

Abstract
Retinal vasculogenesis and ischemic retinopathies provide good model systems for study of vascular development and neovascularization (NV), respectively. Vascular endothelial cell growth factor (VEGF) has been implicated in the pathogenesis of retinal vasculogenesis and in the development of retinal NV in ischemic retinopathies. However, insulin-like growth factor-I and possibly other growth factors also participate in the development of retinal NV and intraocular injections of VEGF antagonists only partially inhibit retinal NV. One possible conclusion from these studies is that it is necessary to block other growth factors in addition to VEGF to achieve complete inhibition of retinal NV. We recently demonstrated that a partially selective kinase inhibitor, PKC412, that blocks phosphorylation by VEGF and platelet-derived growth factor (PDGF) receptors and several isoforms of protein kinase C (PKC), completely inhibits retinal NV. In this study, we have used three additional selective kinase inhibitors with different selectivity profiles to explore the signaling pathways involved in retinal NV. PTK787, a drug that blocks phosphorylation by VEGF and PDGF receptors, but not PKC, completely inhibited retinal NV in murine oxygen-induced ischemic retinopathy and partially inhibited retinal vascularization during development. CGP 57148 and CGP 53716, two drugs that block phosphorylation by PDGF receptors, but not VEGF receptors, had no significant effect on retinal NV. These data and our previously published study suggest that regardless of contributions by other growth factors, VEGF signaling plays a critical role in the pathogenesis of retinal NV. Inhibition of VEGF receptor kinase activity completely blocks retinal NV and is an excellent target for treatment of proliferative diabetic retinopathy and other ischemic retinopathies.
Neovascularization (NV) occurs in wound repair and several pathological processes including tumor growth, arthritis, atherosclerosis, and proliferative retinopathies. Although there are likely to be tissue-specific differences, there are also likely to be shared features, so that new knowledge regarding one of these pathologies may provide insights for the others. Proliferative retinopathies provide good model systems for study of NV, because the new blood vessels can be visualized in vivo and the ocular circulation is well-studied, providing important background information.
The retina is a tissue with very high metabolic activity that is oxygenated from retinal and choroidal circulations, which each originate from branches of the ophthalmic artery. The choroidal circulation is derived from the long and short posterior ciliary arteries, which pierce the sclera and form successively smaller branches that supply the choriocapillaris, fenestrated microvessels separated from the retina by the retinal pigmented epithelium (RPE). The photoreceptor layer of the retina has no blood vessels and receives oxygen by diffusion from the choriocapillaris. The retinal circulation is derived from the central retinal artery, which enters the eye through the optic nerve and branches to form retinal arterioles that run along the surface of the retina and give rise to the superficial capillary bed. The arterioles also send penetrating branches throughout the inner two-thirds of the retina, which form the intermediate and deep retinal capillary beds.
The retinal circulation develops first at the optic nerve and extends to the periphery along the surface of the retina by vasculogenesis, the formation of blood vessels from pre-existent precursor cells. Blood vessels sprout from the superficial retinal vessels and invade the retina by a process referred to as angiogenesis, resulting in formation of the intermediate and deep capillary beds. Therefore, retinal vascular development involves both vasculogenesis and angiogenesis and occurs late, compared to most other developmental processes. It is completed shortly before term in humans; in several species, including rats and mice, it is completed after birth. Hypoxia in the avascular peripheral retina results in up-regulation of vascular endothelial growth factor (VEGF). 1 Hyperoxia inhibits development of retinal blood vessels, and in fact causes them to regress due to apoptosis of vascular endothelial cells. 2 This regression is accompanied by down-regulation of VEGF and is prevented by administration of exogenous VEGF. These data suggest that VEGF plays an important role in retinal vascular development.
Neonatal animals with hyperoxia-induced regression of retinal vessels, when removed from hyperoxia and put back into room air, develop severe retinal hypoxia, dramatic up-regulation of VEGF, and retinal NV. 3,4 This situation models that of retinopathy of prematurity (ROP) in humans, but also shares features with several disease processes in adults in which retinal vessels become damaged and occluded, leading to retinal ischemia. These diseases are collectively referred to as ischemic retinopathies and include branch retinal vein occlusion, central retinal vein occlusion, and proliferative diabetic retinopathy, the most common cause of severe visual loss in people under 60 in developed countries. 5 Hypoxia-induced up-regulation of VEGF has also been implicated in the development of retinal NV in these diseases. 6-11
These data suggest that interruption of VEGF signaling is a good target for pharmacological treatment of retinal NV. This has been borne out by studies in which VEGF antagonists have been injected into the eyes of animals with ischemic retinopathies and have caused partial inhibition of retinal NV. 12,13 Although these studies confirm that VEGF plays a central role, questions remain as to why VEGF antagonists are only partially effective. It may be that the antagonists are not sufficiently potent or effective levels are short-lived. Delivery is an issue because intraocular injections in mouse eyes are technically difficult and potentially unreliable. However, insulin-like growth factor-I (IGF-I) also contributes to retinal NV in ischemic retinopathies; 14 IGF-I, and possibly other growth factors, could be responsible for residual angiogenic activity.
Recently, we demonstrated that a partially selective kinase inhibitor that blocks phosphorylation by VEGF and platelet-derived growth factor (PDGF) receptors and several isoforms of protein kinase C (PKC) completely inhibits retinal NV. 15 In this study, we have used drugs that selectively inhibit tyrosine kinase activity of VEGF and/or PDGF receptors to explore the role of these signaling pathways in retinal vascular development and pathological retinal NV.
Materials and Methods
Drug Treatment of Mice with Ischemic Retinopathy
Ischemic retinopathy was produced in C57BL/6J mice by a method described by Smith et al. 3 Seven-day-old (P7) mice and their mothers were placed in an airtight incubator and exposed to an atmosphere of 75 ± 3% oxygen for 5 days. Incubator temperature was maintained at 23 ± 2°C, and oxygen was measured every 8 hours with an oxygen analyzer. After 5 days, the mice were removed from the incubator and placed in room air, and drug treatment was begun. Drug was dissolved in aqueous buffer or dimethyl sulfoxide (DMSO) depending on solubility and diluted to final concentrations with water; the maximum concentration of DMSO was 1%. Vehicle (aqueous buffer or 1% DMSO) or vehicle containing various concentrations of drug (volume, 10 μl/gram body weight) was placed in the stomach by gavage twice a day. At P17, after 5 days of treatment, mice were sacrificed and their eyes rapidly removed and frozen in optimum cutting temperature embedding compound (OCT; Miles Diagnostics, Elkhart, IN) or fixed in 10% phosphate-buffered formalin and embedded in paraffin. Adult C57BL/6J mice were also treated by gavage with drug or vehicle, sacrificed after 5 days, and their eyes processed for frozen or paraffin sections.
Quantitation of Retinal Neovascularization
Frozen sections (10 μm) of eyes from drug-treated and control mice were histochemically stained with biotinylated Griffonia simplicifolia lectin B4 (Vector Laboratories, Burlingame, CA), which selectively binds to endothelial cells. Slides were incubated in methanol/H2O2 for 10 minutes at 4°C, washed with 0.05 mol/L Tris-buffered saline (TBS), pH 7.6, and incubated for 30 minutes in 10% normal porcine serum. Slides were incubated 2 hours at room temperature with biotinylated lectin and, after rinsing with 0.05 mol/L TBS, incubated with avidin coupled to peroxidase (Vector Laboratories) for 45 minutes at room temperature. After being washed for 10 minutes with 0.05 mol/L TBS, slides were incubated with diaminobenzidine to give a brown reaction product. Some slides were counterstained with hematoxylin and all were mounted with Cytoseal (Stephens Scientific, Riverdale, NJ).
To perform quantitative assessments, 10-μm serial sections were cut through half of each eye, and sections roughly 50 to 60 μm apart were stained with lectin, providing 13 sections per eye for analysis. Lectin-stained sections were examined with an Axioskop microscope (Zeiss, Thornwood, NY), and images were digitized using a 3 CCD color video camera (IK-TU40A, Toshiba, Tokyo, Japan) and a frame grabber. Image-Pro Plus software (Media Cybernetics, Silver Spring, MD) was used to delineate lectin-stained cells on the surface of the retina and their area was measured. The mean of the 13 measurements from each eye was used as a single experimental value.
Drug Treatment of Mice during Retinal Vascular Development
Litters of newborn C57BL/6J mice were divided into treatment and control groups which received daily subcutaneous injections of 10 mg/kg of drug or vehicle, respectively. At P7 or P10, mice were anesthetized with ether and perfused with 1 ml of phosphate-buffered saline containing 50 mg/ml of fluorescein-labeled dextran (2 × 10 6 average molecular weight, Sigma, St. Louis, MO) as previously described. 16 The eyes were removed and fixed for 1 hour in 10% phosphate-buffered formalin. The cornea and lens were removed and the entire retina was carefully dissected from the eyecup, radially cut from the edge of the retina to the equator in all four quadrants, and flat-mounted in Aquamount with photoreceptors facing downward. Flat mounts were examined by fluorescence microscopy and images were digitized using a 3 CCD color video camera (IK-TU40A, Toshiba) and a frame grabber. Image-Pro Plus software (Media Cybernetics) was used to measure the distance from the center of the optic nerve to the leading front of developing retinal vessels in each quadrant and the mean was used as a single experimental value.
Treatment of Rhodopsin/VEGF Transgenic Mice
Rhodopsin/VEGF transgenic mice 16,17 were treated with drug or vehicle by gavage for 2 weeks from P7 to P21. The mice were then anesthetized and perfused with fluorescein-labeled dextran as described above. Retinas were flat mounted with the photoreceptor side up and examined by fluorescence microscopy at 200× magnification, which provides a narrow depth of field, so that when focusing on NV along the outer edge of the retina, the remainder of the retinal vessels are out of focus. This permits easy delineation of the NV. The outer edge of the retina, which corresponds to the subretinal space in vivo, is easily identified; therefore, there is standardization of focal plane from slide to slide. Images were digitized using a 3 CCD color video camera and a frame grabber. Image-Pro Plus was used to delineate each of the lesions and calculate the number in each retina, the area of each lesion, and the total area of NV per retina as previously described. 16
Results
Kinase Inhibitors with Overlapping but Different Activities
The inhibitory profiles of the three partially selective kinase inhibitors used in this study have been published. 18-21 PTK787/ZK 222584 (PTK787) is an excellent inhibitor of phosphorylation by human VEGF receptor 2 and its mouse homologue, Flk-1 (IC50s: 0.10 and 0.27 μmol/L, respectively). It is a less potent inhibitor of VEGF receptor 1 (IC50: 0.49 μmol/L) and also blocks the related tyrosine kinases PDGF β-receptor, c-Kit (the receptor for stem cell factor), and cFms, the receptor for macrophage colony stimulating factor-1 (IC50s: 0.2, 0.38, and 1.2 μmol/L, respectively). In concentrations up to 10 μmol/L, PTK787 does not inhibit kinases from other families, including FGFR1, c-Met, Tie2, EGFR, c-Src, v-Abl, and PKC-α. The dihydrochloride salt of PTK787 was used for all experiments.
CGP 57148 and CGP 53716 are selective inhibitors of the kinase activity of PDGF β-receptors (IC50s: 0.3 and 0.25 μmol/L, respectively).and v-Abl (IC50s: 0.2, 0.38, and 1.2 μmol/L, respectively) and do not inhibit phosphorylation by other kinases, including VEGF receptors or PKCs in concentrations greater than 20 μmol/L. 18-20
PTK787 Inhibits Retinal Neovascularization in Mice with Ischemic Retinopathy
Figure 1, A and B ▶ , shows the appearance of normal griffonia simplicifolia lectin-stained retinal vessels in nonischemic P17 mice. In P17 mice with ischemic retinopathy, there is a marked increase in the area of endothelial cell staining throughout the retina with large clumps of cells constituting NV on the retinal surface (Figure 1, C and D ▶ ; Figure 2E ▶ , columns 2 versus 1). In contrast, P17 mice with ischemic retinopathy treated twice a day by gavage with 25 mg/kg of PTK787 showed a dramatic decrease in the area of endothelial cell staining within the retina (Figure 1E) ▶ . There were vessels in the innermost part of the retina inside the inner limiting membrane adjacent to the vitreous cavity, the region normally occupied by the superficial capillary bed, but there were no vessels on the surface of the retina and the deep capillary beds were absent. The avascular appearance of the retina was very similar to that of mice with ischemic retinopathy treated with 600 mg/kg/day of PKC412, a partially selective kinase inhibitor that blocks phosphorylation by VEGF and PDGF receptors and several isoforms of PKC. 15 Like retinas from mice treated with PKC412, high magnification of retinas from mice treated with PTK787 showed no identifiable endothelial cells above the internal limiting membrane on the surface of the retina (Figure 1F) ▶ . All retinal sections from mice with ischemic retinopathy treated with 50 mg/kg/day of PTK787 had no endothelial cells above the internal limiting membrane, indicating that there was complete inhibition of retinal NV. To assess the effect on retinal vascular development as well as retinal NV, we quantified the total area of endothelial cell staining in the retina in PTK787-treated and control mice by image analysis. There was a dramatic decrease in retinal endothelial cells in mice with ischemic retinopathy treated with 25 mg/kg twice a day (120 μmoles/kg/day) of PTK787 compared to vehicle-treated mice (Figure 2E ▶ , columns 5 and 2, respectively), and a significant decrease from nonischemic mice (Figure 2E ▶ , columns 5 and 1, respectively).
Figure 1.
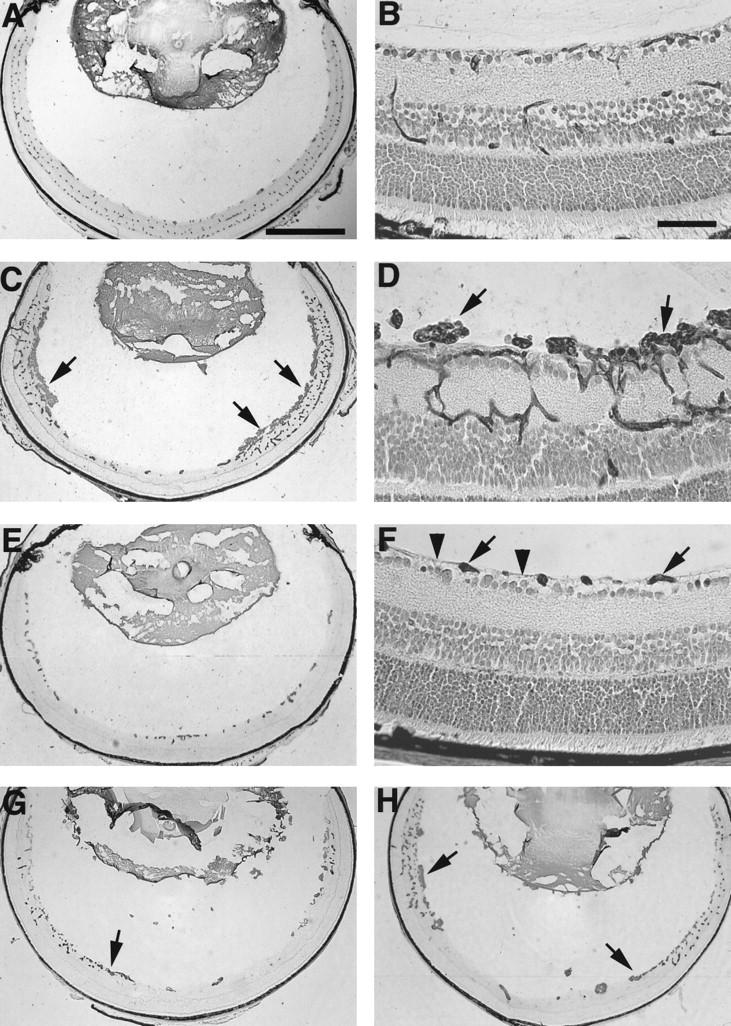
Oral administration of PTK787, an antagonist of VEGF and PDGF receptor tyrosine kinases, inhibits retinal neovascularization in a dose-dependent manner, with complete inhibition at a dose of 50 mg/kg/day. Postnatal day (P) 7 mice were put in high oxygen for 5 days and then, on P12, they were removed to room air and given PTK787 or vehicle by gavage for 5 days. Mice were sacrificed on P17 and retinal frozen section were histochemically stained with the endothelial cell-selective lectin, griffonia simplicifolia I, using the peroxidase-antiperoxidase technique. Retinal blood vessels within the retina and neovascularization on the surface are stained brown with reaction product. The low power views show sections without counterstain, so that all of the staining is due to blood vessels. The high power views are counterstained with H&E, but resolution is sufficient to distinguish the brown-staining vasculature from other retinal cells. A: Normal retinal vessels in a nonischemic P17 mouse. Bar = 0.5 mm. B: High magnification of normal retinal vessels in P17 mouse. Bar = 50 μm. C: A P17 mouse with ischemic retinopathy treated by gavage for 5 days with vehicle alone shows extensive neovascularization on the surface of the retina (arrows). D: High magnification of a P17 mouse with ischemic retinopathy treated with vehicle by gavage for 5 days. There are large fronds of neovascularization above the inner limiting membrane (arrows). E: A P17 mouse with ischemic retinopathy treated by gavage with 25 mg/kg twice a day of PTK787 shows endothelial cells within the superficial part of the retina, with none on the surface of the retina and a striking absence of the deep retinal capillary bed. F: High magnification of the retina from a P17 mouse treated with 25 mg/kg twice a day of PTK787 shows a few clumps of endothelial cells in the superficial part of the retina (arrows) with no neovascularization above the inner limiting membrane (arrowheads) and no deep capillaries. G: A P17 mouse with ischemic retinopathy treated by gavage with 2.5 mg/kg twice a day of PTK787 shows a few areas of neovascularization on the surface of the retina (arrow), but less than that seen in vehicle-treated mice. There is poor development of the deep capillary bed. H: A P17 mouse with ischemic retinopathy treated by gavage with 0.25 mg/kg twice a day of PTK787 shows more neovascularization than that seen in the retinas of mice treated with 2.5 mg/kg (G), but less than that seen in vehicle-treated mice (C). The deep capillary bed is present in the periphery of the retina.
Figure 2.
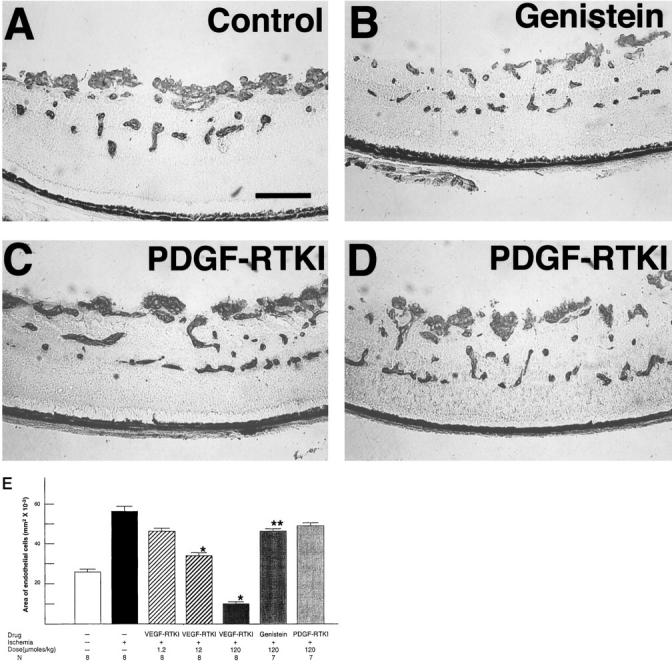
Two PDGF receptor tyrosine kinase inhibitors (RTKIs) do not inhibit retinal neovascularization, suggesting that blockage of VEGF receptor kinase is the activity of PTK787 that is responsible for its neovascularization-inhibiting activity. Vehicle-treated mice with ischemic retinopathy (A) show extensive neovascularization on the surface of the retina, as do mice treated with 120 μmoles/kg/day of CGP 57148 (C) and CGP 53716 (D), two potent PDGF RTKIs that do not inhibit VEGF RTK. Mice treated with 120 μmoles/kg/day of genistein, a nonspecific RTKI, appear to show somewhat less retinal neovascularization (B). E: Image analysis was used to calculate the area of endothelial cell staining per section as described in Materials and Methods for control mice without ischemic retinopathy (column 1) or mice with ischemic retinopathy treated with vehicle (column 2), a range of doses of PTK787, a VEGF and PDGF RTKI (columns 3–5), genistein, a nonselective kinase inhibitor (column 6), or CGP 57148, a PDGF RTKI (column 7). *P < 0.001; ** P < 0.05 for difference from vehicle-treated mice with ischemic retinopathy by t-test for populations with similar variance.
P17 mice with ischemic retinopathy treated twice a day with 2.5 mg/kg of PTK787 showed less NV on the surface of the retina (Figure 1G ▶ , arrows) and decreased staining in the regions of the deep capillary beds compared to vehicle-treated mice (Figure 1, G and C ▶ , respectively). Image analysis demonstrated a statistically significant difference in the area of endothelial cell staining (Figure 2E ▶ , columns 4 versus 2). The retinas of mice with ischemic retinopathy treated orally twice a day with 0.25 mg/kg had the appearance of less endothelial cell staining than retinas of vehicle-treated mice (Figure 1, G ▶ versus C). The difference was not statistically significant by paired t-test, but analysis of variance demonstrated a statistically significant dose-dependant decrease in endothelial cell area for the 3 doses of PTK787 compared to vehicle alone (P < 0.001; Figure 2E ▶ ).
At 120 μmoles/kg/day, a dose equivalent to the highest dose of PTK787 used, CGP 57148 (Figure 2C) ▶ and CGP 53716 (Figure 2D) ▶ , two selective PDGF receptor tyrosine kinase inhibitors (PDGF-RTKI), caused no apparent decrease in NV in mice with ischemic retinopathy compared to vehicle-treated mice (Figure 2A) ▶ . This was confirmed by quantitative analysis in mice treated with CGP 57148 (Figure 2E ▶ , column 7 versus 2). At 120 μmoles/kg/day, genestein, a nonspecific tyrosine kinase inhibitor, caused a small, statistically significant decrease in the area of endothelial cell staining in the retinas of mice with ischemic retinopathy (Figure 2, B ▶ versus A and Figure 2E ▶ , columns 6 versus 2).
PTK787 Inhibits Retinal Vascular Development
Treatment of neonatal mice with PKC412, the inhibitor of VEGF and PDGF receptors and PKC, results in inhibition of retinal vascular development. 15 To determine whether PTK787 inhibits retinal vascular development, neonatal mice were treated with subcutaneous injections of 10 mg/kg of PTK787 or vehicle alone starting on P0, and on P7 or P10 they were perfused with fluorescein-labeled dextran and retinal whole mounts were prepared. At P7, retinal vessels in vehicle-treated mice had almost reached the peripheral edge of retina, but in PTK787-treated mice they had only extended about halfway to the periphery (Figure 3, A and B) ▶ . At P10 in control mice, the superficial capillary bed was complete and extended all the way to the peripheral edge of the retina, and the deep capillary bed was partially developed. But in PTK787-treated mice, the superficial capillary bed had not yet reached the edge of the retina (Figure 3, C and D) ▶ . Measurement of the distance from the optic nerve to the developing edge of retinal vessels at 7 and 10 days showed a highly significant decrease at both time points in PTK787-treated mice compared to controls (Figure 4) ▶ . Therefore, PTK787, like PKC412, inhibits retinal vascular development as well as pathological retinal NV. However, it is interesting to note that at the doses used, both of these drugs delay, but do not permanently prevent, development of the superficial capillary bed, which occurs by vasculogenesis. On the other hand, a 2.5-fold higher dose of PTK787, given between P7 and P17, completely inhibits development of the intermediate and deep capillary beds (Figure 1, E and F) ▶ , which occurs by the process of angiogenesis. Additional studies are needed to determine whether these differences are dose- and/or timing-related, or whether they indicate that angiogenesis is more completely dependent on VEGF signaling than is vasculogenesis.
Figure 3.
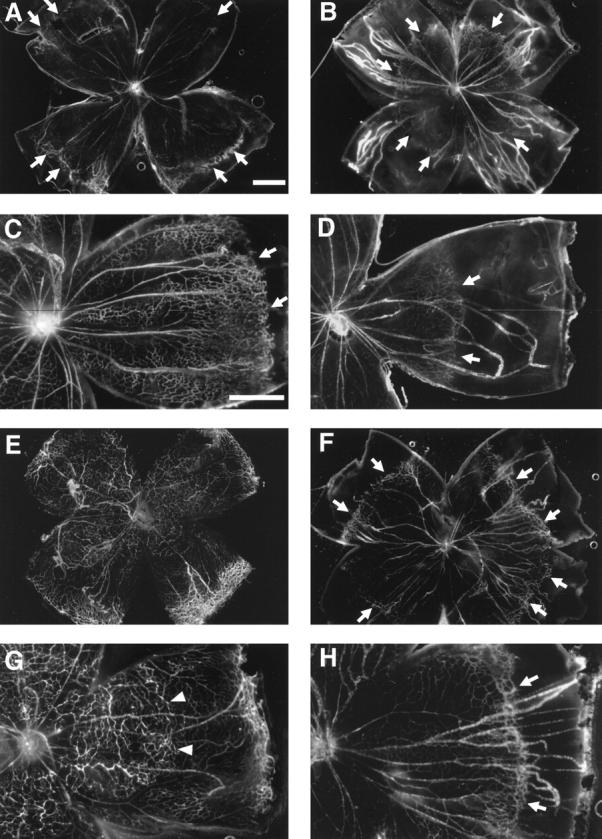
Treatment with PTK787, an antagonist of VEGF and PDGF receptor tyrosine kinases, inhibits development of retinal blood vessels. Mice were treated with vehicle (A, C, E, G) or 10 mg/kg/day of PTK797 (B, D, F, H) starting the day of birth and on postnatal day 7 (P7; A-D) or P10 (E-H) the mice were perfused with fluorescein-labeled dextran and retinal whole mounts were prepared. A and C: At P7, the leading edge of the developing superficial retinal capillary bed (arrows) has almost reached the edge of the retina. B and D: At P7, the edge of developing retinal capillaries (arrows) in PTK797-treated mice, lags far behind those in controls, extending less than halfway to the edge of the retina. E and G: At P10, in vehicle-treated mice, the superficial capillary bed has reached the edge of the retina and the deep capillary beds are developing (arrows, better seen in stereo). F and H: At P10, in PTK797-treated mice, the leading edge of superficial capillaries (arrows) has not yet reached the edge of the retina and no deep capillaries are seen. Bars = 0.5 mm.
Figure 4.

Distance from the center of optic nerve to the leading edge of developing capillaries is significantly less in mice treated with 10 mg/kg PTK787, a VEGF and PDGF receptor tyrosine kinase inhibitor (VEGF-RTKI), compared to vehicle-treated controls. Measurements were made by image analysis in all four quadrants and averaged to give one experimental value per mouse. Each bar is the mean of 5 experimental values. Statistical comparisons were made by paired t-test.
PTK787 Completely Blocks NV in Rhodopsin/VEGF Transgenic Mice
Transgenic mice in which the rhodopsin promoter drives retina-specific expression of VEGF (rho/VEGF mice) develop widespread NV that originates from retinal vessels, extends through the outer nuclear layer, and grows in the subretinal space. 17 In these mice, the expression of VEGF is turned on in photoreceptors around P6 or P7, increases to a steady-state level over a few days, and is sustained for at least several months, the longest time points yet examined. At 3 weeks of age, there is extensive NV in the subretinal space that is easily quantitated on retinal whole mounts. 16 Administration of 25 mg/kg once a day (60 μmoles/kg/day) of PTK787 between 7 and 21 days resulted in complete absence of NV in the subretinal space of rho/VEGF mice (Figure 5) ▶ . Focusing on the outer surface of the retina, which corresponds to the subretinal space, there was no NV in PTK787-treated mice (Figure 5B) ▶ , whereas in vehicle-treated mice, there were numerous areas of NV (Figure 5, A and C) ▶ . Focusing at the level of the deep capillary bed, some abortive buds could be seen extending from retinal vessels in PTK787-treated mice (Figure 5D) ▶ . This indicates that PTK787 is an extremely effective inhibitor of VEGF-induced NV in the retina.
Figure 5.

Oral administration of PTK787 blocks neovascularization in transgenic mice that express VEGF in photoreceptors. Transgenic mice that express VEGF in photoreceptors starting on postnatal day (P) 5 or 6 were treated with 25 mg/kg/day of PTK787 (n = 15) or vehicle (n = 13) between P7 and P21. On P21, mice were perfused with fluorescein-labeled dextran and retinal flat mounts were examined by fluorescence microscopy. A: The retina from a mouse treated with vehicle shows numerous areas of neovascularization (arrows), which appear in the foreground on the outer surface of the retina (subretinal space). The neovascularization is surrounded by retinal pigmented epithelial cells, resulting in the pigmented appearance. B: The retina from a mouse treated with PTK787 shows no areas of neovascularization on the outer surface of the retina. C: High magnification of the retina shown in A demonstrates several clumps of new blood vessels surrounded by retinal pigmented epithelial cells. D: High magnification of the retina shown in B shows no blood vessels on the outer surface of the retina. By focusing within the retina at the level of the deep capillary beds, a few hyperfluorescent lines are seen and may represent sprouts that have not developed into neovascularization. E: The area of neovascularization on the outer surface of the retina was measured by image analysis on P21 in transgenic mice treated with vehicle (n = 13) or PTK787 (n = 15). Statistical comparisons were made by t-test for populations with similar variance.
PTK787 Has No Identifiable Effect on Retinal Vessels in Adult Mice
Adult mice were treated twice a day for 5 days with 25 mg/kg (120 μmoles/kg/day) of PTK787, a dose and duration of dosing that completely inhibit retinal NV in oxygen-induced ischemic retinopathy and a greater dose than that shown to inhibit development of the deep retinal capillary bed. The lectin-stained retinal vessels of PTK787-treated adult mice (Figure 6B) ▶ appeared normal and identical to the retinal vessels of control mice (Figure 6A) ▶ . Image analysis demonstrated no difference in the total area of endothelial cell staining in the retinas of treated versus control mice (Figure 6E) ▶ . Retinas from treated mice stained with H&E were also completely normal and identical in appearance to retinas from control mice (Figure 6, D ▶ versus C). This suggests that treatment with PTK787 for 5 days is not toxic to endothelial cells of mature vessels or to retinal neurons.
Figure 6.
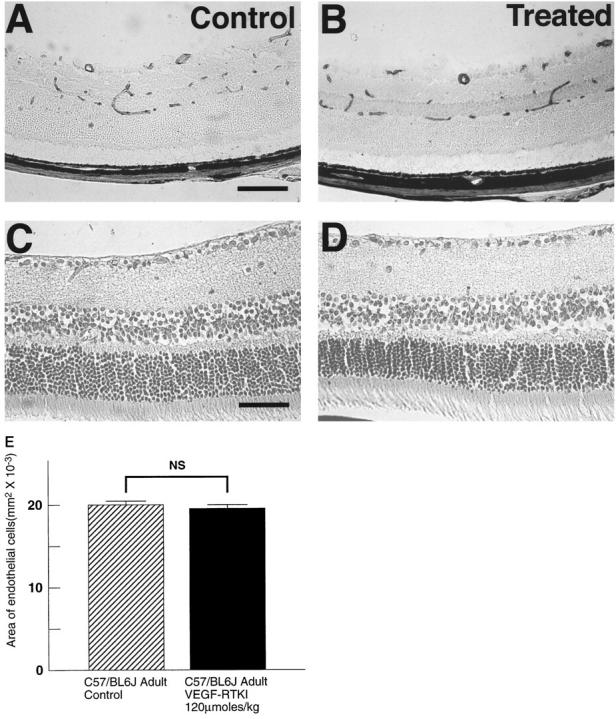
Treatment of adult mice with PTK787 shows no identifiable toxicity to mature retinal blood vessels or retinal neurons. Adult mice were treated twice a day by gavage with vehicle (controls, A and C) or 25 mg/kg (120 μmoles/kg/day) of PTK787 (B and D). After 5 days they were sacrificed and retinal sections were stained with griffonia simplicifolia lectin to stain retinal blood vessels (A and B) or hematoxylin to stain retinal neurons (C and D). Treated mice showed no identifiable changes in retinal blood vessels or retinal neurons compared to controls. E: Image analysis was used to measure the area of endothelial staining for vehicle-treated controls (n = 7) and mice treated with PTK787 (VEGF-RTKI; n = 7). Statistical comparison was made by paired t-test.
Discussion
Identification of stimulatory factors involved in a pathological process provides a means to selectively target those factors to inhibit the process and potentially minimize unwanted effects. This has been the rationale for the quest to identify factors responsible for retinal NV, which has been ongoing since 1948 when such factors were first postulated to exist. 22 Recently, VEGF has been demonstrated to play a role in the development of retinal NV, 6-11,17 but IGF-I has also been implicated. 14 Previous treatment attempts with VEGF antagonists in the mouse model of ischemic retinopathy have resulted in a maximum inhibition of retinal NV of 50%, 12,13 and it is not clear whether this is because VEGF signaling was only partially blocked or whether the residual NV is due to activity of other factors. Intravitreal injection of an anti-VEGF antibody in primates with laser-induced branch retinal vein occlusion completely inhibits iris NV, 23 but it is not clear what relevance this has to treatment of retinal NV. Inhibitors that work by other mechanisms also failed to achieve more than 50% inhibition of retinal NV, 24,25 raising the question of whether this was the maximum effect achievable by pharmacological inhibition.
We have recently demonstrated that it is possible to achieve complete inhibition of retinal NV in the murine model of oxygen-induced ischemic retinopathy by oral administration of a partially selective kinase inhibitor that blocks phosphorylation by VEGF and PDGF receptors, and by serine/threonine kinases of the PKC family. 15 In the present study, we found complete inhibition of retinal NV with a different kinase inhibitor that blocks phosphorylation by VEGF and PDGF receptors, but not PKC. Two drugs that selectively block PDGF receptor kinase activity, but not VEGF receptor kinase or PKC, had no significant effects on retinal NV. By process of elimination, it appears that inhibition of VEGF receptor kinase is the activity responsible for the dramatic inhibition of retinal NV in this model of ischemic retinopathy.
A new model of retinal NV in transgenic mice with photoreceptor-specific expression of VEGF 16,17 differs from the ischemic retinopathy model in that VEGF is the only known stimulus and its secretion is high and sustained, so there is no spontaneous involution of the NV. Oral administration of PTK787 resulted in complete inhibition of NV in the transgenic model, confirming that this drug is a very effective inhibitor of VEGF signaling in the retina. The ability of PTK787 or PKC412 15 to block VEGF signaling in the ischemic retinopathy model also resulted in complete inhibition of retinal NV. This suggests that despite possible contributions from other growth factors, blockade of VEGF receptor signaling is sufficient to completely inhibit retinal NV. The relative lack of efficacy of intraocular injections of VEGF receptor/IgG chimeric proteins 12 or anti-sense VEGF oligomers 13 could be due to delivery problems, or it may be that inhibition of VEGF receptor kinase activity is a more effective way to block VEGF signaling than these two approaches aimed at decreasing the amount of VEGF available to bind to receptors.
Blockade of VEGF signaling with PTK787 or PKC412 15 in neonatal mice inhibits retinal vascular development. This is not surprising in view of previous data demonstrating that VEGF signaling is necessary for the survival of developing retinal vessels. 2 Considering this and our demonstration that retinal vascular development is perturbed in neonatal mice treated with either of two potent VEGF receptor kinase inhibitors, it is likely that any drug that effectively blocks VEGF signaling will have a deleterious effect on retinal vascular development. The lack of a similar effect in the neonatal mouse model of ischemic retinopathy with other VEGF antagonists 12,13 suggests that with those approaches, blockade of VEGF signaling was incomplete. Although there may be a narrow therapeutic window within which it is possible to completely or almost completely inhibit retinal NV without damaging developing retinal vessels, it would be hazardous to treat infants with ROP with highly effective VEGF inhibitors. However, mature retinal vessels are much less dependent on VEGF as a survival factor, 2 and in support of this, we saw no evidence of damage to retinal vessels in adult mice treated with PTK787 or PKC412 for 5 days. This suggests that PTK787, PKC412, or other potent VEGF receptor kinase inhibitors could potentially be useful for treatment of proliferative diabetic retinopathy or other ischemic retinopathies in patients with mature retinas. Clinical trials to investigate safety and efficacy of VEGF kinase inhibitors in patients with proliferative diabetic retinopathy are being planned.
Footnotes
Address reprint requests to Peter A. Campochiaro, M.D., Maumenee 719, The Johns Hopkins University School of Medicine, 600 N. Wolfe Street, Baltimore, MD 21287-9277. E-mail: pcampo@jhmi.edu.
Supported by U. S. Public Health Service grant EY05951 and core grant P30EY1765 from the National Eye Institute, a Lew R. Wasserman Merit Award (to P. A. C.) and an unrestricted grant from Research to Prevent Blindness, Inc., a grant from CIBA Vision, Inc., a Novartis Company, the Rebecca P. Moon, Charles M. Moon, Jr., and Dr. P. Thomas Manchester Research Fund, a grant from Mrs. Harry J. Duffey, a grant from Dr. and Mrs. William Lake, a grant from Project Insight, and a grant from the Association for Retinopathy of Prematurity and Related Diseases. P. A. C. is the George S. and Dolores D. Eccles Professor of Ophthalmology and Neuroscience. CIBA Vision provided partial funding of the study reported in this article.
H. Ozaki’s current address: Fukuoka University School of Medicine, Fukuoka, Japan.
M.-S. Seo’s current address: Chonnam National University Medical School and Hospital, Kwangju, Korea.
P.A. Campochiaro is a consultant to CIBA Vision. The terms of this arrangement are being managed by The Johns Hopkins University in accordance with the conflict of interest policies.
References
- 1.Stone J, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T, Keshet E: Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 1995, 15:4738-4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E: Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995, 1:1024-1028 [DOI] [PubMed] [Google Scholar]
- 3.Smith LEH, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA: Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 1994, 35:101-111 [PubMed] [Google Scholar]
- 4.Penn JS, Tolman BL, Lowery LA: Variable oxygen exposure causes preretinal neovascularization in the newborn rat. Invest Ophthalmol Vis Sci 1993, 34:576-585 [PubMed] [Google Scholar]
- 5.Kahn H, Hiller R: Blindness caused by diabetic retinopathy. Am J Ophthalmol 1974, 78:58-67 [DOI] [PubMed] [Google Scholar]
- 6.Adamis AP, Miller JW, Bernal M-T, D’Amico DJ, Folkman J, Yeo T-K, Yeo K-T: Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol 1994, 118:445-450 [DOI] [PubMed] [Google Scholar]
- 7.Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen MS, Aiello LM, Ferrara N, King GL: Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994, 331:1480-1487 [DOI] [PubMed] [Google Scholar]
- 8.Malecaze F, Clamens S, Simorre-Pinatel V, Mathis A, Chollet P, Favard C, Bayard F, Plouet J: Detection of vascular endothelial growth factor messenger RNA and vascular endothelial growth factor-like activity in proliferative diabetic retinopathy. Arch Ophthalmol 1994, 112:1476-1482 [DOI] [PubMed] [Google Scholar]
- 9.Pe’er J, Shweiki D, Itin A, Hemo I, Gnessin H, Keshet E: Hypoxia-induced expression of vascular endothelial growth factor by retinal cells is a common factor in neovascularizing ocular diseases. Lab Invest 1995, 72:638-645 [PubMed] [Google Scholar]
- 10.Miller JW, Adamis AP, Shima DT, D’Amore PA, Moulton RS, O’Reilly MS, Folkman J, Dvorak HF, Brown LF, Berse B, Yeo T-K, Yeo K-T: Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am J Pathol 1994, 145:574-584 [PMC free article] [PubMed] [Google Scholar]
- 11.Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LEH: Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA 1995, 92:905-909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L, Ferrara N, King GL, Smith LEH: Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA 1995, 92:10457-10461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson GS, Pierce EA, Rook SL, Foley E, Webb R, Smith LES: Oligodeoxynucleotides inhibit retinal neovascularization in a murine model of proliferative retinopathy. Proc Natl Acad Sci USA 1996, 93:4851-4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith LEH, Kopchick JJ, Chen W, Knapp J, Kinose F, Daley D, Foley E, G SR, Schaeffer JM: Essential role of growth hormone in ischemia-induced retinal neovascularization. Science 1997, 276:1706–1709 [DOI] [PubMed]
- 15.Seo M-S, Kwak N, Ozaki H, Yamada H, Okamoto N, Fabbro D, Hofmann F, Wood JM, Campochiaro PA: Dramatic inhibition of retinal and choroidal neovascularization by oral administration of a kinase inhibitor. Am J Pathol 1999, 154:1743-1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tobe T, Okamoto N, Vinores MA, Derevjanik NL, Vinores SA, Zack DJ, Campochiaro PA: Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest Ophthalmol Vis Sci 1998, 39:180-188 [PubMed] [Google Scholar]
- 17.Okamoto N, Tobe T, Hackett SF, Ozaki H, Vinores MA, LaRochelle W, Zack DJ, Campochiaro PA: Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol 1997, 151:281-291 [PMC free article] [PubMed] [Google Scholar]
- 18.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB: Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996, 2:561-566 [DOI] [PubMed] [Google Scholar]
- 19.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Regenass U, Lydon NB: Selective inhibition of the platelet-derived growth factor signal transduction pathway by a protein-tyrosine kinase inhibitor of the 2-phenylaminopyrimidine class. Proc Natl Acad Sci USA 1995, 92:2558-2562 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Myllarniemi M, Calderon L, Lemstrom K, Buchdunger E, Hayry P: Inhibition of platelet-derived growth factor receptor tyrosine kinase inhibits vascular smooth muscle cell migration and proliferation. FASEB J 1997, 11:1119-1126 [DOI] [PubMed] [Google Scholar]
- 21.Wood JM, Bold G, Stover D, Hofmann F, Buchduneger E, Mestan J, Towbin H, Schnell C, Mett H, O’Reilly T, Cozens R, Rosel J, Siemeister G, Menrad A, Schirner M, Thierauch K-H, Schneider MR, Martiny-Baron G, Drevs J, Marme D: A novel and potent inhibitor of VEGF receptor tyrosine kinases impairs VEGF-induced responses and tumor growth after oral administration. Cancer Res (in press) [PubMed]
- 22.Michaelson I: The mode of development of the vascular system of the retina with some observations on its significance for certain retinal diseases. Trans Ophthalmol Soc (UK) 1948, 68:137-180 [Google Scholar]
- 23.Adamis AP, Shima DT, Tolentino MJ, Gragoudas ES, Ferrara N, Folkman J, D’Amore PA, Miller JW: Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization. Arch Ophthalmol 1996, 114:66-71 [DOI] [PubMed] [Google Scholar]
- 24.Luna J, Tobe T, Mousa SA, Reilly TM, Campochiaro PA: Antagonists of integrin α-v β-3 inhibit retinal neovascularization in a murine model. Lab Invest 1996, 75:563-573 [PubMed] [Google Scholar]
- 25.Hammes H, Brownlee M, Jonczyk A, Sutter A, Preissner K: Subcutaneous injection of a cyclic peptide antagonist of vitronectin receptor-type integrins inhibits retinal neovascularization. Nat Med 1996, 2:529-533 [DOI] [PubMed] [Google Scholar]