

Abstract
There are distinct genetic pathways leading to the glioblastoma, the most malignant astrocytic brain tumor. Primary (de novo) glioblastomas develop in older patients and are characterized by epidermal growth factor (EGF) receptor amplification/overexpression, p16 deletion, and PTEN mutations, whereas secondary glioblastomas that progressed from low-grade or anaplastic astrocytoma develop in younger patients and frequently contain p53 mutations. In this study, we assessed the genetic profile of gliosarcoma, a rare glioblastoma variant characterized by a biphasic tissue pattern with alternating areas displaying glial and mesenchymal differentiation. Single-strand conformation polymorphism followed by direct DNA sequencing revealed p53 mutations in five of 19 gliosarcomas (26%) and PTEN mutations in seven cases (37%). Homozygous p16 deletion was detected by differential polymerase chain reaction in seven (37%) gliosarcomas. The overall incidence of alterations in the Rb pathway (p16 deletion, CDK4 amplification, or loss of pRb immunoreactivity) was 53%, and these changes were mutually exclusive. Coamplification of CDK4 and MDM2 was detected in one gliosarcoma. None of the gliosarcomas showed amplification or overexpression of the EGF receptor. Thus gliosarcomas exhibit a genetic profile similar to that of primary (de novo) glioblastomas, except for the absence of EGFR amplification/overexpression. Identical PTEN mutations in the gliomatous and sarcomatous tumor components were found in two cases. Other biopsies contained p16 deletions, an identical p53 mutation, or coamplification of MDM2 and CDK4 in both tumor areas. This strongly supports the concept of a monoclonal origin of gliosarcomas and an evolution of the sarcomatous component due to aberrant mesenchymal differentiation in a highly malignant astrocytic neoplasm.
Glioblastoma multiforme (WHO Grade IV), the most frequent and malignant brain tumor, may arise de novo after a short clinical history without an identifiable less malignant precursor lesion. This type of glioblastoma has been designated primary glioblastoma. Secondary glioblastomas develop more slowly through progression from low-grade (WHO Grade II) or anaplastic astrocytoma (WHO Grade III). 1,2 Primary glioblastomas typically arise in older patients (mean, 55 years), whereas secondary glioblastomas develop in younger patients (mean, 39 years). 3 There is increasing evidence that these two subtypes develop through different genetic pathways. Primary glioblastomas are characterized by epidermal growth factor receptor (EGFR) amplification/overexpression, p16 deletion, and PTEN (MMAC1) mutations. Secondary glioblastomas typically contain p53 mutations but rarely have EGFR amplification/overexpression, p16 deletion, or PTEN mutation. 2-5
We recently reported that the giant cell glioblastoma, a rare glioblastoma variant characterized by the presence of large, bizarre, multinucleated cells, occupies a hybrid position, sharing with primary (de novo) glioblastomas a short clinical history, the absence of a less malignant precursor lesion, and a 30% frequency of PTEN mutations. They have in common with secondary glioblastomas a younger patient age at manifestation and a high frequency of p53 mutations. 6
The gliosarcoma is another morphologically defined glioblastoma variant, originally described in 1895 by Stroebe et al. 7 Gliosarcomas comprise approximately 2% of all glioblastomas 8,9 and are characterized by a biphasic tissue pattern, with areas displaying glial and mesenchymal differentiation. 10,11 Whereas morphological studies suggested an evolution of the sarcomatous component from microvascular proliferations within a highly malignant glioblastoma, two recent genetic studies revealed the presence of identical p53 mutations 12 and similar chromosomal imbalances and cytogenetic alterations 13 in both tumor areas, suggesting a monoclonal origin. In this study, we screened 19 well-documented cases of gliosarcoma for a variety of genetic alterations in an attempt to identify the genetic profile of gliosarcoma as compared to other glioblastoma subtypes and to elucidate the histogenesis of the sarcomatous component present in this neoplasm.
Materials and Methods
Tumor Samples
The surgical specimens were obtained from a total of 19 patients diagnosed in the University Hospitals of Zürich (Switzerland), Porto (Portugal), and Ribeirão Preto, São Paulo (Brazil). Gliosarcomas were diagnosed according to the WHO classification of brain tumors. 10 Care was taken to include only classical cases, showing the typical biphasic pattern with alternating areas of glial and mesenchymal differentiation. High-grade gliomas with a mesenchymal component that might have resulted from infiltration of the dura were excluded. The areas with glial differentiation usually expressed glial fibrillary acidic protein (GFAP) and showed necrosis and/or vascular endothelial proliferation. The sarcomatous portions showed strong reticulin staining as well as signs of malignant transformation (eg, nuclear atypia, mitotic activity, and necrosis; Figure 1 ▶ ). Glioblastomas with focal sarcomatous appearance but without reticulin staining were not included. The age and sex of patients are shown in Table 1 ▶ . Eleven patients were male and eight were female (M/F ratio, 1.4). The mean age of patients at first diagnosis of gliosarcoma was 56 ± 12 years (range, 32–76 years).
Figure 1.

Histological features of gliosarcoma. The sarcomatous component shows a dense reticulin network (A) but lacks GFAP immunoreactivity, which is strongly expressed in the gliomatous component (B). Magnification, ×150.
Table 1.
Genetic Profile of Gliosarcomas
| Patient no. | Age/sex | Biopsy/diagnosis | Location | Area | p53 miscoding mutation | PTEN miscoding mutation | p16/sts ratio | CDK4/IFGN ratio | MDM2/DR ratio | EGFR/CF ratio | Immunohistochemistry | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p53 | MDM2 | EGFR | RB | |||||||||||
| 200 | 72 /M | 1st/GS | F | S | — | — | 0.38 | 0.95 | 1.17 | 1.25 | − | ++ | − | ++ |
| 206 | 72 /M | 1st/GS | T | S | Exon 7-codon 245 (GGC→GAC, Gly→Asp) | — | 0.04 | 0.63 | 0.79 | 0.8 | ++ | − | − | ++ |
| 207 | 55 /F | 1st/GS | T,P | S | Exon 6-codon 197 (GTG→GTGTG, 2bp insertion, stop at codon 246) | Exon 5-codon 111 (TGG→TAG, Trp→stop) | 0.44 | 1.06 | 1.02 | 0.99 | − | − | − | ++ |
| 208 | 76 /F | 1st/GS | O | S | —* | — | 0.16 | 2.33 | 1.04 | 1.25 | − | + | − | ++ |
| 209 | 32 /M | 1st/GS | B,S | S | —* | Exon 5-codon 88 (TAT→TCT, Tyr→Ser) | 0.51 | 1.30 | 1.03 | 0.94 | − | ++ | − | +++ |
| 210 | 51 /M | 1st/GS | T | S | Exon 6-codon 190 (CCT→TCT, Prol→Ser) | — | 1.10 | 0.92 | 1.40 | 0.96 | ++ | ++ | − | − |
| 52 /M | 2nd/GS | T | S | Exon 5-codon 151 (CCC→TCC, Prol→Ser) | — | 0.32 | 0.83 | 1.22 | 1.08 | +++ | − | − | ++ | |
| G | Exon 5-codon 151 (CCC→TCC, Prol→Ser) | — | 0.50 | 0.44 | 0.78 | 0.85 | +++ | − | − | − | ||||
| 211 | 51 /M | 1st/GS | P | S | —* | — | 0.17 | 1.34 | 1.49 | 1.67 | +++ | + | − | ++ |
| 212 | 51 /F | 1st/GS | T,O | S | nd | Exon 1-codon 17(CAA→CCA, Gln→Pro) | nd | nd | nd | nd | nd | nd | nd | nd |
| 52 /F | 2nd/GS | T,O | S | —* | Exon 1-codon 17(CAA→CCA, Gln→Pro) | 0.31 | 0.95 | 0.88 | 0.76 | ++ | + | + | ++ | |
| 213 | 61 /M | 1st/GS | — | S | — | — | 0.08 | 0.61 | 0.79 | 0.75 | nd | nd | nd | nd |
| G | — | — | 0.31 | 0.56 | 1.06 | 0.81 | nd | nd | nd | nd | ||||
| 214 | 37 /F | 1st/GS | T | S | — | — | 0.04 | 0.74 | 0.59 | 1.23 | nd | nd | nd | nd |
| G | — | — | 0.01 | 0.79 | 0.60 | 1.18 | nd | nd | nd | nd | ||||
| 215 | 43 /F | 1st/GS | C | S | — | Exon 5-codon 107 (GAT→GCT, Asp→Ala) | 0.75 | 1.19 | 1.09 | 1.35 | ++ | + | − | ++ |
| G | — | Exon 5-codon 107 (GAT→GCT, Asp→Ala) | 0.65 | 1.08 | 1.02 | 0.83 | +++ | + | − | ++ | ||||
| 216 | 41 /M | 1st/AA | T,P | G | — | nd | nd | nd | nd | nd | nd | nd | nd | nd |
| 42 /M | 2nd/GBM | T | G | — | nd | nd | nd | nd | nd | nd | nd | nd | nd | |
| 42 /M | 3rd/GS | B,S | S | Exon 8-codon 274 (GTT→GCT, Val→Ala) | — | 0.59 | 0.96 | 1.90 | 0.73 | +++ | ++ | − | ++ | |
| 217 | 52 /M | 1st/GS | T | S | — | — | 0.33 | 2.3 | 1.43 | 0.83 | − | + | − | +++ |
| 218 | 57 /M | 1st/GS | T | S | — | Exon 8-codon 274 (TGG→TGA, Trp→stop) | 0.60 | 1.23 | 1.29 | 0.88 | ++ | + | − | ++ |
| G | — | Exon 8-codon 274 (TGG→TGA, Trp→stop) | 0.78 | 1.24 | 1.47 | 1.28 | ++ | + | − | ++ | ||||
| 219 | 67 /M | 1st/GS | C | S | — | Exon 8-codon 274 (TGC→TAC, Trp→stop) | 0.17 | 0.34 | 0.49 | 0.58 | ++ | − | − | +++ |
| 220 | 63 /F | 1st/GS | T | S | — | — | 0.62 | 3.08 | 5.78 | 1.15 | − | +++ | − | +++ |
| G | — | — | 0.45 | 3.44 | 9.24 | 1.18 | − | +++ | − | +++ | ||||
| 221 | 56 /M | 1st/GBM | P,O | G | nd | nd | 0.14 | nd | nd | nd | nd | nd | nd | nd |
| 2nd/GS | P,O | S | — | — | 0.02 | 0.98 | 0.96 | 0.83 | + | ++ | − | +++ | ||
| 222 | 68 /F | 1st/GS | T | S | Exon 7-codon 237 (ATG→ATA, Met→Ile) | Exon 2-codon 48 (AAC→GAC, Asn→Asp) | 0.8 | 3.10 | 0.97 | 1.08 | ++ | − | − | +++ |
| 223 | 58 /F | 1st/GS | T | S | — | — | 0.48 | 0.71 | 0.74 | 1.13 | − | − | − | ++ |
| G | — | — | 0.58 | 0.80 | 0.68 | 1.06 | − | − | − | ++ |
*Previously reported by Biernat et al. 12 nd, not determined; —, negative; GS, gliosarcoma; GBM, glioblastoma multiforme; AA, anaplastic astrocytoma; S, sarcomatous component; G, gliomatous component; O, occipital; P, parietal; F, frontal; BS, brainstem; C, cerebrum. The results of immunohistochemistry were recorded as negative (−), positive in <5% of cells (+), in 5–50% of cells (++), and in >50% of cells (+++). Differential PCR data (in bold) indicate amplification or deletion (see Materials and Methods).
In seven cases, we were able to microdissect the glial and mesenchymal areas (cases 210, 213, 214, 215, 218, 220, and 223), and DNA was extracted separately from these two portions. Areas were sufficiently large for manual dissection after microscopic identification and labeling of the respective tumor component.
Of 12 patients for whom we could obtain detailed clinical data, 10 were diagnosed with gliosarcoma at the first biopsy; the mean preoperative clinical history was 12 ± 16 weeks. In one case (case 221), the first biopsy was histologically classified as glioblastoma (preoperative history, 4 months), and the second biopsy as gliosarcoma. In another case (case 216), the first biopsy showed an anaplastic astrocytoma, the second biopsy a glioblastoma, and only the third biopsy showed the typical features of gliosarcoma.
Polymerase Chain Reaction-Single-Strand Conformational Polymorphism Analysis and Direct Sequencing for p53 Mutations
DNA was extracted as previously described. 14 Mutations in exon 5–8 of the p53 gene were screened using polymerase chain reaction-single-strand conformational polymorphism (PCR-SSCP) as previously described. 3 Samples that showed a mobility shift in the PCR-SSCP analysis were further analyzed by direct DNA sequencing. Primer sequences for PCR and DNA sequencing were described previously. 3
PCR-SSCP Analysis and Direct DNA Sequencing for PTEN Mutations
Prescreening for mutations in exons 1–9 of the PTEN gene was carried out by PCR-SSCP as previously described. 6 Samples that showed a mobility shift in the SSCP analysis were further analyzed by direct DNA sequencing as previously described. 6 In some cases, individual abnormally shifted SSCP bands were cut directly from the dried gels, placed in 100 μl of distilled water, incubated at 80°C for 15 minutes and centrifuged briefly; 1 μl of the supernatant was used for PCR. Sequencing primers used were as follows: 5′-CTC TCC TCC TTT TTC TTC A-3′ (sense) and 5′-AGA AAG GTA AAG AGG AGC AG-3′ (antisense) for exon 1; 5′-TTT CAG ATA TTT CTT TCC TTA-3′ (sense) and 5′-TGA AAT AGA AAA TCA AAG CAT-3′ (antisense) for exon 2; 5′-TAA AGC TGG AAA GGG ACG AA-3′ (sense) and 5′-TAT CAT TAC ACC AGT TCG TC-3′ (antisense) for exon 5; 5′-TTT TTT TTT AGG ACA AAA TGT TT-3′ (sense) and 5′-TCA CAT ACA TAC AAG TCA CCA AC-3′ (antisense) for exon 8.
Differential PCR for p16 Homozygous Deletion and EGFR, CDK4, and MDM2 Amplification
To assay p16 homozygous deletions in gliosarcomas, differential PCR was carried out, using the STS reference sequence, as reported by Ueki et al, 15 with some modifications. 4 The average p16/STS ratio, using normal blood DNA, was 1.04, with a standard variation of 0.15. Values of less than 0.2 for the p16/STS ratio indicated deletions of the p16 gene. 4 Two primary glioblastomas, which showed a ratio of less than 0.2 in the previous study, 4 served as positive controls for p16 deletion.
To detect CDK4 amplification, differential PCR was carried out as described previously. 4 Interferon γ (IFNγ) was used as a reference gene. The value for normal blood DNA was 1.07, with a standard variation of 0.19. A value of more than 2.7 for the CDK4/IFNG ratio was regarded as positive for CDK4 amplification. This value was calculated according to the method of Rollbrocker et al. 16 One primary glioblastoma, which showed a ratio higher than 2.7 in a previous study, 4 served as the positive control for CDK4 amplification.
MDM2 amplification was detected by differential PCR analysis as previously described. 17 Dopamine receptor (DR) was used as the reference gene. The MDM2/DR ratio from normal blood DNA was 0.91, with a standard variation of 0.4. A value of more than 3.02 for the MDM2/DR ratio was regarded as positive for MDM2 amplification. Two primary glioblastomas, which showed a ratio higher than 3.02 in a previous study, 17 served as positive controls for MDM2 amplification.
To detect EGFR amplification, differential PCR with the cystic fibrosis (CF) reference gene was carried out as described previously, 18 with some modifications. 5 The mean EGFR/CF ratio, from DNA from the peripheral blood of healthy adults, was 1.2, with a standard variation of 0.20. The threshold value 2.94 was regarded as evidence of EGFR amplification, according to the method of Rollbrocker et al. 16 One primary glioblastoma, which showed EGFR amplification in a previous study, 5 was used as a positive control.
Immunohistochemistry
The sections were deparaffinized in xylene and rehydrated in graded ethanol. The endogenous peroxidase was blocked by incubation in 0.3% H2O2 solution in methanol for 30 minutes.
For p53 immunohistochemistry, the sections were boiled three times for 5 minutes in 10 mmol/L sodium citrate buffer (pH 6.0) in a microwave oven. The incubations of anti-human p53 monoclonal antibody (PAb 1801; Genosys Biotechnologies, Cambridge, UK; diluted 1:1000). were carried out overnight at 4°C after blocking of nonspecific binding with 5% skimmed milk for 60 minutes.
For MDM2 immunohistochemistry, the sections were boiled in 10 mmol/L sodium citrate buffer (pH 6.0) for 10 minutes in a steam cooker, subsequently incubated in 5% skimmed milk for 1 hour at room temperature, then incubated overnight at 4°C with the monoclonal antibody to MDM2 (clone IF2; Oncogene Research Products, Cambridge, MA; diluted at 1:2000).
For EGFR immunohistochemistry, sections were pretreated with 0.1% trypsin in 0.1% CaCl2 (pH 7.8) for 15 minutes at 37°C and then incubated in 5% skimmed milk for 60 minutes. Sections were then reacted overnight at 4°C with EGFR monoclonal antibody (NCL-EGFR; Novocastra Laboratories, Newcastle, UK), which recognizes the EGFR ligand binding domain (dilution 1:100).
For RB immunohistochemistry, the sections were boiled three times for 5 minutes in 10 mmol/L sodium citrate buffer (pH 6.0) in a microwave oven. The sections were allowed to cool at room temperature. Sections were incubated overnight at 4°C with the RB monoclonal antibody (clone G3–245; PharMingen, San Diego, CA; diluted 1:100), which recognizes an epitope between amino acids 300 and 380 and both phosphorylated and underphosphorylated RB protein.
For c-MET immunohistochemistry, the sections were boiled for 10 minutes in 10 mmol/L sodium citrate buffer (pH 6.0) in a steam cooker and subsequently incubated in 5% skimmed milk overnight at room temperature. Sections were then incubated for 1 hour at room temperature with the c-MET monoclonal antibody (NCL-cMET, diluted 1:100; Novocastra Laboratories).
The reaction was visualized using the Vectastain ABC Kit and diaminobenzidine (Vector Laboratories, Burlingame, CA). Sections were counterstained with hematoxylin. Fractions of positive cells were recorded as follows: −, negative; +, <5%; ++, 5–50%; +++, >50%.
Results
p53 Mutations and p53 Protein Accumulation
Miscoding p53 mutations were found in five of 19 (26%) gliosarcomas analyzed (Table 1) ▶ . One tumor (case 211) contained a silent mutation (GTG→GTA, Val→Val) in codon 173. In case 210 (Table 1) ▶ , the first biopsy showed the missense mutation CCC→TCT in codon 190; the second biopsy contained a different mutation, CCC→TCC, in codon 151, which was present in both the gliomatous and sarcomatous components (Table 2) ▶ . In all cases, the wild-type base was present along with the mutated base. In case 216, the mutation was present only in the third biopsy but not in the first and second biopsies. Of five gliosarcomas with a p53 mutation, four showed nuclear accumulation of p53 protein in a variable fraction of neoplastic glial and mesenchymal cells (Table 1) ▶ . One gliosarcoma (case 207) contained a 2-bp insertion mutation, resulting in a stop codon, and did not show p53 immunoreactivity (Table 1) ▶ .
Table 2.
Genetic Alterations in Sarcomatous and Gliomatous Components of Gliosarcomas
| Patient no. | Biopsy | Area | Genetic alteration |
|---|---|---|---|
| 210 | 2nd | S | p53 mutation (codon 151, CCC→TCC) |
| G | p53 mutation (codon 151, CCC→TCC) | ||
| 213 | 1st | S | p16 deletion |
| G | — | ||
| 214 | 1st | S | p16 deletion |
| G | p16 deletion | ||
| 215 | 1st | S | PTEN mutation (codon 107, GAT→GCT) |
| G | PTEN mutation (codon 107, GAT→GCT) | ||
| 218 | 1st | S | PTEN mutation (codon 274, TGG→TGA) |
| G | PTEN mutation (codon 274, TGG→TGA) | ||
| 220 | 1st | S | CDK4 amplification, MDM2 amplification |
| G | CDK4 amplification, MDM2 amplification | ||
| 223 | 1st | S | — |
| G | — |
S, sarcomatous component; G, gliomatous component; —, no genetic alterations.
PTEN Mutations
SSCP followed by direct DNA sequencing revealed that seven of 19 (37%) gliosarcomas contained a PTEN mutation (Table 1) ▶ . Of these, three mutations were in exon 5 (phosphatase domain), two were in exon 8, and one each were in exons 1 and 2. Three were nonsense mutations leading to a truncated protein, and four were missense mutations. In all cases, the wild-type base was also detectable. In case 212, the same mutation was present in both primary and second biopsies. In cases 215 and 218, identical mutations were detected in gliomatous and sarcomatous areas (Tables 1 and 2 ▶ ▶ and Figure 2 ▶ ).
Figure 2.
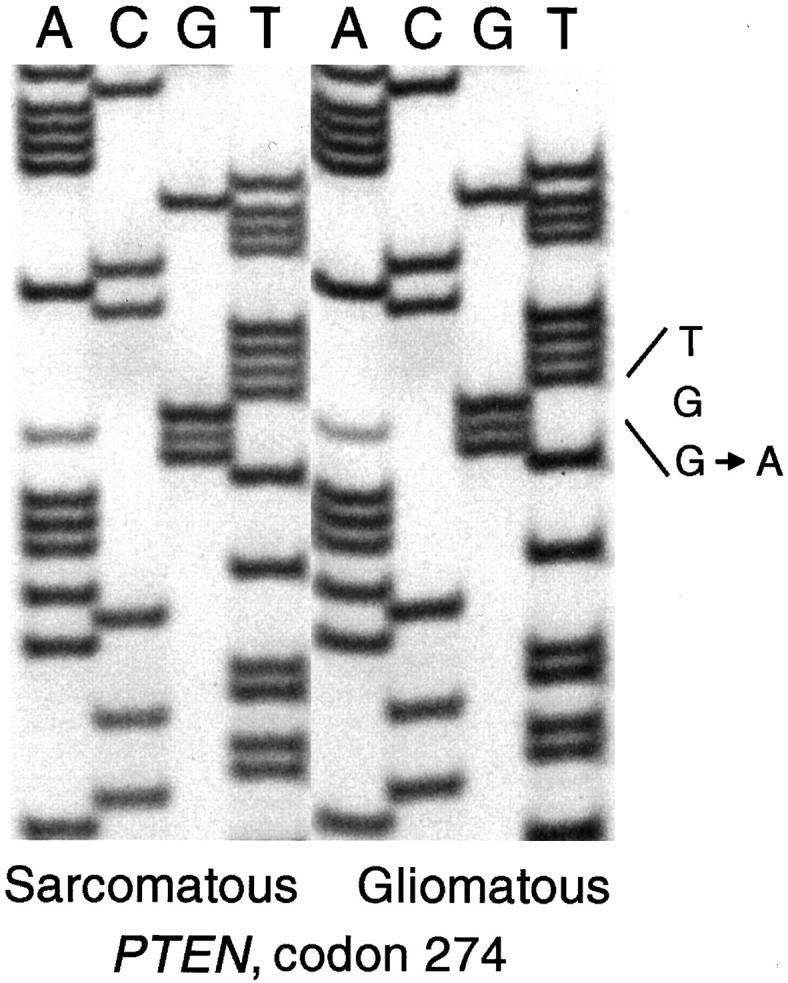
Sequencing gels showing an identical PTEN mutation in codon 274 (TGG→TGA, Trp→Stop) in microdissected sarcomatous and gliomatous areas of a gliosarcoma (case 218).
p16 Deletion, CDK4 Amplification, and RB Expression
In seven cases (37%), differential PCR revealed a homozygous p16 deletion (Table 1) ▶ . In one tumor (case 214), the p16 deletion was detected in both gliomatous and sarcomatous areas (Tables 1 and 2 ▶ ▶ and Figure 3 ▶ ). In another case (case 221), a p16 deletion was detected in the first (glioblastoma) and second (gliosarcoma) biopsies. Differential PCR further revealed amplification of CDK4 in one gliosarcoma (case 220, Table 1 ▶ ), again in both gliomatous and sarcomatous areas (Figure 3) ▶ .
Figure 3.

Differential PCR assay for p16 homozygous deletion and CDK4 and MDM2 amplification. Both gliomatous (G) and sarcomatous (S) areas in case 220 show a significantly increased signal intensity of the CDK4 and MDM2 bands (*) when compared to the respective reference sequence (IFNG, DR), suggesting coamplification of these genes. In case 214 (bottom), both tumor components show a reduced p16 signal (*) when compared to the reference sequence (9q STS), suggesting a homozygous p16 deletion. N, normal DNA; M, molecular size marker.
Strong immunoreactivity to RB was observed in 5–50% of neoplastic cells in 10 cases and in more than 50% of neoplastic cells in six cases. In one case (case 210, Table 1 ▶ ) RB expression was not detectable.
A cell cycle-related gene alteration (p16 deletion, CDK4 amplification, or loss of RB expression) was found in 10 of 19 (53%) gliosarcomas, but these were mutually exclusive, ie, no biopsy contained more than one of these alterations.
Amplification and Overexpression of the MDM2, EGFR, and c-MET Genes
MDM2 amplification was detected by differential PCR in one biopsy (case 220), which also showed CDK4 amplification (Tables 1 and 2 ▶ ▶ and Figure 3 ▶ ). In this tumor, MDM2 overexpression was detected immunohistochemically in more than 50% of neoplastic cell nuclei of both gliomatous and sarcomatous areas.
Differential PCR did not reveal EGFR amplification in any of the 19 cases, and EGFR overexpression was also absent immunohistochemically (Table 1) ▶ .
c-MET immunoreactivity presented as strong cytoplasmic and plasma membrane staining in glioma cells in gliomatous areas, but not in sarcomatous areas in all gliosarcomas analyzed.
Discussion
The term gliosarcoma was introduced in 1898 by Stroebe. 7 Fifty years later, Feigin et al 19 defined it as a glioblastoma subtype in which proliferating vessels had acquired the features of a sarcoma. Some studies showed expression of monohistiocytic markers, suggesting that gliosarcomas develop from histiocytes, whereas others suggested an origin from fibroblasts, pluripotent mesenchymal cells of the perivascular adventitia or perivascular spaces. 20,21 The expression of α-smooth muscle actin in sarcomatous portions pointed to vascular smooth muscle as the potential origin of the mesenchymal tissue component. 22 More recent investigations suggested a common origin of for the two tissue components; the sarcomatous areas result from advanced glioma progression with acquisition of a mesenchymal phenotype. 23,24 This view is strongly supported by genetic analyses, including the present study. Using interphase cytogenetic analysis, Paulus et al 25 detected similar cytogenetical abnormalities in the gliomatous and sarcomatous components of two gliosarcomas, but Biernat et al 12 were the first to prove a monoclonal origin by demonstrating the presence of identical p53 mutations in the two tumor areas. Similar genetic alterations in both tumor components were subsequently reported by Boerman et al, 13 using comparative genomic hybridization (CGH), cytogenetic analysis, fluorescence in situ hybridization, and microsatellite analysis.
The present study extends these findings to a variety of other gene alterations (Table 2) ▶ . We detected in gliomatous and sarcomatous tumor areas identical PTEN mutations (two cases), a p53 mutation (one case), homozygous p16 deletion (one case), and coamplification of MDM2 and CDK4 (one case). In one biopsy (case 213), only the sarcomatous area showed an unequivocal p16 deletion, whereas in the gliomatous portion the p16/sts ratio was 0.31 and thus did not reach the criterion of p16 deletion. This may be due to an admixture of DNA from nonneoplastic neural tissue. Taken together, these data firmly establish the gliosarcoma as a monoclonal tumor with focal aberrant mesenchymal differentiation. Identical genetic alterations have also been detected in both the carcinomatous and sarcomatous components of uterine carcinosarcomas, 26 and in epithelial and stromal components of pulmonary carcinosarcomas. 27
In this as well as in previous studies, 11 gliosarcoma typically developed in older patients (Table 3) ▶ and was diagnosed at first biopsy after a short clinical history, suggesting that these tumors developed de novo, ie, without an identifiable, less malignant precursor lesion. Occasionally (case 221) the histological features of gliosarcoma appeared in the second biopsy of a primary glioblastoma. Perry et al 28 reported that 25 of 32 cases (78%) were diagnosed as gliosarcoma in the first biopsy, whereas seven (22%) developed after irradiation for glioblastoma. Rarely, gliosarcomas develop through progression from low-grade 25 or anaplastic astrocytoma ( Ref. 29 and case 216 of this study).
Table 3.
Clinical and Genetic Data of Glioblastoma Subtypes
| Primary glioblastoma | Gliosarcoma | Giant cell glioblastoma | Secondary glioblastoma | |
|---|---|---|---|---|
| Clinical onset | De novo | De novo | De novo | Secondary |
| Preoperative clinical history | 1.7 months3 | 3 months* | 1.6 months6 | 53 m. from low-grade astrocytoma3 |
| 25 m. from anaplastic astrocytoma3 | ||||
| Sex ratio (M/F) | 1.43 | 1.4* | 1.26,44 | 0.83 |
| 1.811 | ||||
| Age of diagnosis | 563,46 | 56* | 446,44 | 403,46 |
| 5311 | ||||
| p53 mutation | 2/19 (11%)3 | 8/35 (23%)*12,43 | 31/37 (84%)6,44 | 20/30 (67%)3 |
| PTEN mutation | 9/28 (32%)5 | 8/21 (38%)*45 | 6/22 (27%)6,45 | 1/25 (4%)5 |
| p16 deletion | 10/28 (36%)4 | 7/19 (37%)* | 1/37 (3%)6,44 | 1/23 (4%)4 |
| MDM2 amplification | 2/29 (7%)17 | 1/19 (5%)* | 0/18 (0%)6 | 0/27 (0%)17 |
| EGFR amplification | 11/28 (39%)5 | 1/22 (4%)*32 | 2/37 (5%)6,44 | 0/22 (0%)5 |
| CDK4 amplification | 1/28 (4%)4 | 2/19 (10%)* | 1/19 (5%)44 | 3/23 (13%)4 |
*This study.
Superscripts are numbers from the list of references at the end of this paper.
The present study shows that gliosarcomas contain some genetic alterations similar to those typically encountered in primary glioblastomas, ie, frequent p16 deletions (37%) and PTEN mutations (37%) (Table 3) ▶ . The unexpected and most striking difference is the complete absence of amplification or overexpression of the EGFR gene, a hallmark of primary (de novo) glioblastomas. 2,3,5 The absence of EGFR amplification in gliosarcomas may affect their biological behavior, but large clinical trials showed no significant difference in prognosis between gliosarcomas and ordinary glioblastomas. 9,28 The reason for selective aberrant mesenchymal differentiation in a subset of glioblastomas without EGFR amplification remains to be elucidated.
The most common changes in gliosarcomas detected in cytogenetic studies included gains of chromosome 7 (EGFR gene locus) and loss of chromosome 10, followed by deletions of the long arm of chromosomes 13 and 9. 13,30-32 The absence of EGFR amplification in this study suggests that other protooncogenes on chromosome 7 may be involved in the evolution of this glioblastoma variant. CDK6, PDGF-A and c-MET genes on chromosome 7 have been reported to be amplified or overexpressed in malignant gliomas. 33-36 c-MET immunohistochemistry in these study shows that c-MET is overexpressed in gliomatous but not in sarcomatous components in gliosarcomas. It remains to be clarified whether amplification of CDK6, PDGF-A genes is involved in the development of gliosarcomas.
The frequency of p53 mutations in gliosarcomas was 26% and thus was significantly lower than in secondary glioblastomas (67%, P = 0.0086) but somewhat higher than in our cohort of patients with primary glioblastomas, but the difference was not significant (26 versus 11%, P = 0.405). 3
The 12q13–14 chromosomal region contains several genes (MDM2, CDK4, sarcoma amplified sequence SAS and GLI) that have been reported to be coamplified in sarcomas 37 and glioblastomas. 38 In this study, one gliosarcoma (case 220) showed coamplification of MDM2 and CDK4 in gliomatous and sarcomatous tumor areas.
The progression of cells from G1 to S phase is regulated by cyclin-dependent kinases (CDKs), their inhibitors, and the retinoblastoma protein (pRB). In a simplified model, p16 protein binds to CDK4 and inhibits the formation of CDK4/cyclin D complex. When activated, this complex phosphorylates the RB protein, thereby inducing the release of the E2F transcription factor, which in turn activates genes involved in the late G1 and S phases. 39,40 Homozygous p16 deletion, CDK4 amplification, and loss of RB expression are frequent in glioblastomas. 4,41,42 In this study, approximately one-half of gliosarcomas showed aberrant expression in one of these genes (Table 1) ▶ . The frequency of p16 homozygous deletion in seven (37%) gliosarcomas is similar to that in primary glioblastomas. 4 Our finding that in gliosarcomas homozygous p16 deletion, CDK4 amplification, and loss of RB expression were mutually exclusive corresponds to similar observations in other glioblastomas 4,15,42 and indicates that altered expression of any of these genes may lead to loss of cell cycle control.
In conclusion, gliosarcomas exhibit clinical features and a genetic profile similar to those of primary (de novo) glioblastomas, ie, advanced patient age, short clinical history, and frequent p16 deletions and PTEN mutations. The unexpected and most striking difference is the absence of amplification/overexpression of the EGFR gene, a genetic hallmark of primary glioblastomas. The presence of identical genetic alterations in both gliomatous and sarcomatous components strongly supports the concept of a monoclonal origin of gliosarcomas.
Acknowledgments
We are grateful to Dr. Leila Chimelli, Pathology Department, University of Ribeirão Preto, São Paulo, Brazil, for providing one of the gliosarcoma biopsies. We thank Ms. N. Lyandrat and Ms. M. Laval for excellent technical assistance. R. M. R. was supported by a grant from the Fundação para a Ciência e Tecnologia (PRAXIS XXI), Portugal.
Footnotes
Address reprint requests to Dr. Hiroko Ohgaki, Unit of Molecular Pathology, International Agency for Research on Cancer, 69372 Lyon cedex 08, France. E-mail: ohgaki@iarc.fr.
Supported by a grant from the Foundation for Promotion of Cancer Research, Japan.
References
- 1.Kleihues P, Burger PC, Plate KH, Ohgaki H, Cavenee WK: Glioblastoma. Kleihues P Cavenee WK eds. Pathology, Genetics of Tumours of the Nervous System. 1997, :pp 16-24 International Agency for Research on Cancer, Lyon [Google Scholar]
- 2.Kleihues P, Ohgaki H: Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-Oncology 1999, 1:44-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watanabe K, Tachibana O, Sato K, Yonekawa Y, Kleihues P, Ohgaki H: Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 1996, 6:217-224 [DOI] [PubMed] [Google Scholar]
- 4.Biernat W, Tohma Y, Yonekawa Y, Kleihues P, Ohgaki H: Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol 1997, 94:303-309 [DOI] [PubMed] [Google Scholar]
- 5.Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y, Kleihues P, Ohgaki H: PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol 1998, 57:684-689 [DOI] [PubMed] [Google Scholar]
- 6.Peraud A, Watanabe K, Schwechheimer K, Yonekawa Y, Kleihues P, Ohgaki H: Genetic profile of the giant cell glioblastoma. Lab Invest 1999, 79:123-129 [PubMed] [Google Scholar]
- 7.Stroebe H: Uber Entstehung und Bau der Hirngliome. Beitr Pathol Anat 1895, 18:405-486 [Google Scholar]
- 8.Burger PC, Scheithauer BW: Tumors of the Central Nervous System. 1994. Armed Forces Institute of Pathology, Washington
- 9.Meis JM, Martz KL, Nelson JS: Mixed glioblastoma multiforme and sarcoma. A clinicopathologic study of 26 radiation therapy oncology group cases. Cancer 1991, 67:2342-2349 [DOI] [PubMed] [Google Scholar]
- 10.Kleihues P, Burger PC, Scheithauer BW: The new WHO classification of brain tumours. Brain Pathol 1993, 3:255-268 [DOI] [PubMed] [Google Scholar]
- 11.Biernat W, Hegi M, Aguzzi A, Kleihues P: Gliosarcoma: Pathology and Genetics of Tumours of the Nervous System. Edited by P Kleihues, WKCavenee. Lyon, International Agency for Research on Cancer, 1997, pp 27–28
- 12.Biernat W, Aguzzi A, Sure U, Grant JW, Kleihues P, Hegi ME: Identical mutations of the p53 tumor suppressor gene in the gliomatous and the sarcomatous components of gliosarcomas suggest a common origin from glial cells. J Neuropathol Exp Neurol 1995, 54:651-656 [DOI] [PubMed] [Google Scholar]
- 13.Boerman RH, Anderl K, Herath J, Borell T, Johnson N, Schaeffer-Klein J, Kirchhof A, Raap AK, Scheithauer BW, Jenkins RB: The glial and mesenchymal elements of gliosarcomas share similar genetic alterations. J Neuropathol Exp Neurol 1996, 55:973-981 [DOI] [PubMed] [Google Scholar]
- 14.Brüstle O, Ohgaki H, Schmitt HP, Walter GF, Ostertag H, Kleihues P: Primitive neuroectodermal tumors after prophylactic central nervous system irradiation in children. Association with an activated K-ras gene. Cancer 1992, 69:2385-2392 [DOI] [PubMed] [Google Scholar]
- 15.Ueki K, Ono Y, Henson JW, Efird JT, von Deimling A, Louis DN: CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res 1996, 56:150-153 [PubMed] [Google Scholar]
- 16.Rollbrocker B, Waha A, Louis DN, Wiestler OD, von Deimling A: Amplification of the cyclin-dependent kinase 4 (CDK4) gene is associated with high cdk4 protein levels in glioblastoma multiforme. Acta Neuropathol (Berl) 1996, 92:70-74 [DOI] [PubMed] [Google Scholar]
- 17.Biernat W, Kleihues P, Yonekawa Y, Ohgaki H: Amplification and overexpression of MDM2 in primary (de novo) glioblastomas. J Neuropathol Exp Neurol 1997, 56:180-185 [DOI] [PubMed] [Google Scholar]
- 18.Hunter SB, Abbott K, Varma VA, Olson JJ, Barnett DW, James CD: Reliability of differential PCR for the detection of EGFR and MDM2 gene amplification in DNA extracted from FFPE glioma tissue. J Neuropathol Exp Neurol 1995, 54:57-64 [DOI] [PubMed] [Google Scholar]
- 19.Feigin IM, Allen LB, Lipkin L, Gross SW: The endothelial hyperplasia of the cerebral blood vessels and its sarcomatous transformation. Cancer 1958, 11:264-277 [DOI] [PubMed] [Google Scholar]
- 20.Grant JW, Steart PV, Aguzzi A, Jones DB, Gallagher PJ: Gliosarcoma: an immunohistochemical study. Acta Neuropathol (Berl) 1989, 79:305-309 [DOI] [PubMed] [Google Scholar]
- 21.Kochi N, Budka H: Contribution of histiocytic cells to sarcomatous development of the gliosarcoma. An immunohistochemical study. Acta Neuropathol (Berl) 1987, 73:124-130 [DOI] [PubMed] [Google Scholar]
- 22.Haddad SF, Moore SA, Schelper RL, Goeken JA: Vascular smooth muscle hyperplasia underlies the formation of glomeruloid vascular structures of glioblastoma multiforme. J Neuropathol Exp Neurol 1992, 51:488-492 [DOI] [PubMed] [Google Scholar]
- 23.Meis JM, Ho KL, Nelson JS: Gliosarcoma: a histologic and immunohistochemical reaffirmation. Mod Pathol 1990, 3:19-24 [PubMed] [Google Scholar]
- 24.Jones H, Steart PV, Weller RO: Spindle-cell glioblastoma or gliosarcoma? Neuropathol Appl Neurobiol 1991, 17:177-187 [DOI] [PubMed] [Google Scholar]
- 25.Paulus W, Bayas A, Ott G, Roggendorf W: Interphase cytogenetics of glioblastoma and gliosarcoma. Acta Neuropathol (Berl) 1994, 88:420-425 [DOI] [PubMed] [Google Scholar]
- 26.Wada H, Enomoto T, Fujita M, Yoshino K, Nakashima R, Kurachi H, Haba T, Wakasa K, Shroyer KR, Tsujimoto M, Hongyo T, Nomura T, Murata Y: Molecular evidence that most but not all carcinosarcomas of the uterus are combination tumors. Cancer Res 1997, 57:5379-5385 [PubMed] [Google Scholar]
- 27.Holst VA, Finkelstein S, Colby TV, Myers JL, Yousem SA: p53 and K-ras mutational genotyping in pulmonary carcinosarcoma, spindle cell carcinoma, and pulmonary blastoma: implications for histogenesis. Am J Surg Pathol 1997, 21:801-811 [DOI] [PubMed] [Google Scholar]
- 28.Perry JR, Ang LC, Bilbao JM, Muller PJ: Clinicopathologic features of primary and postirradiation cerebral gliosarcoma. Cancer 1995, 75:2910-2918 [DOI] [PubMed] [Google Scholar]
- 29.Sreenan JJ, Prayson RA: Gliosarcoma: a study of 13 tumors, including p53 and CD34 immunohistochemistry. Arch Pathol Lab Med 1997, 121:129-133 [PubMed] [Google Scholar]
- 30.Bigner SH, Mark J, Bigner DD: Cytogenetics of human brain tumors. Cancer Genet Cytogenet 1990, 47:141-154 [DOI] [PubMed] [Google Scholar]
- 31.Jenkins RB, Kimmel DW, Moertel CA, Schultz CG, Scheithauer BW, Kelly PJ, Dewald GW: A cytogenetic study of 53 human gliomas. Cancer Genet Cytogenet 1989, 39:253-279 [DOI] [PubMed] [Google Scholar]
- 32.Bigner SH, Wong AJ, Mark J, Muhlbaier LH, Kinzler KW, Vogelstein B, Bigner DD: Relationship between gene amplification and chromosomal deviations in malignant human gliomas. Cancer Genet Cytogenet 1987, 29:165-170 [DOI] [PubMed] [Google Scholar]
- 33.Costello JF, Plass C, Arap W, Chapman VM, Held WA, Berger MS, Huang HJ, Cavenee WK: Cyclin-dependent kinase 6 (CDK6) amplification in human gliomas identified using two-dimensional separation of genomic DNA. Cancer Res 1997, 57:1250-1254 [PubMed] [Google Scholar]
- 34.Westermark B, Heldin CH, Nister M: Platelet-derived growth factor in human glioma. Glia 1995, 15:257-263 [DOI] [PubMed] [Google Scholar]
- 35.Koochekpour S, Jeffers M, Rulong S, Taylor G, Klineberg E, Hudson EA, Resau JH, Vande WG: Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res 1997, 57:5391-5398 [PubMed] [Google Scholar]
- 36.Wullich B, Sattler HP, Fischer U, Meese E: Two independent amplification events on chromosome 7 in glioma: amplification of the epidermal growth factor receptor gene and amplification of the oncogene MET. Anticancer Res 1994, 14:577-579 [PubMed] [Google Scholar]
- 37.Nilbert M, Rydholm A, Mitelman F, Meltzer PS, Mandahl N: Characterization of the 12q13–15 amplicon in soft tissue tumors. Cancer Genet Cytogenet 1995, 83:32-36 [DOI] [PubMed] [Google Scholar]
- 38.Collins VP: Gene amplification in human gliomas. Glia 1995, 15:289-296 [DOI] [PubMed] [Google Scholar]
- 39.Sherr CJ: Cancer cell cycles. Science 1996, 274:1672-1677 [DOI] [PubMed] [Google Scholar]
- 40.Mulligan G, Jacks T: The retinoblastoma gene family: cousins with overlapping interests. Trends Genet 1998, 14:223-229 [DOI] [PubMed] [Google Scholar]
- 41.Schmidt EE, Ichimura K, Reifenberger G, Collins VP: CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res 1994, 54:6321-6324 [PubMed] [Google Scholar]
- 42.Ichimura K, Schmidt EE, Goike HM, Collins VP: Human glioblastomas with no alterations of the CDK2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene 1996, 13:1065-1072 [PubMed] [Google Scholar]
- 43.Frankel RH, Bayona W, Koslow M, Newcomb EW: p53 mutations in human malignant gliomas: comparison of loss of heterozygosity with mutation frequency. Cancer Res 1992, 52:1427-1433 [PubMed] [Google Scholar]
- 44.Meyer-Puttlitz B, Hayashi Y, Waha A, Rollbrocker B, Bostrom J, Wiestler OD, Louis DN, Reifenberger G, von Deimling A: Molecular genetic analysis of giant cell glioblastomas. Am J Pathol 1997, 151:853-857 [PMC free article] [PubMed] [Google Scholar]
- 45.Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von Deimling A: PTEN mutations in gliomas and glioneuronal tumors. Oncogene 1998, 16:2259-2264 [DOI] [PubMed] [Google Scholar]
- 46.von Deimling A, von Ammon K, Schoenfeld D, Wiestler OD, Seizinger BR, Louis DN: Subsets of glioblastoma multiforme defined by molecular genetic analysis. Brain Pathol 1993, 3:19-26 [DOI] [PubMed] [Google Scholar]