

Abstract
The complement system constitutes a series of enzymatic steps involved in the inflammatory response and is activated in Alzheimer’s disease (AD). Using Down’s syndrome (DS) brains as a temporal model for the progression of AD, we examined components of the complement cascade and their relationship to other principal events in AD pathology: Aβ42 deposition, neuritic changes, neurofibrillary tangles (NFTs), and gliosis (reactive astrocytes, activated microglia). Adjacent sections of frontal cortex from 24 DS subjects ranging in age from 12 to 73 years were immunohistochemically examined for immunoreactivity (IR) of classical complement proteins (Clq and C3), markers indicating activation of complement (C4d and C5b-9), the complement inhibitor apolipoprotein J (apo J), and markers of AD neuropathology. Aβ42-labeled diffuse plaques were first detected in a 12-year-old DS subject and were not labeled by any of the complement antibodies. Colocalization of Aβ42 with Clq, C3, C4d, and/or apo J was first detected in compacted plaques in the brain of a 15-year-old DS patient with features of mature AD pathology, such as reactive astrocytes, activated microglia, dystrophic neurites, and a few NFTs. IR for C4d and C5b-9 (membrane attack complex, MAC) was observed in small numbers of plaque-associated dystrophic neurites and in focal regions of pyramidal neurons in this 15-year-old. The only other young (≤30 years) DS brain to show extensive complement IR was that of a 29-year-old DS subject who also displayed the full range of AD neuropathological features. All middle-aged and old DS brains showed IR for Clq and C3, primarily in compacted plaques. In these cases, C4d IR was found in a subset of Aβ42 plaques and, along with C5b-9 IR, was localized to dystrophic neurites in a subset of neuritic plaques, neurons, and some NFTs. Our data suggest that in AD and DS, the classical complement cascade is activated after compaction of Aβ42 deposits and, in some instances, can progress to the local neuronal expression of the MAC as a response to Aβ plaque maturation.
Alzheimer’s disease (AD) is characterized by two major neuropathological features: amyloid (senile) plaques and neurofibrillary tangles (NFTs). NFTs comprise paired helical filaments (PHF), abnormal cytoplasmic fibers that result from the hyperphosphorylation of the microtubule-associated protein tau. 1-3 Senile plaques are primarily composed of amyloid β-protein (Aβ), which is proteolytically generated from the β-amyloid precursor protein (βAPP). 4 The βAPP gene is encoded on chromosome 21. Down’s syndrome (DS) subjects have three copies of chromosome 21 and thus an additional copy of the βAPP gene, leading to increased production of Aβ very early in life and the development of Alzheimer-type neuropathology in young and middle-aged subjects. 5,6 The development of amyloid deposits was described in teenage DS subjects by silver staining techniques. 7 Subsequently, sensitive antibodies to Aβ42 were used to detect abundant diffuse Aβ42 plaques (thioflavin S negative) as early as 12 years in DS. 8 Typically, by around 30–40 years of age, and infrequently in younger subjects, a subset of plaques in DS brains shows fibrillar Aβ, ie, thioflavin-positive amyloid; the number of such fibrillar plaques increases with age. 8 Many of these plaques are associated with local gliosis and neuronal changes such as altered axons and dendrites (dystrophic neurites) and NFTs. The presence of senile plaques (plaques containing Aβ fibrils associated with neuritic changes), NFTs, and progressive dementia with behavioral changes 9 in DS subjects all suggest that DS brains can provide a powerful model for studying the temporal development of AD-type neuropathology. 10,11
In AD brains, compacted Aβ plaques are often associated with such inflammatory markers as activated microglia, reactive astrocytes, and complement proteins, including Clq, C3, C9, C3d, and C4d. 12-15 The complement cascade comprises a series of enzymatic steps that play a role in the immune response. 16 Clq, C3, and C9 are three proteins involved in the beginning, middle, and end, respectively, of the classical complement cascade. Whereas the classical complement pathway is more commonly activated by the binding of Clq to the Fc portion of an immunoglobulin, aggregated Aβ but not monomeric Aβ 17 has been reported to activate the classical complement cascade by directly binding to Clq in vitro. 18-24 Synthetic preaggregated Aβ42 peptides were observed to bind Clq much more effectively than Aβ40 peptides, and the binding site of the Aβ42 peptides was localized to the Clq A chain collagen-like region, residues 14–26. 19 Binding of Aβ to Clq has been shown to enhance Aβ aggregation because of the complementary spacing in the structures of the two proteins. 20 Moreover, in situ evidence for the activation of the classical complement pathway has been observed in AD brain. 15,18,25-27 This cascade marks cells for attack by macrophages and causes the release of various proteins to serve as anaphylotoxins that further stimulate the immune response. The cascade ends in the assembly of two molecules of preassembled C5b, 6, 7 with two molecules of C8 and subsequently 12–18 molecules of C9 to form the membrane attack complex (MAC), C5b-9, which creates a leaky pore in the plasma membrane and leads to lysis of target cells. 28 C4d and C3d constitute by-products of degradation of C4b and C3b on the cell surface and indicate complement activation; immunoreactivity (IR) for each has been observed in senile plaques, dystrophic neurites, and NFTs in AD. 28 The presence of C4d is indicative of activation of the classical pathway, whereas C3d is indicative of either the classical or alternative complement pathways. 29 C5b-9 IR has been detected in dystrophic neurites in senile plaques (but not in association with extracellular amyloid) and in NFTs in AD. 26,28 Apo-J, an inhibitor of the membrane attack complex, has been shown to colocalize with senile plaques in AD and DS cerebral cortex. 30 Furthermore, apo J has been observed to complex with soluble Aβ in cerebrospinal fluid. 31 The activation of the alternative complement pathway by Aβ peptides has also been described. 32
The purpose of the current study was to characterize the temporal appearance of several complement proteins (Clq, C3, C4d, and C5b-9) as well as the complement inhibitor, apo J, in DS brain and to relate this process to other AD neuropathological changes: Aβ42 deposits, dystrophic neurites, reactive astrocytes, and activated microglia. Thus the activation, progression, and completion of the classical complement cascade were examined in a temporal series of DS brains of increasing ages and compared to the classical changes of AD.
Materials and Methods
Patient Groups
Autopsied brains from 24 DS patients were examined. These brains are part of a collection of DS tissue obtained by us over the past 13 years. The cases were divided into young (12–30 years), middle aged (31–50 years), and old (51–73 years) for the purpose of temporal comparison. Tissues from the young cases were kindly provided by Dr. Krystyna Wisniewski (Institute for Basic Research in Developmental Disabilities, Staten Island, NY).
Tissue Preparation
Blocks of DS frontal cortex were briefly fixed (1–48 hours) or routinely fixed (more than 48 hours up to 1 year) in 10% neutral buffered formalin. These fixation times are respectively designated “short” and “long” in Table 2 ▶ . Young cases were fixed “very long,” ie, a period of formalin fixation exceeding 1 year. The exact formalin fixation times of the young cases are unknown but exceed 1 year and, in some instances, may exceed 10 or more years. After fixation, the brain tissue was dehydrated and embedded in paraffin. Eight-micron serial sections were cut, dried, and baked at 60°C for 1 hour.
Table 2.
Complement Protein and AD Marker Immunoreactivities in Young, Middle-Aged, and Old DS Brain
| No. | Age group | Conditions | Immunoreactivity | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Formalin | Age (years) | Aβ42* | PHF tau/ NF | GFAP* | Ferritin* | C1q* | C3* | C4d | C5b-9 | Apo-J | ||
| 1 | Young | 330 -82 | Very long | 12 | + | − | − | − | − | − | − | +/− gn | − |
| 2 | 340-82 | Very long | 14 | − | − | − | − | − | − | − | +/− gn | − | |
| 3 | 329-82 | Very long | 15 | + | + | + | + | + | + | + pl, fn | + fn | + | |
| 4 | 337-82 | Very long | 16 | + | − | +/− | +/− | +/− | +/− | +/− gn | + gn | − | |
| 5 | 299-82 | Very long | 17 | + | − | +/− | − | − | − | +/− gn | +/− gn | − | |
| 6 | 307-82 | Very long | 21 | + | − | − | +/− | − | − | − | + gn | − | |
| 7 | 327-82 | Very long | 23 | − | − | − | − | − | − | +/− gn | − | ||
| 8 | 341-82 | Very long | 27 | + | − | +/− | − | − | − | +/− gn | + gn | + | |
| 9 | 305-82 | Very long | 29 | + | + | + | + | + | + | + pl, fn, dn, NFTs | + fn, NFTs | + | |
| 10 | Middle | A85 -140 | Long | 36 | + | +/− | +/− | + | + | + | − | + fn, NFTs | + |
| 11 | 96 -3535 | Long | 38 | + | + | + | + | + | + | Ne | ne | + | |
| 12 | A93-137 | Long | 46 | + | + | + | + | + | + | + pl, fn | + fn | + | |
| 13 | A91 -143 | Long | 47 | + | + | + | + | + | + | + pl, fn | +/− gn | + | |
| 14 | A93 -76 | Long | 49 | + | + | + | + | + | + | + fn | + fn, NFTs | + | |
| 15 | Old | X1809 | Short | 52 | + | + | + | + | + | + | + pl, dn, NFTs | + fn, gn | + |
| 16 | A89 -312 | Short | 55 | + | + | + | + | + | + | +pl, dn, NFTs | + fn, gn | + | |
| 17 | A94 -92 | Long | 57 | + | + | + | + | + | + | + fn, NFTs | +/− gn | + | |
| 18 | A89 -366 | Short | 59 | + | + | + | + | + | + | + pl, dn, few NFTs | +/− gn | + | |
| 19 | A94 -74 | Long | 60 | + | + | + | + | + | + | + pl, dn, NFTs | + fn, dn, NFTs | + | |
| 20 | A92 -69 | Long | 61 | + | + | + | + | + | + | + fn, dn, NFTs | + fn, NFTs | + | |
| 21 | A92 -328 | Long | 64 | + | + | + | + | + | + | + fn, dn, NFTs | +fn, NFTs | + | |
| 22 | A94 -18 | Long | 65 | + | + | + | + | + | + | + fn NFTs | + fn, NFTs | + | |
| 23 | A98 -23 | Long | 65 | + | + | + | + | + | + | + fn, dn, NFTs | + fn, dn, NFTs | + | |
| 24 | A93 -52 | Long | 73 | + | + | + | + | + | + | + pl, dn, NFTs | + fn, dn, NFTs | + |
Very long, fixed 1–10 or more years; long, fixed 2 weeks to 1 year; short, fixed 1–3 hours; ne, not examined; PHF, paired helical filament; NF, neurofilament; *, plaque-associated staining; +, positive staining; +/−, weak staining; −, no staining; pl, plaque labeling; fn, focal neuronal staining; gn, general weak neuronal staining; dn, dystrophic neurites associated with plaques; NFTs, neurofibrillary tangles.
Antibodies and Histological Stains
The antibodies listed in Table 1 ▶ were used to immunostain the DS brain sections. Tests were performed on briefly, routinely, and long-term-fixed, paraffin-embedded AD tissue to determine the best staining conditions for each antibody. 21F12 (gift of Elan Pharmaceuticals, San Francisco, CA) is a mouse monoclonal antibody that selectively recognizes Aβ peptides ending at residue 42 and was used to detect diffuse and compacted Aβ plaques. 33 A monoclonal antibody directed against paired helical filament (PHF) tau protein, AT8 (Innogenetics, Belgium), was used to detect NFTs, neuropil threads, and plaque-associated dystrophic neurites. A monoclonal antibody directed against phosphorylated neurofilament protein, SMI-34 (Sternberger Monoclonals, Baltimore, MD), was also used to detect NFTs and dystrophic neurites. Rabbit anti-human glial fibrillary acidic protein (GFAP) (Dako, Carpinteria, CA) was used to label reactive astrocytes. Rabbit anti-human ferritin (Sigma Chemical Company, St. Louis, MO) was used to detect microglia associated with Aβ plaques in all DS cases. Mouse anti-human HLA-DR (Neomarkers Corp., Fremont, CA) was used to detect activated microglia in middle-aged and older DS cases but was nonreactive in the very long-term-fixed young DS cases. Rabbit anti-human Clq (Dako) was used to detect Clq. Goat anti-human C3 (Quidel Corp., San Diego, CA) was used to label C3. Mouse anti-human C4d (Quidel Corp.) was used to label C4d. Mouse anti-human SC5b-9 (Quidel Corp.), directed at a neoepitope on C9 that is present only when the membrane attack complex has been assembled, was used to detect C5b-9. 34 Goat anti-human apo J (Rockland, Gilbertsville, PA) was used to detect apo J in single- and double-labeling experiments. Cell nuclei were counterstained with hematoxylin.
Table 1.
Conditions, Sources, and Manufacturers of Antibodies Used
| Antibody/(antigen) | Labeling in AD and DS Brains | Species | Dilution | Pretreatment on paraffin sections | Source |
|---|---|---|---|---|---|
| 21F12 (Aβ 31–42) | Aβ42; plaques, some vascular deposits | Mouse anti-human | 1:1000 | Formic acid | Elan Pharmaceuticals, San Francisco, CA |
| AT8 (PHF tau) | Neurofibrillary tangles and dystrophic neurites | Mouse anti-human | 1:25 | Microwave | Innogenetics, Belgium (Polymedco, Cortlandt, NY) |
| SMI-34 (phosphorylated neurofilaments) | Neurofibrillary tangles and dystrophic neurites | Mouse anti-human | 1:5000 | Microwave/formic acid | Sternberger Monoclonals, Baltimore, MD |
| GFAP | Reactive astrocytes | Rabbit anti-human | 1:1000 | None | Dako, Carpinteria, CA |
| HLA-DR | Plaque-associated activated microglia | Mouse anti-human | 1:100 | Microwave | Neomarkers Corp., San Diego, CA |
| Ferritin | Plaque-associated activated microglia | Rabbit anti-human | 1:250 | Microwave | Sigma Chemical Company, St. Louis, MO |
| Clq | Clq | Rabbit anti-human | 1:500 | Microwave (y); microwave/proteinase K (m, o) | Dako, Carpinteria, CA |
| C3 | C3 | Goat anti-human | 1:1000 | Microwave (y); microwave/proteinase K (m, o) | Quidel Corp., San Diego, CA |
| C4d | C4d | Mouse anti-human | 1:250 | Microwave/formic acid | Quidel Corp., San Diego, CA |
| SC5b-9 | C5b-9 | Mouse anti-human | 1:250 | Microwave/formic acid | Quidel Corp. San Diego, CA |
| Apo-J | Apo-J | Goat anti-human | 1:200 | Microwave/proteinase K | Rockland, Gilbertsville, PA |
Immunohistochemistry
Single Primary Antibody Labeling Experiments
Sections were deparaffinized in Histoclear (National Diagnostics, Atlanta, GA) and rehydrated. Endogenous peroxidase activity was quenched by incubation of the sections in 0.3% hydrogen peroxide in methanol for 5 minutes. The sections were washed in water for 5 minutes. Appropriate pretreatments were carried out for each primary antibody as designated in Table 1 ▶ . Formic acid pretreatment consisted of the application of 88% formic acid to sections for 15 minutes, followed by a 5-minute water wash. Microwave pretreatment consisted of heating sections in a citrate buffer (BioGenex, San Ramon, CA) until the solution came to a boil. The heat level was then reduced to provide gentle cyclic boiling for an additional 5 minutes. Sections were cooled to room temperature and washed in water. Proteinase K pretreatment entailed applying a 1:100 dilution of proteinase K (Dako) in 50 mmol/L Tris buffer (50 mmol/L Tris-HCl, pH 7.6) onto sections for 8 minutes. Sections were then rinsed in water briefly and washed in Tris-buffered saline (TBS) for 5 minutes. After these various pretreatments, all sections were blocked for 20 minutes in 10% goat serum (for GFAP and ferritin), 10% horse serum (for 21F12, AT8, SMI-34, and HLA-DR), or 3% bovine serum albumin (BSA) (for apo J, Clq, C3, C4d, and C5b-9). Sections were incubated with primary antibody overnight at 4°C. The horseradish peroxidase avidin-biotin complex system (rabbit, mouse, or goat Elite ABC kits; Vector Laboratories, Burlingame, CA) and diaminobenzidine (DAB) (Sigma Immunochemicals, St. Louis, MO) were used to visualize the bound antibodies. To minimize variability in staining intensity for each antibody used, sections from all of the DS cases were immunostained simultaneously. Sections were counterstained with hematoxylin, differentiated with acid alcohol, dehydrated, cleared in Histoclear, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA).
Double Primary Antibody Labeling Experiments
In a few DS cases of varying age, double-label immunostaining was carried out to assess colocalization between selected proteins more directly. To detect two primary antibodies on one section, the procedure described in the previous section was followed for the first antibody. After DAB development, the sections were rinsed in water for 5 minutes. Any necessary pretreatments for the second antibody were carried out, and the sections were rinsed in water for 5 minutes. After blocking with 3% BSA for 20 minutes, the sections were incubated in the second primary antibody overnight at 4°C. After a 30-minute incubation with the corresponding secondary antibody, the alkaline phosphatase kit (AP ABC kit; Vector Laboratories) was used with a red substrate (Alkaline Substrate Kit 1; Vector Laboratories) to visualize the second primary antibody. Sections were counterstained in hematoxylin, differentiated with acid alcohol, dehydrated, and coverslipped with Permount.
Results
Temporal Progression of AD Pathology in DS Brains
Our 24 DS cases were divided into young (12–30 years), middle aged (31–50 years), and old (51–73 years) for the purpose of temporal comparison (Table 2 ▶ and Figure 1 ▶ ). Diffuse Aβ42 plaques were first observed in the 12-year-old DS brain (not shown but illustrated in Ref. 8 ). GFAP-reactive astrocytes associated with Aβ42 plaques and some dystrophic neurites were first labeled in the 15-year-old DS case (not shown). This 15-year-old patient had exceptionally compacted Aβ42 plaques, whereas the other teenage DS cases had overwhelmingly diffuse (noncompacted) Aβ42 plaques; the number of compacted amyloid plaques in these young cases was judged in part from previous thioflavin S staining experiments (not shown). The presence of tau-positive dystrophic neurites in compacted Aβ42 plaques was first observed in the 15-year-old DS brain, whereas NFTs were first observed in the 29-year-old case. None of the other young cases had neuritic plaques or NFTs (Table 2) ▶ .
Figure 1.

Immunolabeling of complement proteins Clq and C3 in young (29 years), middle-aged (47 years), and old (73 years) Down’s syndrome (DS) patients. Serial sections show the association of these proteins with Aβ42 plaque deposits, activated microglia (ferritin), reactive astrocytes (GFAP), and dystrophic neurites (SMI-34). * indicates the same microvessels observed in serial sections a–f, g–l, or m–r. In all three cases, immunoreactivities to complements Clq and C3 were found to colocalize with many Aβ42 plaques; staining was strongest in the most compacted plaques. In the young patient, strong GFAP (e) and weak microglial (c) and neuritic (f) staining was detected in the same region bearing Aβ42- (a) and complement- (b and d) IR plaques (arrows in a–f). Note: The majority of young DS subjects in this study, bearing exclusively diffuse Aβ42, had no complement IR. In the middle-aged and older DS subjects, complement staining frequently colocalized with Aβ, activated microglia, reactive astrocytes, and dystrophic neurites (arrows in g–r). Bar, 100 μm.
GFAP-positive astrocytes near blood vessels were labeled in all DS cases. However, among the young cases, only the 15- and 29-year-old DS cases showed abundant GFAP-positive astrocytes associated with Aβ42 plaques (Figure 1e ▶ and Table 2 ▶ ). The 16-, 17-, and 27-year-old cases showed a very small number of reactive astrocytes. The amount of GFAP-positive astrocytes associated with blood vessels was greater in the 12-year-old subject with Aβ42 plaques than in the 23-year-old subject, who did not show any Aβ42 plaques. Plaque-associated reactive astrocytes were readily observed in all middle-aged and old DS subjects (Figure 1, k and q ▶ , and Table 2 ▶ ).
A ferritin antibody was used to label microglia in all of the DS cases, because the long fixation precluded using the HLA-DR antibody in the young cases. Ferritin-positive microglia, associated with a small subset of compacted Aβ42 plaque deposits, were first observed in the brain of the 15-year-old DS subject. Plaque-associated ferritin-positive microglia were more frequently observed in the 29-year-old DS subject (Figure 1c) ▶ . None of the other young cases showed plaque-associated ferritin microglia (Table 2) ▶ , but all young cases had ferritin-positive microglia near blood vessels and in white matter. Activated microglia, frequently associated with Aβ42 plaques, were labeled by both the ferritin and HLA-DR antibodies in all middle-aged and old DS brains (Figure 1, i and o) ▶ . As with ferritin at all ages, HLA-DR-positive microglia were also intensely labeled in white matter and blood vessels in many middle-aged and old DS subjects. Furthermore, HLA-DR- and ferritin-positive microglia were observed to have varying morphologies in gray matter. The more lightly labeled microglia tended to have longer processes and small cell bodies (ie, resting microglia), whereas the more intensely labeled microglia had shorter processes and larger cell bodies (ie, activated microglia; Figure 1i ▶ ). Microglia in the latter form were more often associated with Aβ42 plaques than were other labeled microglia.
Temporal Deposition of Complement Protein in DS Brains
Clq and C3 Immunoreactivities
Clq IR in plaques was first detected in the 15-year-old DS brain and was closely associated with compacted Aβ42 staining (Table 2) ▶ . Clq-labeled deposits were not seen in the brain of the 12-year-old DS patient, our youngest, who had overwhelmingly diffuse Aβ42 plaques. Weak Clq IR was detected in a small subset of semicompacted Aβ42 plaques in the 16-year-old patient. The 29-year-old patient was the only other young subject to show Clq IR deposits (Figure 1b) ▶ ; these deposits colocalized strongly with reactive astrocytes but only moderately with microglia, C3, and dystrophic neurites (Figure 1, b–f) ▶ . The 15-year-old and 29-year-old DS patients showed more compacted Aβ42-IR plaques, some with activated glia and/or degenerating neurites, than the other young patients.
All middle-aged and old DS patients showed Clq deposits strongly associated with the large majority of compacted Aβ42 plaque deposits (Figure 1, g and h ▶ , and Figure 1, m and n ▶ ). These patients showed more Clq IR than the two young patients (aged 15 and 29 years), reflective of the increase in compacted, mature plaques in the older cases. Clq IR colocalized closely with Aβ42 plaques in all cortical layers, although some diffuse subpial and white matter Aβ42 deposits appeared to lack Clq IR. Overall, Clq IR was associated with the largest number of Aβ42 plaques when compared to the immunoreactivities of the other complement proteins.
In serial sections, Clq IR frequently overlapped with C3 IR, activated microglia, reactive astrocytes, and plaque-associated dystrophic neurites in middle-aged and old DS patients (Figure 1, h–l ▶ , and Figure 1, n–r ▶ ). The colocalization of Clq with activated microglia and dystrophic neurites in plaques was confirmed by double-labeling sections from a subset of patients from each age group; results for the 29-year-old and the 73-year-old DS patients are illustrated in Figure 2 ▶ . Clq IR was found in many plaques containing activated microglia in their centers (Figure 2, a and e) ▶ . In addition, Clq IR overlapped with neuritic plaques in these two patients (Figure 2, b and f) ▶ .
Figure 2.

Compacted plaques, frequently containing activated microglia in their centers and dystrophic neurites, were often immunoreactive for complements Clq and C3. Double labeling of Clq and C3, each with tau and ferritin, is shown for a 29-year-old (left) and a 73-year-old (right) DS patient. Left: Microglia (brown in a) and dystrophic neurites (brown in b) were detected in many Clq-labeled plaques (red in a and b) in adjacent sections of frontal cortex of the 29-year-old DS patient. C3-labeled plaques (red in c and d) frequently contained microglia (brown in c) and dystrophic neurites (brown in d) in adjacent sections 50 μm from those shown in a and b. Note: The majority of young DS patients in this study, bearing exclusively diffuse Aβ42 plaques and lacking plaque-associated activated microglia and dystrophic neurites, had no complement immunoreactivity (see Table 2 ▶ ). Right: Staining of serial sections of frontal cortex of the 73-year-old DS patient showed colocalization of Clq (red in e and f) and C3 (red in g and h) with microglia (brown in e and g) and dystrophic neurites (brown in f and h). Scale bars, 100 μm.
C3 IR was first detected in the brain of the 15-year-old DS patient (Table 2) ▶ and was closely associated with compacted Aβ42 IR plaques. Weak C3 IR was observed in a minority of Aβ42 plaques in the 16-year-old patient. In the 29-year-old patient, C3 IR was seen in the centers of a subset of Aβ42 plaques and colocalized to some degree with Clq, microglia, reactive astrocytes, and a few dystrophic neurites (Figure 1, a–f) ▶ . None of the remaining young patients showed C3 plaque staining. Middle-aged and old DS cases showed C3 IR in Aβ42 plaques, in association with Clq, activated microglia, reactive astrocytes, and dystrophic neurites (Figure 1, g–l ▶ , and Figure 1, m–r ▶ ). C3 IR was observed in a smaller subset of Aβ42 plaques than was Clq IR. C3 IR was absent from most of the subpial and deep cortical Aβ42 deposits found mainly in old DS cases. Double-labeling experiments in a subset of patients from each age group confirmed the colocalization of C3 with activated microglia and neuritic plaques, as shown in Figure 2 ▶ for the 29-year-old (c and d) and the 73-year-old (g and h) patients.
C4d and C5b-9 IR
C4d IR was first detected in a small number of compacted plaques and focally in scattered groups of neurons in the 15-year-old DS patient (Table 2) ▶ . Weak neuronal C4d IR was also observed in the 16-year-old and 17-year-old patients. A few compacted plaques, dystrophic neurites, and NFTs, in addition to abundant neuronal staining, were detected using the C4d monoclonal in the 29-year-old patient (Figure 3d) ▶ . C4d IR in amyloid deposits and plaque-associated dystrophic neurites was observed in roughly half of the middle-aged and old DS cases; however, focal neuronal and some NFT staining was detected in most of these cases (Figure 3i ▶ and Table 2 ▶ ). C4d plaque staining overlapped with Aβ42 and was frequently accompanied by the presence of microglia and dystrophic neurites (Figure 3 ▶ , a–d, f–i). After we doubly pretreated the long-term and very long-term-fixed tissues with microwave heating followed by formic acid, focal neurons and/or NFTs were detected using the C5b-9 antibody in those brains bearing consolidated Aβ plaques, including that of the 15-year-old and the 29-year-old (Table 2 ▶ and Figure 3, e and j ▶ ) patients. General, low-level neuronal C5b-9 IR was observed in most DS brains, regardless of age; however, with the appearance of compacted plaques, dystrophic neurites, NFTs, and gliosis, a more intense focal labeling of C5b-9 in neurons was detected. As with C4d, the presence of multiple markers of AD pathology, including Aβ, ferritin, and tau, was observed in the same vicinity as the C5b-9 IR in adjacent sections (Figure 3) ▶ . C5b-9-IR dystrophic neurites were observed in plaques in five of six available briefly fixed brain tissues and in two long-term-fixed middle-aged and old DS brain tissues (Table 2) ▶ .
Figure 3.
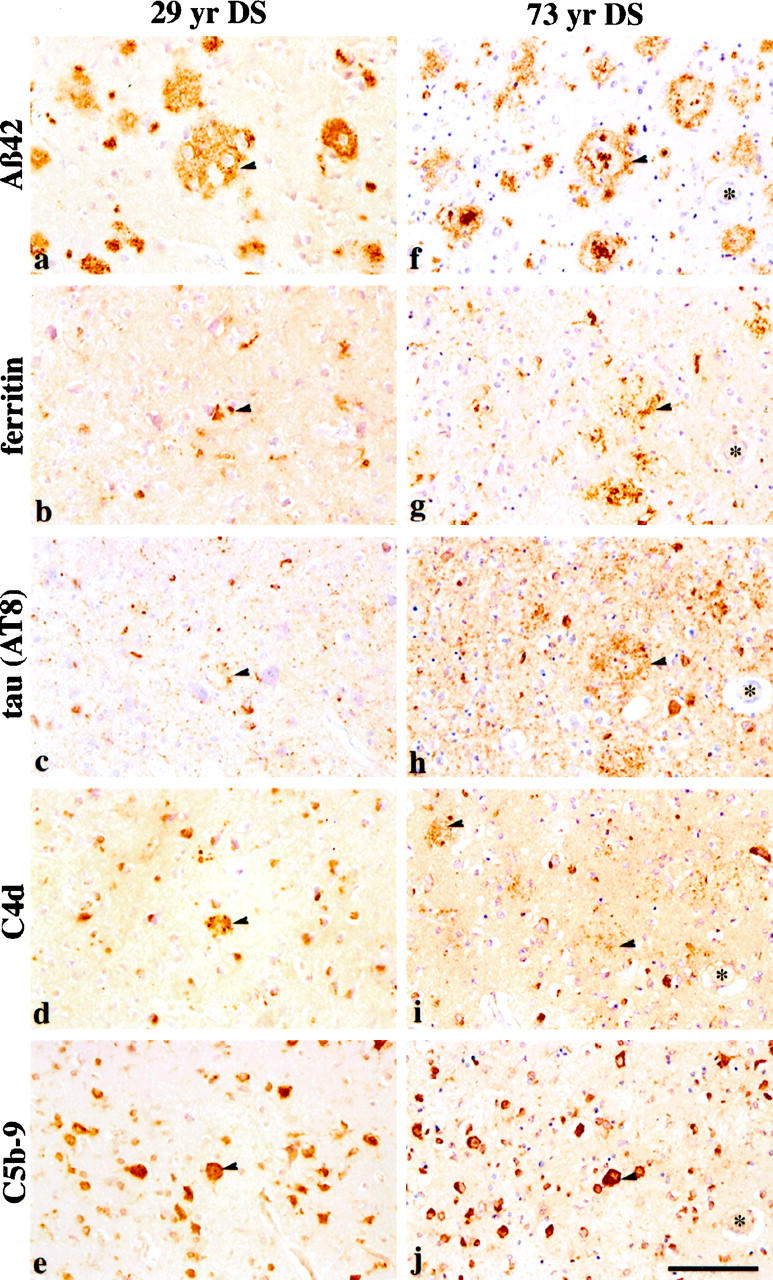
Immunolabeling of the activated complement components, C4d, and the membrane attack complex, C5b-9, was compared with other AD neuropathological markers in serial sections of frontal cortex of a 29-year-old DS subject (left, a–e) and a 73-year-old DS subject (right, f–j). Left: Arrows indicate colocalization of an Aβ42 plaque (a), microglia (b), a few tau-positive dystrophic neurites (c), C4d-positive neuritic staining (d), and C5b-9 neuronal labeling (e). Right: In the older DS patient, colocalization of Aβ42 (f) with microglia (g), tau (h), C4d (i), and C5b-9 (h) was also observed (arrows in f–j). Note that the sections from the older patient (right) show quantitatively more staining for microglia, tau, C4d, and C5b-9 compared with those of the younger patient (left). Scale bar, 100 μm.
Temporal Deposition of apo J in DS Brain
Apo J IR was first observed within compacted Aβ42 plaques in the 15-year-old DS patient (Table 2) ▶ . Apo J-IR deposits were observed in three young patients (15, 27, and 29 years); all three of these cases had moderate to numerous compacted Aβ42 plaques. Apo J IR was observed in all middle-aged and old DS subjects. In general, the relative abundance of apo J IR most closely resembled that of Clq IR. Apo J IR colocalized with Aβ42-IR plaques in all cortical layers. Double-labeling experiments demonstrated the colocalization of apo J with Aβ42 plaques and plaque-associated reactive astrocytes in brains of the 15-, 46-, and 65-year-old DS patients (Figure 4, a, c, and e) ▶ . Reactive astrocytes were often observed surrounding the apo J-IR deposits, with their processes extending into these deposits (Figure 4, b, d, and f) ▶ .
Figure 4.
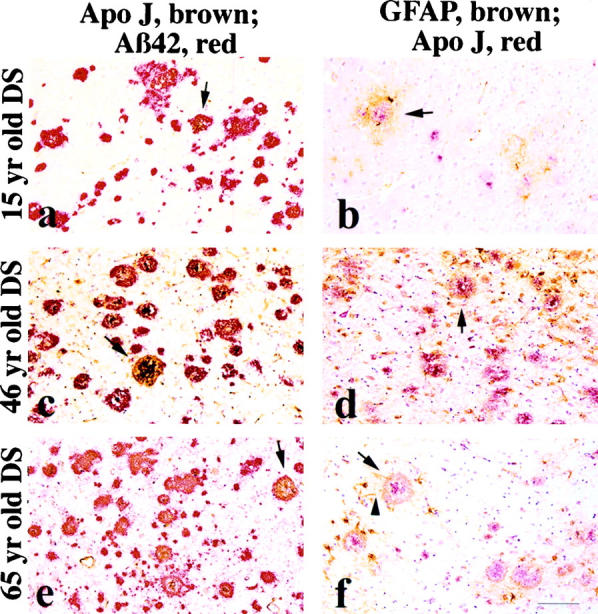
Left: Double labeling of apo-J (brown) with Aβ42 (red) was examined in young (15 years), middle-aged (46 years), and old (65 years) DS patients (a, c, and e, respectively). Colocalization was observed at all three ages, but middle-aged and old subjects showed much more than the young subject (eg, arrows in a, c, and e). Right: Double labeling with antibodies to apo-J (red) and GFAP (reactive astrocytes; brown) shows the presence of reactive astrocytes surrounding apo-J IR deposits at all three ages (arrows in b, d, and f, respectively). Note how the reactive astrocytes extend their processes toward and into the apo-J IR plaques (arrow in f). Bar, 100 μm.
Discussion
Aβ42-IR diffuse plaques were detected in the majority of our young DS patients; little or no gliosis, neuritic dystrophy, NFTs, or complement IR was observed in these cases. In two young patients (ages 15 and 29 years) and in all middle-aged and old DS patients, Clq IR, apo J IR, and reactive astrocytes were associated with a large subset of Aβ-IR plaques. C3 IR and activated microglia labeled a moderate subset of Aβ42 plaques. C4d IR was associated with the smallest subset of Aβ42 plaques and, in addition, labeled dystrophic neurites and some NFTs. C5b-9 IR did not colocalize with extracellular Aβ42 but was detected focally in subsets of neurons and in some dystrophic neurites and NFTs.
The results of this study suggest that the presence of complement IR in plaques is dependent on the state of compaction and maturation of Aβ42-IR plaques; those brains with more compacted Aβ plaques, dystrophic neurites, and plaque-associated gliosis showed more complement IR. Thus, the more severe the AD pathology, regardless of age, the greater the inflammatory response. Several observations support this conclusion. First, abundant Clq and C3 deposits were detected in only two of the young DS cases (15 and 29 years). These two patients showed more severe AD neuropathology, such as compacted plaques, reactive astrocytes, activated microglia, and neuritic changes, than did the other young DS patients (which had predominantly diffuse plaques), and the two patients appear to represent an accelerated form of the AD process in DS. All middle-aged and old DS patients had many compacted Aβ42 plaques and abundant Clq and C3 plaque IR. Second, diffuse white matter Aβ42 plaques observed in several old DS patients did not have associated complement Clq IR, despite the presence of strong Clq staining in other, more compacted plaques in both gray and white matter in the same section. In addition, diffuse subpial deposits of Aβ42 were most prevalent in old DS patients and were not Clq immunoreactive. The absence of Clq subpial staining in tissue that was otherwise clearly labeled by the Clq antibody supports the result obtained in young DS brains, in which complement staining was not observed in those cases bearing only diffuse Aβ42 plaques. These results are in agreement with previous work by Afagh et al, in which the authors describe an absence of Clq IR in thioflavin-negative diffuse plaques in the brains of AD patients and nondemented controls. 15 In addition to plaque labeling by Clq antibodies, others have reported the staining of dystrophic neurites and some NFTs, using other Clq antibodies. 28 We were unable to detect Clq labeling of such structures; however, fixation techniques and the use of a different antibody may account for the reduced sensitivity we observed.
Compared to Clq-labeled plaques in the 15- and 29-year-old and older patients, fewer plaques were IR for C3. Lower sensitivity of the antibody on long-fixed paraffin sections may be a reason for this diminution in C3 staining relative to that of Clq. A strong association between C3 IR in plaques and activated microglia was observed in the two young cases and in all middle-aged and old DS cases. Activated microglia (with shortened, thickened processes and large cell bodies) were often associated with Aβ plaques and stained more intensely than resting microglia with antibodies to HLA-DR and ferritin. Previous work by Walker et al showed that cultured microglia from postmortem human brain express complement proteins, suggesting microglia as a potential source for these proteins. 35
C4d and C5b-9 immunoreactive dystrophic neurites and NFTs, indicators of classical complement activation, were detected in the aforementioned two young patients (aged 15 and 29 years) and in most of the middle-aged and old DS cases. C4d IR was also observed in a subset of senile plaques in many of these cases. In many cases of all ages, both C4d and C5b-9 antibodies demonstrated broadly distributed, weak neuronal staining, primarily of pyramidal neurons, even in the absence of glial activation or visible neurodegenerative changes. In those cases having more severe AD pathology, focal groups of neurons, in addition to nearby dystrophic neurites and NFTs, were stained much more intensely, and this may indicate some intermediate stage of cell lysis and neurodegeneration. Previously, Terai et al 36 showed that pyramidal neurons in AD brain express the complement proteins involved in the classical pathway. However, because C4d is a by-product of degradation that signals complement activation and C5b-9 is assembled from several complement proteins, it seems unlikely that the neuronal staining we observed is due to actual localization of C4d and C5b-9 with each of the weakly labeled neurons. Instead, doubly pretreating the strongly fixed sections with microwave heating and formic acid may have allowed some cross-reactivity of the antibodies with other complement proteins or with other neuronal proteins. The intense labeling of C4d and C5b-9 in a subset of dystrophic neurites associated with senile plaques and in NFTs suggests that the classical complement pathway was activated and progressed to completion in those structures. However, the levels of “specific” C4d and C5b-9 staining were low relative to that of Clq and C3, thus indicating that the initiation of the complement cascade by the binding of Clq to Aβ does not necessarily progress to the MAC. This conclusion is in agreement with previous in vitro studies by Cadman et al, 37 in which the authors found that high levels of Aβ peptides, able to activate early complement cascade components, showed lower levels of MAC formation than expected.
Overall, our data suggest that complement proteins are abundant in compacted Aβ plaques. Aβ can directly activate the complement cascade via binding to Clq in in vitro experiments. 18-24 The prevalence of Clq in Aβ plaques suggests that the complement cascade is available for activation. The observation of abundant C3 IR in Aβ plaques suggests that the components necessary for progression of the cascade are available and in the right location. C4d and C5b-9 IRs in the brains of two young and all middle-aged and old DS patients suggest that the complement cascade is activated and can progress to completion. Moreover, the large number of activated microglia, reactive astrocytes, and dystrophic neurites, as markers of inflammation and potential sources of complement proteins, suggests that the cascade is active in the AD-type lesions that occur in DS.
Apo J, a known inhibitor of C5b-9, showed much more IR than either C4d or C5b-9. This result is supportive of the role of apo J in inhibiting the complement cascade end product, C5b-9 (membrane attack complex; MAC), and provides indirect evidence of complement activation in AD. Apo J IR colocalized with many Aβ plaques. In addition to inhibiting MAC formation, it may perform other functions. As a minor species of high-density lipoproteins, it is thought to carry out lipid transport functions. 38 Its secretion from reactive astrocytes may be an early immune response, as apo J has been observed to slow the aggregation of Aβ in vitro. 39,40
Our results suggest that AD and DS brains provide a primed environment for complement-mediated inflammation. The potential negative effects of an active complement cascade in the brain are several. First, Clq has been shown to enhance Aβ aggregation and promote nucleation. 20 Such events would be predicted to accelerate plaque maturation, including associated inflammatory responses, and to reduce the likelihood of clearance of Aβ. Second, the vast proliferation of C3 molecules could mark healthy neighboring cells near the plaque for phagocytosis. Third, if the C5b-7 complex, the precursor form of the MAC that attaches to available cell membranes, is not near a target cell, it will engage in the cell membrane of a neighboring cell, causing cell lysis. This process is called bystander lysis. 16,41 The complement cascade is thought to be activated because of Aβ plaque deposition but presumably is not capable of eliminating the extracellular amyloid deposits, which lack cellular membranes and other features that are required for cell lysis by MAC. However, C5b-9 IR has been observed in dystrophic neurites within Aβ plaques, a more reasonable target of complement-mediated cell lysis. 23,26,42 Furthermore, the complement cascade can excite the respiratory burst apparatus in microglia 43,44 which can then activate them to create free radicals. This highly oxidative environment appears to be conducive to neuronal degeneration. Thus therapeutic interventions aimed at slowing or halting this cascade of inflammation in response to compacted Aβ in the brain may be of value in treating or preventing AD.
Footnotes
Address reprint requests to Dr. Cynthia A. Lemere, Center for Neurologic Diseases, Harvard Institutes of Medicine, Room 622, 77 Avenue Louis Pasteur, Boston, MA 02115. E-mail: lemere@cnd.bwh.harvard.edu.
Supported by National Institutes of Health grants AG06173 and AG15408.
References
- 1.Kosik KS, Joachim CL, Selkoe DJ: Microtubule-associated protein, tau, is a major antigenic component of paired helical filaments in Alzheimer’s disease. Proc Natl Acad Sci USA 1986, 83:4044-4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung Y-C, Zaidi MS, Wisniewski HM: Microtubule-associated protein tau: a component of Alzheimer paired helical filaments. J Biol Chem 1986a, 261:6084-6089 [PubMed] [Google Scholar]
- 3.Goedert M: Tau mutations cause frontotemporal dementias. Neuron 1998, 21:955-958 [DOI] [PubMed] [Google Scholar]
- 4.Selkoe DJ: Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399:A23-A31 [DOI] [PubMed] [Google Scholar]
- 5.Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, Martins R, Hockey A, Montgomery P, Beyreuther K, Masters CL: Amyloid A4 protein, and its precursor in Down’s syndrome and Alzheimer’s disease. N Engl J Med 1989, 320:1446-1452 [DOI] [PubMed] [Google Scholar]
- 6.Mann DMA: Cerebral amyloidosis, aging and Alzheimer’s disease: a contribution from studies on Down’s syndrome. Neurobiol Aging 1989, 10:397-399 [DOI] [PubMed] [Google Scholar]
- 7.Wisniewski K, Wisniewski H, Wen G: Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 1985, 17:278-282 [DOI] [PubMed] [Google Scholar]
- 8.Lemere CA, Blustzjan JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ: Sequence of deposition of heterogeneous amyloid β-peptides and Apo E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 1996, 3:16-32 [DOI] [PubMed] [Google Scholar]
- 9.Lai F, Williams RS: A prospective study of Alzheimer’s disease in Down syndrome. Arch Neurol 1989, 46:849-853 [DOI] [PubMed] [Google Scholar]
- 10.Mann DMA, Yuonis N, Jones D, Stoddart RW: The time course of pathological events in Down’s syndrome with particular reference to the involvement of microglial cells and deposits of β/A4. Neurodegeneration 1992, 1:201-215 [Google Scholar]
- 11.Tagliavini F, Giaccone G, Linoli G, Frangione B, Bugiani O: Cerebral extracellular preamyloid deposits in Alzheimer’s disease, Down syndrome and nondemented elderly individuals. Prog Clin Biol Res 1989, 317:1001-1005 [PubMed] [Google Scholar]
- 12.Eikelenboom P, Stam FC: Immunoglobulins and complement factors in senile plaques: an immunoperoxidase study. Acta Neuropathol 1982, 57:239-242 [DOI] [PubMed] [Google Scholar]
- 13.McGeer PL, McGeer EG: Complement proteins and complement inhibitors in Alzheimer’s disease. Res Immunol 1992, 143:621-624 [DOI] [PubMed] [Google Scholar]
- 14.Rogers J, Schultz J, Brachova L, Lue L-F, Webster S, Bradt B, Cooper NR, Moss DE: Complement activation and β-amyloid-mediated neurotoxicity in Alzheimer’s disease. Res Immunol 1992, 143:624-630 [DOI] [PubMed] [Google Scholar]
- 15.Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ: Localization and cell association of Clq in Alzheimer’s disease brain. Exp Neurol 1996, 138:22-32 [DOI] [PubMed] [Google Scholar]
- 16.Kuby J, Ed: The Complement System. Immunology. New York, W. H. Freeman and Company, 1998, pp. 335–355
- 17.Snyder SW, Wang GT, Barrett L, Ladror US, Casuto D, Lee CM, Krafft GA, Holzman RB, Holzman TF: Complement Clq does not bind monomeric β-amyloid. Exp Neurol 1994, 128:136-142 [DOI] [PubMed] [Google Scholar]
- 18.Rogers J, Cooper NR, Websger S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, Lieberburg I: Complement activation by β-amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1992, 89:10016-10020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang H, Burdick D, Glabe C, Cotman C, Tenner A: β-amyloid activates complement by binding to a specific region of the collagen-like domain of the Clq A chain. J Immunol 1994, 152:5050-5059 [PubMed] [Google Scholar]
- 20.Webster S, Glabe C, Rogers J: Multivalent binding of complement protein Clq to the amyloid β-peptide promotes the nucleation phase of Aβ aggregation. Biochem Biophys Res Commun 1995, 217:869-875 [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Frederickson RCA, Brunden KR: Neuroglial-mediated immunoinflammatory responses in Alzheimer’s disease: complement activation and therapeutic approaches. Neurobiol Aging 1996, 17:781-787 [DOI] [PubMed] [Google Scholar]
- 22.Velazquez P, Cribbs D, Poulos T, Tenner A: Aspartate residue 7 in amyloid β-protein is critical for classical complement pathway activation: implications for Alzheimer’s disease pathogenesis. Nature Med 1997, 3:77-79 [DOI] [PubMed] [Google Scholar]
- 23.Webster S, Bonnell B, Rogers J: Charge-based binding of complement component Clq to the Alzheimer amyloid β-peptide. Am J Pathol 1997, 150:1531-1536 [PMC free article] [PubMed] [Google Scholar]
- 24.Cribbs DH, Velazquez P, Soreghan B, Glabe CG, Tenner AJ: Complement activation by cross-linked truncated and chimeric full-length β-amyloid. NeuroReport 1997, 8:3457-3462 [DOI] [PubMed] [Google Scholar]
- 25.Eikelenboom P, Hack CE, Rozemuller JM, Stam FC: Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch B Cell Pathol 1989, 56:259-262 [DOI] [PubMed] [Google Scholar]
- 26.McGeer PL, Akiyama H, Itagaki S, McGeer EG: Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 1989, 107:341-346 [DOI] [PubMed] [Google Scholar]
- 27.Yasojima K, Schwab C, McGeer EG, McGeer PL: Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am J Pathol 1999, 154:927-936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGeer PL, Akiyama H, Itagaki S, McGreer EG: Immune system response in Alzheimer’s disease. Can J Neurol Sci 1989, 16:516-527 [DOI] [PubMed] [Google Scholar]
- 29.Ayakima H, Yamada T, Kawamata T, McGeer PL: Association of amyloid P component with complement proteins in neurologically diseased brain tissue. Brain Res 1991, 548:349-352 [DOI] [PubMed] [Google Scholar]
- 30.Kida E, Miura-Choi N, Wisniewski KE: Deposition of apolipoproteins E and J in senile plaques is topographically determined in both Alzheimer’s disease and Down’s syndrome brain. Brain Res 1995, 685:211-216 [DOI] [PubMed] [Google Scholar]
- 31.Ghiso J, Matsubara E, Koudinov A, Choi-Miura N, Tomita M, Wisniewski T: The cerebrospinal-fluid soluble form of Alzheimer’s amyloid β is complexed to SP-40,40 (apolipoprotein J), an inhibitor of the complement membrane-attack complex. Biochem J 1993, 293:27-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bradt BM, Kolb WP, Cooper NR: Complement-dependent pro-inflammatory properties of the Alzheimer’s disease β-peptide. J Exp Med 1998, 188:431-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L: Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94(4):1550–1555 [DOI] [PMC free article] [PubMed]
- 34.Kolb WB, Morrow PR, Jensen FC, Tamerius JD: Development of a highly sensitive capture EIA for the quantitation of the SC5b-9 complex in human plasma using a monoclonal antibody reactive with a poly-C9 neoantigenic determinant. Complement 1988, 5:213 [Google Scholar]
- 35.Walker DG, Kim WU, McGeer PL: Complement and cytokine gene expression in cultured microglia derived from postmortem human brains. J Neurosci Res 1995, 40:478-493 [DOI] [PubMed] [Google Scholar]
- 36.Terai K, Walker DG, McGeer EG, McGeer PL: Neurons express proteins of the classical complement pathway in Alzheimer disease. Brain Res 1997, 769:385-390 [DOI] [PubMed] [Google Scholar]
- 37.Cadman ED, Puttfarcken PS: β-amyloid peptides initiate the complement cascade without producing a comparable effect on the terminal pathway in vitro. Exp Neurol 1997, 146:388-394 [DOI] [PubMed] [Google Scholar]
- 38.May P, Finch CE: Sulfated glycoprotein 2: new relationships to this multifunctional protein to neurodegeneration. Trends Neurosci 1992, 15:391-396 [DOI] [PubMed] [Google Scholar]
- 39.Oda T, Pasinetti GM, Osterburg HH, Anderson C, Johnson SA, Finch CE: Purification and characterization of brain clusterin. Biochem Biophys Res Commun 1994, 204:1131-1136 [DOI] [PubMed] [Google Scholar]
- 40.Matsubara E, Soto C, Governale S, Frangione B, Ghiso J: Apolipoprotein J and Alzheimer’s amyloid β solubility. Biochem J 1996, 316:671-679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGeer PL, McGeer EG: Neuroimmune mechanisms in the pathogenesis of Alzheimer’s disease. Alzheimer’s Disease. Edited by Z. S. Khachaturian and T. S. Radebaugh. CRC Press, Boca Raton, FL, 1996, pp. 217–225
- 42.McGeer PL, Walker DJ, Akiyama H, Kawamata T, Guan AL, Parker CJ, Okada N, McGeer EG: Detection of the membrane inhibitor of reactive lysis (CD59) in diseased neurons of Alzheimer brain. Brain Res 1991, 544:315-320 [DOI] [PubMed] [Google Scholar]
- 43.Klegeris A, McGeer PL: Inhibition of respiratory burst in macrophages by complement receptor blockade. Eur J Pharmacol 1994, 260:273-277 [DOI] [PubMed] [Google Scholar]
- 44.McGeer PL, McGeer EG: The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Rev 1995, 21:195-218 [DOI] [PubMed] [Google Scholar]