whereas there is oliguria and, therefore, oligohydramnios in the donor. Major polyhydramnios may cause extreme premature labor. Severe oligohydramnios results in the stuck twin phenomenon, a condition in which the donor appears tightly wrapped in his amniotic sac. In the most severe cases, the recipient may die from cardiac failure and the smaller twin from anemia and hypoxia, or both twins may die as a result of premature labor. In either twin, severe ischemic brain damage may result from acute hemodynamic changes.
Over the past decade, the survival rate of TTS babies has increased owing to improved neonatal resuscitation and to prenatal therapy based on amniodrainage, 1-3 coagulation of the intertwin placental shunts, 4-6 selective feticide, 7-9 or amniotomy. 10 However, TTS still carries a perinatal mortality as high as 50% with a short-term neurological morbidity of 5 to 18%, 2,5,8 underscoring the need for further research.
Previous studies suggest that TTS has a complex pathophysiology that is derived primarily from an unbalanced blood flow from the donor to the recipient through arteriovenous placental anastomoses. In the recipient twin, polyuria is thought to result from hypervolemia 11,12 and could be partly mediated by the activation of atrial–natriuretic-factor production. 13 In addition, a recent case report suggested that systemic hypertension may occur in the recipient during the fetal stage. 14 In contrast, the diuresis of the donor decreases, possibly as a consequence of hypovolemia. Therefore, the regulation of fetal volemia and diuresis could be central to the pathophysiology of TTS .
We speculated that the activation of the renin angiotensin system (RAS) in the donor could be one of the noxious adaptive changes in TTS. To test this hypothesis, we conducted a retrospective postmortem study in 21 monochorionic twin pairs who died from TTS and in 39 controls. The morphology of the fetal kidneys and the level of renin expression were studied as markers of the potential involvement of the RAS in the cascade of hemodynamic and metabolic changes induced by the syndrome.
Materials and Methods
Between 1988 and 1998, 21 pairs of monochorionic twins with a prenatal history of severe TTS were referred to two laboratories of fetal pathology (Cochin Hospital, 9 cases; Necker Enfants-Malades Hospital, 12 cases). All kidneys were examined by one of the authors (M. C. G.).
Gestational age at clinical presentation ranged from 17 to 28 menstrual weeks and, at delivery, from 19 to 30 weeks. There were 13 terminations of pregnancy, 3 miscarriages at ≤24 weeks, 4 premature deliveries with neonatal deaths of both twins, and 1 premature delivery with intrauterine death of the donor and neonatal death of the recipient.
The prenatal diagnosis of TTS was based on the occurrence of major polyhydramnios in one twin of a monochorionic diamniotic pregnancy. Nine recipients were hydropic, and eight had cardiomyopathy without hydrops. The donor had oligohydramnios in 20 cases, 7 of which presented with a stuck-twin syndrome (Table 1) ▶ . The diagnosis of a monochorionic-diamniotic twin pregnancy was confirmed by a gross examination of the placenta and by a histological study of the intertwin membrane. In all cases, postmortem findings confirmed the diagnosis of TTS, the donor being growth retarded with a small heart and kidneys while the recipient twin was larger, with an enlarged heart and kidneys (Table 2) ▶ . There were no associated malformations.
Table 1.
Obstetrical History
| Amniotic fluid volume | Recipient | Donor |
|---|---|---|
| Polyhydramnios/stuck twin (n = 7) | Hydropic (n = 5) | IUGR (n = 5) |
| Cardiomyopathy, intrauterine death (n = 1) | IUGR (n = 1) | |
| Normal heart (n = 1) | IUD of donor (n = 1) | |
| Polyhydramnios/oligohydramnios (n = 13) | Hydropic (n = 3) | IUGR (n = 3) |
| Hydropic, IUD (n = 1) | IUD (n = 1) | |
| Cardiomyopathy (n = 6) | IUGR (n = 6) | |
| IUD (n = 1) | Neonatal death (n) | |
| Normal heart (n = 2) | IUGR (n = 2) | |
| Polyhydramnios/normal AFV (n = 1) | Cardiomyopathy (n = 1) | IUGR (n = 1) |
IUGR, intrauterine growth restriction; IUD, intrauterine death; AFV, amniotic fluid volume.
Table 2.
Body, Heart, and Kidney Weights: Donor versus Recipient
| Parameter | Donor | Recipient | Recipient/donor |
|---|---|---|---|
| Body weight (g) | 476 ± 49 | 645 ± 74 | 1.4 ± 0.06 |
| Heart weight (g) | 3.2 ± 0.37 | 8.6 ± 1.23 | 2.8 ± 0.25 |
| Kidney weight (g) | 3.45 ± 0.50 | 6 ± 0.90 | 2.0 ± 0.20 |
The recipient/donor weight ratios were not calculated in cases with intrauterine death of one twin. Results are expressed as mean ± SEM.
Normal kidneys obtained from 21 singleton fetuses (18–30 weeks), 7 twins, and 2 triplet fetuses (18–28 weeks) served as controls. Control kidneys from singleton fetuses were obtained at an autopsy after the spontaneous abortion of 5 apparently normal fetuses or the termination of pregnancy for extrarenal anomalies (16 fetuses). None of these fetuses had polyhydramnios or oligohydramnios. The termination of the twin pregnancy was performed because of chemotherapy for a tumor in the mother in one case and severe extrarenal malformations of the fetuses in another. No specific cause was found for the abortion of the five other twin pairs with a dichorionic, diamniotic placenta. Spontaneous abortion also occurred in two triplet pregnancies, one at 22 and one at 27 weeks, the former of which was due to chorioamniotitis. The kidneys from only two of the triplet fetuses were available for immunohistochemical studies. There were no symptoms of TTS in any case of multiple pregnancies. Amniotic fluid abundance was normal. No significant difference in body weight was observed between twins or triplets. The fetal kidneys were processed as described below
Morphology
Both kidneys of each twin were fixed in 10% buffered formalin. Frontal corticomedullary sections were taken through the hilus and embedded in paraplast. Sections (4 μm thick) were stained with trichrome-light green, trichrome-safran, or periodic acid/Schiff reagent.
Immunohistochemistry
Immunostaining was performed on formalin-fixed paraffin-embedded tissues by the avidin-biotin method and with the Universal Immunostaining Streptavidin-Peroxidase Kit (Immunotech, Marseille, France). Good conservation of the kidneys allowed their study in 19 twin pairs. Deparaffinized sections were rehydrated, washed in a phosphate-buffered saline buffer for 5 minutes, treated 5 minutes by 3% H2O2 in methanol to block any endogenous-peroxidase activity, and washed in a buffer for 2 minutes. Then they were incubated sequentially for 10 minutes with the protein-blocking agent and for 60 minutes with the primary antibody (see next section). After washing in a phospate-buffered saline, the sections were incubated for 30 minutes with the polyvalent secondary biotinylated antibody. Washed sections were incubated for 45 minutes with the streptavidin-peroxidase reagent. After washing, they were incubated for 10 to 20 minutes with the freshly prepared chromogen solution (H2O2 + the chromogen 3-amino-9 ethylcarbazole). The sections were counterstained with hematoxylin and mounted in aqueous media. The control sections were processed as above, but the primary antibodies were omitted.
Rabbit polyclonal anti-renin antibodies were raised against human renin purified from a juxtaglomerular cell tumor. 15 Their specificity was previously established. 16 The antiserum with the highest antirenin titer, a gift from P. Corvol (Institut National de la Santé et de la Recherche Médicale Unité 36, Paris), was selected for the study. We used a monoclonal anti-α-smooth muscle actin (SMA; Sigma Chemical, St. Louis, MO), an Iopath monoclonal antibody anti-epithelial membrane antigen (EMA), which is a marker for distal and collecting tubules (Immunotech, Marseille, France), and a monoclonal antibody anti-CD15, which recognizes an early myeloid differentiation antigen and is a marker for proximal tubules (PTs; Immunotech). Antibodies were used at the following concentrations: anti-EMA, pure (prediluted by the manufacturer); anti-CD15, 1/100; anti-SMA, 1/500; and anti-renin, 1/500.
A semiquantitative evaluation of renin content was performed on the strictly corticomedullary sections taken through the hilus. Developing nephrons were not considered. We recorded the number of positive juxtaglomerular apparatus (JGA) for the 100 glomeruli examined and the number of renin-positive cells per section of JGA. We also evaluated the presence of renin-positive cells in the wall of afferent arterioles at a distance from glomeruli and in the interlobular arteries. Normal kidneys from single or multiple pregnancies served as controls.
In Situ Hybridization
The renin cDNA clone pGRh14 17 was a gift from J. M. Gasc. 18 Recombinant plasmids were linearized with the restriction enzymes HindIII or Sac. The in vitro transcription was carried out in the presence of digoxigenin (DIG)-uridine 5′-triphosphate, using the RNA polymerases T7 or SP6 probes to produce antisense or sense probes, by the manufacturer’s instructions (Boehringer Mannheim, Mannheim, Germany). These probes were DNase I-treated, purified, and stored at −80°C.
In situ hybridization was performed in seven twin pairs. Sections (6 μm thick) mounted on silanated slides were deparaffinized, postfixed in 4% paraformaldehyde before digestion for 20 minutes with proteinase K (Sigma), and again postfixed in paraformaldehyde. After dehydration and air-drying, the sections were prehybridized, drained, and hybridized overnight in a humid chamber at 52°C with DIG-labeled RNA sense or antisense probes (5–10 ng/ml) in the prehybridization buffer. Slides were washed at room temperature in solutions of various degrees of stringency (from 5× standard saline citrate with 50% formamide at 55°C to 0.1× standard saline citrate). DIG-labeled probes were detected by immunohistochemical methods with the Nonradioactive Nucleic Acid Detection Kit (Boehringer Mannheim). Briefly, slides were washed for 5 minutes at room temperature (RT) in a washing buffer and incubated for 30 minutes with a 0.5% Boehringer blocking reagent in the same buffer. They were then incubated for 1 hour at RT with alkaline phosphatase-conjugated sheep anti-DIG antibody at 1/500 dilution. After washing, the color was developed with the chromogenic agents nitroblue tetrazolium, 5-bromo-4-chloro-3-indolyl phosphate, and 2 mmol/L levamisole in the dark at RT. After the color was fully developed, all of the sections were washed at the same time with the buffer (100 mmol/L Tris-HCl, 1 mmol/L ethylenediaminetetraacetic acid, pH 8) to allow a comparison of the hybridization signal and then mounted in an aqueous medium.
A semiquantitative evaluation of renin expression was performed by the same method as described for the assessment of renin protein. Four normal fetal kidneys served as controls.
Results
Morphology and Immunochemistry
Normal Controls (18–30 Weeks)
In the 39 control fetuses, the various steps of nephron differentiation were observed from the outer to the deep cortex. Undifferentiated mesenchyme and immature structures were located beneath the capsula, and the most mature nephrons were seen at the juxtamedullary junction. Even at the early development stages, the different tubular segments could be identified by immunostaining. PTs were labeled with CD15 antibodies from the glomerular capillary stage onwards, whereas distal tubules and collecting ducts were stained with anti-EMA antibodies. The vascular smooth muscle cells strongly expressed α-SMA. No expression was detected in the interstitial or blastema cells, whereas occasional expression could be seen in a few glomerular mesangial cells.
Donor Twins (18–30 Weeks)
In 10 of the 21 fetuses (20–30 weeks), renal tubular dysgenesis (RTD) was observed (Figure 1a) ▶ . The cortical tubules were lined by small cuboidal or columnar epithelial cells, none showing an apical brush border characteristic of the proximal tubules. With the CD15 antibody, a marker of PT, there was no or only very occasional labeling of the small sections of poorly differentiated tubules of the deep cortex. Conversely, strong and uniform tubular apical labeling was seen with the anti-EMA antibody, which normally recognizes distal tubules and collecting ducts (Figure 1b) ▶ . Glomeruli were crowded together because of the poor differentiation of tubules. They appeared ischemic, with small glomerular tufts within relatively large Bowman’s capsules, and the capillary loops lined by podocytes were closely distributed at the periphery of the tuft. The increase in glomerular size from the superficial to the deep cortex normally associated with maturation was not observed. The large arteries were normal, but interlobular and preglomerular arteries showed muscular-wall thickening also evidenced by α-SMA labeling (Figure 2a) ▶ . Occasionally, mesangial cells of developing glomeruli were α-SMA-positive. In the medulla, the interstitial mesenchyme was strikingly increased, resulting in an abrupt corticomedullary delimitation (Figure 1a) ▶ .
Figure 1.

a: Light microscopy. Periodic acid-Schiff. Donor twin at 20 weeks’ gestation. RTD characterized by the absence of identifiable proximal tubules, retraction of glomerular tufts, and abundance of cortical and medullary interstitial cells. b and c: Immunolabeling, anti-EMA antibody (28-week twin pair). b: Donor twin. Most tubules are EMA-positive, between densely packed glomeruli. c: Recipient twin. Normal labeling of distal and collecting ducts and absence of labeling of proximal tubules. Comparison between b and c shows the reduced thickness of the cortex in the donor twin, owing to the absence of identifiable proximal tubules. Micrographs b and c, taken at the same magnification, were from a 28-week twin pair. Original magnification, ×80. Scale bar, 100 μm.
Figure 2.
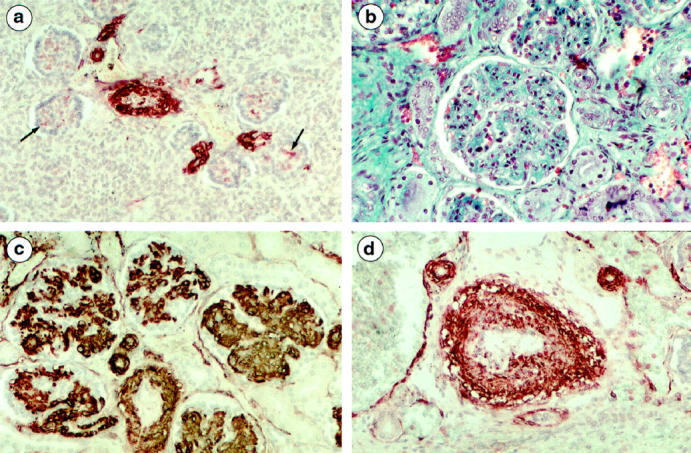
a: Donor twin (21 weeks) by α-SMA immunolabeling. Strong labeling of arterial smooth muscle cells and very occasional labeling of mesangial cells (arrow) in glomeruli of the deep cortex. b: Recipient twin (30 weeks) by light microscopy. Striking glomerular enlargement due to mesangial-cell proliferation and hypertrophy. c and d: Recipient twins (21 and 26 weeks) by α-SMA immunolabeling. c: Strong glomerular expression of α-SMA emphasizing the hypertrophy and subendothelial expansion of mesangial cells. d: Labeling of an arcuate artery, showing the irregular thickening and vacuolization of smooth muscle cells. Micrographs a and c, taken at the same magnification, were from a 21-week twin pair. Original magnification, ×190.
In 5/21 fetuses (19.8–29.2 weeks), the differentiation of PTs corresponding to the most mature nephron was normal, whereas lesions of RTD were observed in the intermediate cortex. In 3/21 fetuses, there was a diffuse but less severe atrophy of PT, resembling the so-called “endocrinoid kidney” characterized by the small size and clear cytoplasm of PTT. The remaining three donors had normal kidneys.
Recipient Twins (21 Fetuses, 18–30 Weeks)
In all cases, the recipient’s kidneys were enlarged (Table 2) ▶ and congested. Hemorrhagic infarction was present in 19/21 cases. It was extensive in four fetuses, involved diffusely the corticomedullary junctions in four, and was focal in 11. Glomeruli were strikingly enlarged due to the diffuse mesangial matrix expansion and mesangial hypercellularity (Figure 2b) ▶ . Hypertrophy always predominated in the most mature glomeruli located in the deep cortex. Focal (three cases) or diffuse (seven cases) glomerular lesions were superimposed on glomerulomegaly and congestion. They consisted in thickening and the double-contour appearance of the capillary wall due to mesangial interposition and to the presence of clear subendothelial deposits. Focal mesangiolysis was observed in three of the seven cases with diffuse glomerular lesions. With anti-α-SMA antibodies, all mesangial cells and their subendothelial expansion were strongly labeled (Figure 2c) ▶ . Thickening of preglomerular and interlobular arteries was observed in 11 fetuses, due to smooth muscle cell hypertrophy and focal vacuolation (Figure 2d) ▶ . Tubular differentiation was normal with positive CD15 labeling of proximal segments and EMA labeling of distal and collecting tubules.
Renin Expression
Immunohistochemistry
Normal fetal kidneys. In 18 of the 21 singleton fetuses, a few cells (1–4) in 5 to 10% of the JGA were renin-positive, whereas renin labeling was observed in 10 to 20% of JGA in three fetuses (Table 3) ▶ . In twin pregnancies, renin-positive cells were observed in 10 to 25% of JGA in the fetal kidneys of eight twin pairs, with usually less than four renin-positive cells by positive JGA (Figure 3a) ▶ . No mesangial cell was renin-positive. In each fetus of one twin pair, 30 to 40% of JGA were labeled with a variable number (1 to 8) of positive cells in each JGA. These 22-week-old fetuses with dichorionic, diamniotic placenta were female and 590 and 500 g, respectively, at birth, with morphologically normal kidneys and no sign of TTS.
Table 3.
Percentage of Renin-Positive Juxtaglomerular Apparatus
| Subject | Subjects (n) in each percentage range | ||||
|---|---|---|---|---|---|
| None | 5–10% | 10–30% | 30–40% | 40–90% | |
| Singleton controls | — | 18 | 3 | — | — |
| Twin controls | — | — | 16 | 2 | — |
| Donor twins | — | — | 5 | — | 14 |
| Recipient twins | 19 | — | 1 | — | — |
Figure 3.

Renin expression. a: Control twin (22 weeks) by Renin immunolabeling. Renin-positive cells are observed in about 20% of JGA. b: Recipient twin (28 weeks) by Renin immunolabeling. All JGA are renin-negative. c: Recipient twin (27 weeks) by in situ hybridization. Expression of renin is detected in very rare JGA cells (arrow). d–f: Donor twins; d and e, by renin immunolabeling. d: Most juxtaglomerular apparatus are renin-positive (28 weeks). e: High magnification clearly shows the presence of renin in arteriolar smooth muscle cells and in glomerular mesangial cells (arrow; 22 weeks). f: In situ hybridization with DIG-labeled RNA antisense probe. Renin transcripts are present in arteriolar smooth muscle cells and in glomerular cells (arrow; 28 weeks). Micrographs b, d, and f were from a 28-week twin pair. Original magnifications: ×80 (a-d, f); ×320 (e).
Donor twins (19 fetuses, 19.8–30 weeks). Immunolabeling with anti-renin antibodies showed that renin-positive JGA were present in 20 to 30% of glomeruli in twin donors (21 to 27 weeks) from five pregnancies (Table 3) ▶ . The number of renin-positive cells by JGA was less than four in three donors and reached six cells in some JGA sections in the other two. In 14 twin donors, 40 to 90% of glomeruli had renin-positive JGA, with more than four renin-positive cells by positive JGA (Figure 3d) ▶ . The labeling was always prominent in mature nephrons. In these patients, the renin-positive cells were also frequently observed in the muscular layer of the afferent arterioles at a distance from the glomeruli and in the interlobular arteries in three patients. In addition, positive mesangial cells were observed in eight of these patients and were very abundant in the five patients with 80 to 90% of renin-positive JGA (Figure 3e) ▶ .
Recipient twins (20 fetuses, 18–30 weeks). Immunolabeling with anti-renin antibodies detected no renin-positive cells in 19 recipients (Figure 3b ▶ ; Table 3 ▶ ) . Conversely, in one fetus with severe arteriolar thickening, renin-positive cells were present in the JGA of about 20% of the glomeruli. It is interesting that only the developing glomeruli had positive JGA, which were associated with the presence of renin-positive cells in numerous sections of arterioles located in the outer cortex. JGA of the enlarged glomeruli of the deep cortex were renin-negative.
In Situ Hybridization
Normal fetal kidneys (four kidneys). Renin expression was observed in the JGA at the vascular poles of 10 to 30% of the mature glomeruli. In these four fetuses, renin protein was seen in 10 to 20% of JGA. Renin transcripts were also present in the dispersed cells of preglomerular arteries.
Donor twins (seven kidneys). A strong expression of renin was observed in the JGA of 30 to 90% of the glomeruli, with more than six cells labeled in most JGA. The extensive expression of renin messenger RNA (mRNA) was also observed in afferent arteriole segments at a distance from the glomeruli. Correlations between protein and mRNA expression were as follows: 30% of the glomeruli had RNA-positive JGA in one fetus expressing the protein in 40% of JGA; 50 to 60% of JGA were mRNA positive in the four fetuses expressing the renin protein in 20, 30, 40, and 50% of JGA, respectively; and 90% of JGA were positive in the two patients showing the renin protein in 80 and 90% of JGA, respectively (Figure 3f) ▶ . In these two patients, the renin transcript, as well as the protein, were also observed focally in the mesangial cells and in the smooth muscle cells of interlobular arteries.
Recipient twins (seven kidneys). In 6/7 fetuses tested, there was either no renin expression (2 cases) or minor renin expression restricted to about 10% of JGA (four cases; Figure 3c ▶ ). However, in the kidney of the one fetus expressing renin protein, renin transcripts were also detected in JGA of 30% of glomeruli, mostly in the developing nephrons located in the intermediate and outer cortex. They were also observed in the smooth muscle cells of arterioles of the superficial cortex.
Discussion
Striking differences in kidney changes were observed between the donor and recipient twins. Most donors had renal tubular dysgenesis. This lesion, characterized by the complete or nearly complete absence of identifiable proximal tubules and the ischemic appearance of the glomeruli, has already been reported in donor twins. 19 It has also been observed as a congenital autosomal, recessive disorder responsible for oligohydramnios 20,21 and in fetuses exposed in utero to angiotensin-converting enzyme inhibitors. 22 Because donor twins are thought to suffer from chronic hypovolemia, it may be assumed that the RTD lesions of these fetuses result from chronic hypoperfusion of the kidney, as previously suggested in stuck twins who died from TTS. 19,23 This hypothesis is also supported by the observation of similar tubular changes in children with postnatal renal ischemia. 24,25 Up-regulation of renin synthesis is another marker of chronic renal hypoperfusion. Using the immunohistochemistry and in situ hybridization methods, we demonstrated a strong increase in renin protein and mRNA content in the donor kidneys. Not only were the number of renin-positive JGA and the number of positive cells in each JGA increased compared with the normal singleton or twin fetuses, but renin synthesis was also detected in the mesangial cells, a quite unusual location, in about one third of donors.
The renin-angiotensin system is highly active during fetal life, 26 and is regulated by the same mechanisms as in postnatal life. One major mechanism stimulating renin synthesis and release is volume depletion and decreased renal perfusion pressure. 27,28 The endpoint of the stimulation of the renin-angiotensin cascade is the generation of angiotensin II, a potent vasoconstrictor peptide whose expected beneficial effects are to restore extracellular volume through stimulation of aldosterone release and to maintain peripheral blood pressure by acting directly on the vascular smooth muscle cells. However, these adaptive mechanisms may fail to compensate volume depletion in the donor. In this case, angiotensin II-induced intrarenal vasoconstriction may act as an additional deleterious factor by further reducing renal blood flow and fetal diuresis, which in turn aggravates oligohydramnios.
The recipient kidneys were large and congested, showing more or less extensive hemorrhagic infarction. The proximal tubule differentiation was normal, whereas prominent changes involved the glomeruli and, to a lesser extent, arteries. Glomeruli were strikingly enlarged due to mesangial matrix expansion. Mesangial hypercellularity and hypertrophy were outlined by an increased expression of α-SMA. α-SMA is a marker of mesangial cell proliferation and activation in human and experimental glomerulonephritis. 29,30 In the human fetal kidney, it is normally expressed by mesangial cells in the developing glomeruli as soon as the mesangium and capillary loops become identifiable. 31 Its expression decreases substantially in mature human fetal glomeruli. 31 The strong mesangial expression in the recipient fetuses may result from hypertension as up-regulation of α-SMA by mesangial cells is observed in hypertension-injured glomeruli. 29,32
Half of the recipient twins had additional glomerular lesions consisting of the presence of clear subendothelial deposits associated with focal or diffuse peripheral mesangial interposition between the glomerular basement membrane and endothelial cells, causing a double-contour appearance of the capillary wall. These lesions are reminiscent of those observed in polycythemia or in hypertension-induced thrombotic microangiopathy in children. 24,33 The occurrence of focal mesangiolysis in some fetuses and of severe arterial and arteriolar changes strongly suggests that not only polycythemia but also hypertension participates in the development of renal lesions. Hypertension has been documented recently in a recipient fetus by ultrasonography. 14 Hypertension could be a direct consequence of hypervolemia, but other pathogenic factors may be involved and particularly the inappropriate production of angiotensin II.
Down-regulation of renin synthesis is expected in hypervolemic recipients, resulting from the inhibiting action of high-perfusion pressure on renal baroreceptors. Actually, no renin protein was detected in 20/21 recipient fetuses, whereas none or a reduced number of JGA expressed the renin transcripts, suggesting a double transcriptional and post-transcriptional regulation of renin synthesis. In the fetus with severe hypertensive-glomerular and arteriolar lesions, renin expression, observed in the distal part of the renal vasculature (JGA of the developing nephrons and arterioles of the outer cortex), was probably secondary to arteriolar changes. Plasma renin concentration or activity has not been measured in this series of patients. However, we may speculate that circulating renin from the donor is transferred to the recipient through placental vascular anastomoses. Indeed, renin activity was previously detected in both twins in three cases of TTS. 13 This inappropriate presence of renin in hypervolemic fetuses, with the eventual increase of angiotensin II generation and aldosterone synthesis, represents a potential factor in aggravating hypervolemia and its renal and extrarenal consequences for the recipient, including systemic hypertension.
In conclusion, in TTS the renal lesions observed in donors and recipients are partly due to hemodynamic changes in both fetuses: hypovolemia in the donor and hypervolemia/polycythemia in the recipient. Hypovolemia-induced increased renin synthesis in the donor may negatively affect both fetuses by aggravating hypoperfusion in the donor and increasing blood pressure and volemia in the recipient. Further prospective studies are needed to evaluate the precise role of vasoactive hormones in the cascade of events leading from twin-to-twin transfusion to severe renal dysfunction and extrarenal complications in both fetuses.
Footnotes
Address reprint requests to Dr Marie-Claire Gubler, INSERM U423, Tour Lavoisier, Hôpital Necker Enfants Malades, 149, rue de Sèvres, 75743 Paris Cedex 15, France. E-mail: gubler@necker.fr.
Supported by the Institut National de la Santé et de la Recherche Médicale, the Association Claude-Bernard, and the Association pour l’Utilisation du Rein Artificiel.
References
- 1.Montan S, Jörgensen C, Sjöberg NO: Amniocentesis in treatment of acute polyhydramniosis in twin pregnancies. Acta Obstet Gynecol Scand 1985, 64:537-539 [DOI] [PubMed] [Google Scholar]
- 2.Mahony BS, Petty CN, Nyberg DA, Luthy DA, Hickok DE, Hirsch JH: The “stuck twin” phenomenon: ultrasonographic findings, pregnancy outcome, and management with serial amniocenteses. Am J Obstet Gynecol 1990, 163:1513-1522 [DOI] [PubMed] [Google Scholar]
- 3.Reisner D, Mahony B, Petty C, Nyberg D, Flint Porter T, Zingheim R, Williams M, Luthy D: Stuck twin syndrome: outcome in thirty-seven consecutive cases. Am J Obstet Gynecol 1993, 169:991–995 [DOI] [PubMed]
- 4.De Lia JE, Kulhmann RS, Harstad TW, Cruikshank DP: Fetoscopic laser ablation of placental vessels in severe previable twin-twin transfusion syndrome. Am J Obstet Gynecol 1995, 172:1202-1211 [DOI] [PubMed] [Google Scholar]
- 5.Ville Y, Hecher K, Gagnon A, Sebire N, Hyett J, Nicolaides K: Endoscopic laser coagulation in the management of severe twin-to-twin transfusion syndrome. Br J Obstet Gynaecol 1998, 105:446-453 [DOI] [PubMed] [Google Scholar]
- 6.Hecher K, Plath H, Bregenzer T, Hansman M, Hackelöer BJ: Endoscopic laser surgery versus serial amniocentesis in the treatment of severe twin-twin transfusion syndrome. Am J Obstet Gynecol 1999, 180:717-724 [DOI] [PubMed] [Google Scholar]
- 7.Dommergues M, Mandelbrot L, Delezoide AL, Aubry MC, Fermont L, Mahieu-Caputo D, Dumez Y: Twin-to-twin syndrome: selective feticide by embolization of the hydropic fetus. Fetal Diagn Ther 1995, 10:26-31 [DOI] [PubMed] [Google Scholar]
- 8.Deprest J, Audibert F, Van Schoubroeck D, Hecher K, and Mahieu-Caputo D: Bipolar coagulation of the umbilical cord in complicated monochorionic twin pregnancy. Am J Obstet Gynecol (in press) [DOI] [PubMed]
- 9.Quintero RA, Romero R, Reich H, Goncalves L, Johnson MP, Carrero C, Evans MI: In utero percutaneous umbilical cord ligation in the management of complicated monochorionic multiple gestations. Ultrasound Obstet Gynecol 1996, 8:16-22 [DOI] [PubMed] [Google Scholar]
- 10.Saade Saade GR, Belfort MA, Berry DL, Bui TH, Montgomery LD, Johnson A, O’Day M, Olson GL, Lindholm H, Garoff L, Moise KJ Jr. Amniotic septostomy for the treatment of twin oligohydramnios-polyhydramnios sequence. Fetal Diagn Ther 1998, 13:86–93 [DOI] [PubMed]
- 11.Berry SM, Puder KS, Bottoms SF, Uckele JE, Romero R, Cotton DB: Comparison of intrauterine hematologic and biochemical values between twin pairs with and without stuck twin syndrome. Am J Obstet Gynecol 1995, 172:1403-1410 [DOI] [PubMed] [Google Scholar]
- 12.Talbert DG, Bajoria R, Sepulveda W, Bower S, Fisk NM: Hydrostatic and osmotic pressure gradients produce manifestations of fetofetal transfusion syndrome in a computerized model of monochorial twin pregnancy. Am J Obstet Gynecol 1996, 174:598-608 [DOI] [PubMed] [Google Scholar]
- 13.Wiecaker P, Wilhelm C, Prompeler H, Petersen KG, Schillinger H, Breckwoldt M: Pathophysiology of polyhydramnios in twin transfusion syndrome. Fetal Diagn Ther 1992, 7:87-92 [DOI] [PubMed] [Google Scholar]
- 14.Baud O, Lebidois J, Van Peborgh P, Ville Y: Fetal and neonatal hypertension in twin-twin transfusion syndrome: a case report. Fetal Diagn Ther 1998, 12:223-226 [DOI] [PubMed] [Google Scholar]
- 15.Galen FX, Devaux C, Guyenne T, Menard J, Corvol P: Multiple forms of human renin: purification and characterization. J Biol Chem 1979, 254:4848-4855 [PubMed] [Google Scholar]
- 16.Galen FX, Guyenne TT, Devaux C, Auzan C, Corvol P, Menard J: Direct immunoassay of human renin. J Clin Endocrinol Metab 1979, 48:1041-1043 [DOI] [PubMed] [Google Scholar]
- 17.Soubrier F, Panthier JJ, Corvol P, Rougeon F: Molecular cloning and nucleotide sequence of a human renin cDNA fragment. Nucleic Acids Res 1983, 11:7181-7190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schütz S, Le Moullec JM, Corvol P, Gasc JM: Early expression of all the components of the renin-angiotensin-system in human development. Am J Pathol 1996, 149:2067-2079 [PMC free article] [PubMed] [Google Scholar]
- 19.Barr M, Sedman AB, Heidelberger KP: Renal tubular dysgenesis in twins. Pediatr Nephrol 1998, 12:408-413 [DOI] [PubMed] [Google Scholar]
- 20.Allanson JE, Hunter AGW, Mettler GS, Jimenez C: Renal tubular dysgenesis: a not uncommon autosomal recessive syndrome. A review. Am J Med Genet 1992, 43:811-814 [DOI] [PubMed] [Google Scholar]
- 21.Gubler MC, Sarrut S, Imbert MC, Narcy F, Lacoste M, Guicharnaud L, Gasc JM, Mounier F: Renal tubular dysgenesis (RTD), an autosomal recessive disorder and the renin-angiotensin system. J Am Soc Nephrol 1993, 4:263. [DOI] [PubMed] [Google Scholar]
- 22.Pryde PG, Sedman AB, Nugent CE, Barr M: Angiotensin-converting enzyme inhibitor fetopathy. J Am Soc Nephrol 1993, 3:1575-1582 [DOI] [PubMed] [Google Scholar]
- 23.Altemani A, Vassalo J, Billis A: Congenital focal glomerular lesions in only one monozygotic twin related to a probable twin transfusion syndrome. Histopathology 1986, 10:991-994 [DOI] [PubMed] [Google Scholar]
- 24.Helmchen U, Wenzel UO: Benign and malignant nephrosclerosis and renovascular disease. ed 2 Tisher CC Brenner BM eds. Renal Pathology with Clinical and Functional Correlations, 1994, :pp 1201-1236 JB Lippincott, Philadelphia [Google Scholar]
- 25.Landing BH, Ang SM, Herta N, Larson EF, Turner M: Labeled lectin studies of renal tubular dysgenesis and renal tubular atrophy of postnatal renal ischemia and end-stage kidney disease. Pediatr Pathol 1994, 14:87-99 [DOI] [PubMed] [Google Scholar]
- 26.Taylor M, Peart S, Porter K, Zondek L, Zondek T: Concentration and molecular forms of active and inactive renin in human fetal kidney, amniotic fluid and adrenal gland: evidence for renin-angiotensin system hyperactivity in second trimester of pregnancy. J Hypertens 1986, 4:121-129 [DOI] [PubMed] [Google Scholar]
- 27.Gomez RA, El-Dahr S, Chevalier RL: Vasoactive hormones. ed 4 Barratt TM Avner ED Harmon WE eds. Pediatric Nephrology, 1999, :pp 83-99 Lippincott-Williams & Wilkins, Baltimore [Google Scholar]
- 28.Robillard JE, Nakamura KT: Neurohumoral regulation of renal function during development. Am J Physiol 1988, 254:F771-F779 [DOI] [PubMed] [Google Scholar]
- 29.Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Pritzl P, Floege J, Schwartz SM: Renal injury from angiotensin II-mediated hypertension. Hypertension 1992, 19:464-474 [DOI] [PubMed] [Google Scholar]
- 30.Alpers CE, Hudkins KL, Gown AM, Johnson RJ: Enhanced expression of “muscle specific” actin in glomerulonephritis. Kidney Int 1992, 41:1134-1142 [DOI] [PubMed] [Google Scholar]
- 31.Alpers CE, Seifert RA, Hudkins KL, Johnson RJ, Bowen-Pope DF: Developmental patterns of PDGF B-chain, PDGF-receptor, and α-actin expression in human glomerulogenesis. Kidney Int 1992, 42:390-399 [DOI] [PubMed] [Google Scholar]
- 32.Kimura K, Suzuki N, Ohba S, Nagai R, Hiroi J, Mise N, Tojo A, Nagaoka A, Hirata Y, Goto A, Yazaki Y, Omata M: Hypertensive glomerular damage as revealed by the expression of α-smooth muscle actin and non-muscle myosin. Kidney Int 1996, 55(Suppl):S169-S172 [PubMed] [Google Scholar]
- 33.Weidner N, Buckalew VM: Sickle cell anemia, sickle cell trait, and polycythemic states. Renal Pathology with Clinical and Functional Correlations, ed. 2. Edited by CC Tisher, BM Brenner. Philadelphia, JB Lippincott, 1994, pp 1491–1510