

Abstract
Genetic alterations of pancreatic intraductal lesions adjacent to invasive ductal carcinoma were investigated. We submitted nine foci of ordinary epithelium, 12 foci of nonpapillary hyperplasia, 12 foci of papillary hyperplasia (pap HP), 66 foci of severe ductal dysplasia, and 27 invasive foci from a total of 10 pancreatic carcinomas for genetic analysis. All foci were individually microdissected and allelic losses of 3p, 4q, 5q, 6q, 8p, 9p, 10q, 11q, 13q, 16q, 17p, and 18q were studied. All invasive and severely dysplastic intraductal foci exhibited loss of heterozygosity (LOH) at more than one chromosomal locus. For each case, allelic loss was frequently observed on 9p (severe ductal dysplasia 90%, invasion 100%), 17p (severe ductal dysplasia 80%, invasion 80%), and 18q (severe ductal dysplasia 88%, invasion 88%). Ninety-four percent of severe ductal dysplasia and 96% of invasive foci had multiple LOH. Seventeen percent of nonpapillary hyperplasia and 33% of pap HP showed LOH. Only one focus of pap HP showed multiple LOH. The patterns of allelic loss identified in severe ductal dysplasia were generally conserved in synchronous infiltrating tumors, supporting the paradigm that infiltrating tumors are clonally derived from severe ductal dysplasia. In eight of 10 cases, however, we found frequent genetic heterogeneity in the intraductal lesion, suggestive of genetic progression or diversion. These findings indicate that invasive pancreatic carcinoma evolves through successive and divergent genetic changes with selection of aggressive subclones in the intraductal component.
Pancreatic ductal carcinoma carries a very poor prognosis because early detection is difficult and effective treatment is not established. The mortality rate of pancreatic carcinoma has been increasing recently in eastern Europe, North America, and in Japan. 1,2 A molecular understanding of pancreatic tumorigenesis is critical for developing efficient detection methods and treatment protocols. Molecular studies of pancreatic carcinoma have advanced recently. The oncogene K-ras mutation has been described as its earliest genetic change. 3-8 A number of tumor suppressor genes, such as p53, p16, and DPC4, have also been found to play a critical role in the clonal progression of pancreatic tumorigenesis. 9-20 Using frozen tissue, cell lines, and xenografts, invasive pancreatic carcinoma has been found to have allelic loss on many chromosomal arms. 9,21-25 Among these, 1p, 9p, 17p, and 18q have been reported to be deleted in more than 60% of cases, followed by 3p, 6p, 6q, 8p, 10q, 12q, 13q, 21q, and 22q, which have been reported to be deleted in 40 to 60% of pancreatic carcinomas. These chromosomal loci may harbor unidentified tumor suppressor genes critical to the development of pancreatic carcinoma.
A diverse spectrum of intraductal epithelial changes, ie, nonpapillary hyperplasia (non-pap HP), papillary hyperplasia (pap HP), atypical hyperplasia/severe dysplasia, and so forth, has been described around invasive pancreatic carcinomas. Mucous cell hyperplasia has been reported to be the most important precursor in the histogenesis of human pancreatic ductal carcinoma. 26-32 However, there have been only a few genetic studies of these pancreatic intraductal preinvasive lesions which correspond to morphology. Using microdissection techniques and extensive allelotyping, Fujii et al 23 showed genetic progression and heterogeneity in intraductal papillary-mucinous neoplasms of the pancreas; however, the allelic loss pattern was distinct from solid infiltrating carcinomas. Moskaluk et al 6 examined p16 and K-ras mutations in pancreatic intraductal neoplasia accompanied by invasive pancreatic ductal carcinoma, and found frequent p16 alterations to be high-risk precursors of invasive foci.
In the present study, we microdissected multiple foci of ordinary epithelium, non-pap HP, pap HP, severe ductal dysplasia, and invasive foci (invasion, infiltrating components) of solid-type (common type) pancreatic ductal carcinoma. Polymerase chain reaction (PCR)-based microsatellite assays were used to detect loss of heterozygosity (LOH) on chromosome arms 3p, 4q, 5q, 6q, 8p, 9p, 10q, 11q, 13q, 16q, 17p, and 18q. Using this approach, we found evidence of genetic progression and divergence in the intraductal clonal development of solid-type pancreatic ductal carcinoma into the invasive phenotype.
Materials and Methods
Patients
Patient demographics are summarized in Table 1 ▶ . Patients included six men and four women. The mean age at the time of diagnosis was 66 (range, 51 to 79) years old. The TNM staging of these pancreatic carcinomas ranged from II T2N0 M0/G1 to III T3N1 M0/G1 (Table 1) ▶ . 33
Table 1.
Patient Demographics and Microdissected Foci
| Case | Age | Sex | Tumor status* | Site | Number of dissected foci | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of dissected foci | Ordinary epithelium | Hyperplasia† | Severe ductal dysplasia† | Invasion† | ||||||
| Nonpapillary | Papillary | |||||||||
| G2 | 71 | F | III T2N1M0/G1 | Head | 13 | 1 | 1 | 8 | 3 | |
| G4 | 64 | M | II T2N0M0/G1 | Head | 16 | 3 | 1 | 3 | 8 | 1 |
| G5 | 66 | F | II T2N0M0/G2 | Head | 19 | 1 | 1 | 15 | 2 | |
| G8 | 67 | M | III T2N1M0/G3 | Head | 13 | 2 | 1 | 9 | 1 | |
| G21 | 51 | M | II T2N0M0/G2 | Head | 11 | 1 | 1 | 2 | 5 | 2 |
| G24 | 74 | M | III T2N1M0/G2 | Head | 16 | 1 | 13 | 2 | ||
| G25 | 79 | M | III T2N1M0/G1 | Body and tail | 11 | 1 | 1 | 5 | 4 | |
| G30 | 64 | F | III T3N1M0/G1 | Body and tail | 16 | 2 | 4 | 7 | 3 | |
| G31 | 65 | M | II T2N0M0/G2 | Head | 8 | 3 | 2 | 2 | ||
| G33 | 62 | F | II T2N0M0/G1 | Head | 22 | 3 | 11 | 8 |
*Tumor grade and stage were evaluated using established TNM criteria.33
†According to AFIP.31
Tissue Samples
Ten cases of paraffin-embedded tissues diagnosed as severe ductal dysplasia (in situ carcinoma or atypical ductal lesion) adjacent to invasive pancreatic ductal carcinoma were obtained from 166 cases of surgically resected pancreatic ductal carcinoma files between 1976 and December 1997 in the First Department of Pathology at Niigata University School of Medicine. In all pancreatic carcinomas, a formalin-fixed specimen of the whole pancreas was cut into 4- to 5-mm stepwise tissue blocks along the main pancreatic duct and embedded in paraffin.
All pathology was reviewed for consistency of diagnosis using published criteria proposed by the Armed Forces Institute of Pathology (AFIP) for ordinary epithelium, non-pap HP, pap HP, severe ductal dysplasia (carcinoma in situ) which shows severe cellular atypia of duct cells with or without papillary proliferation, and invasive foci. 31,32 Intraductal lesions were defined as lesions whose walls had fine circular elastic fibers on Victoria-Blue staining, especially in the main pancreatic duct or in the first and second branches of pancreatic ducts which have accessory mucous glands.
For each case, appropriate tissue blocks were selected, and 3-μm sections were cut and stained with hematoxylin and eosin (H&E) and with antibodies against p53 (PAb 1801 mouse monoclonal antibody; Oncogene Science, Inc., Manhasset, NY) and Ki 67 (antibody MIB-1; Immnotech, Marseille, France). Multiple serial sections 10-μm thick were cut, deparaffinized, stained with H&E, visualized with an inverted microscope, and microdissected using a 26-gauge needle. Two to 15 foci of severe ductal dysplasia, one to six foci of pap HP or non-pap HP, one to three foci of ordinary epithelium, and one to eight invasive carcinoma foci were individually microdissected for each case. The cellular composition of all microdissected foci were regarded as cytologically homogeneous. Most microdissected intraductal lesions were cytologically homogeneous and the entire ductal epithelium was microdissected. However, when a single dysplastic ductal section contained both severely dysplastic and focally nonatypical hyperplastic epithelial cells, only dysplastic cells were microdissected. Normal control tissue (acinar cells) was obtained from nontumorous lobules. Tissue was digested overnight in buffer containing 0.5% Nonidet P-40 and 200 μg/ml of proteinase K at 50°C. A total of 126 foci (8 to 17 foci in each case) including nine ordinary epithelium, 12 non-pap HP, 12 pap HP, 66 severe ductal dysplasia (carcinoma in situ), and 27 invasive foci from 10 cases were microdissected, and allelic status was analyzed for 12 chromosomal arms using 24 microsatellite markers.
Microsatellite Markers
PCR amplification of microsatellite markers was performed using primers for 3p (D3S1234, D3S1293, and D3S1286), 4q (D4S424 and D4S473), 5q (D5S421 and 5S1956), 6q21–25 (D6S264, D6S255, D6S311, and D6S473), 8p (D8S255, D8S261, and D8S264), 9p21 (D9S1748 and D9S1749), 10q (D10S219, D10S547, and D10S574), 11q23.3 (D11S29), 13q12–22 (D13S166, D13S168, and D13S171), 16q12–24 (D16S265 and D16S541), 17p12–14 (D17S122, D17S786, CHRNB1, TP53, and D17S1866), and 18q (D18S46, D18S474, D18S487, and D18S55). All primers were obtained from Research Genetics (Huntsville, AL). Encompassed in these chromosomal regions are several candidate suppressor genes for pancreatic ductal carcinoma, including BRCA2 (13q), Rb1 (13q), p53 (17p), 9-13 MKK4 (17p), 34,35 E-cadherin (16q), DCC (18q), DPC4 (18q), 13,18-20 APC (5q), 36,37 and p16/MTS1 (9p). 13-17,38
LOH Analysis
PCR reactions contained 1 μl of DNA lysate, 0.4 μCi [γ−32p]ATP-radiolabeled microsatellite primer, 0.2 mmol/L dNTP, 10 mmol/L Tris-HCl, pH 8.3, 1.5 mmol/L MgCl2, 50 mmol/L KCl, and 0.4 U Taq polymerase in a total reaction volume of 10 μl. The hot-start method of PCR was used to eliminate nonspecific amplification. The samples underwent 35 cycles of PCR amplification (94°C for 30 seconds, 58°C for 40 seconds, and 72°C for 60 seconds) followed by a 2-minute extension at 72°C. PCR products were separated on a 5% denaturing polyacrylamide-urea-formamide gel and visualized using a phosphoimager (Bas 2500, Bio Imaging System; Fuji Film, Tokyo, Japan). LOH was determined from a >75% reduction in the intensity in one of the two alleles compared with the normal control.
Elimination of PCR Artifacts
A number of previous studies suggested frequent PCR artifacts from paraffin-embedded materials, especially when the number of cells analyzed are small. To exclude all artifactually created allelic imbalances, we used at least 100 cells per PCR reaction. On average, 100 to 200 cells were included in each 10 μl of PCR reaction. Normal controls also contained approximately 100 to 300 nontumor cells per PCR reaction. Because the degree of DNA degradation affects the amplification efficiency and the chance of artifacts, cases and foci which show suboptimal PCR amplifications as well as nonreproducible allelic patterns were excluded. Thus, of the initial 33 cases collected, only 10 cases were selected for the present analysis. Only microdissected foci with reproducible amplifications were included in the present analysis. Because the size of PCR products larger than 200 bp are prone to nonreproducible amplification artifacts, most PCR primers in the present study were chosen to amplify <200 bp. 39-42 All PCR reactions which showed homogeneous and heterogeneous LOH were repeated at least three times in duplicate reactions to confirm the findings and to exclude spurious PCR reactions. If necessary, microdissection was repeated. 23,43,44 Heterogeneous patterns of allelic losses were also confirmed by other microsatellite markers located on the same chromosomal arms showing the same allelic loss patterns.
Results
Comparison of Allelic Loss in Hyperplasia versus Severe Ductal Dysplasia versus Invasive Foci
The frequency of LOH for each case is shown in Table 2 ▶ . When allelic loss of at least one foci was scored as LOH for each case, the most frequent allelic losses (>60%) were detected on 9p (non-pap HP 11%, pap HP 17%, severe ductal dysplasia 90%, invasion 100%), followed by 17p (HP 0%, severe ductal dysplasia 80%, invasion 80%), 18q (HP 0%, severe ductal dysplasia 88%, invasion 88%), and 6q (HP 0%, severe ductal dysplasia 63%, invasion 50%). Therefore, 9p− and 17p− followed by 18q− and 6q− were the important allelic losses in pancreatic ductal carcinoma, not only in invasive foci but also in severe ductal dysplasia.
Table 2.
Frequency of LOH in Ordinary Epithelium, Hyperplasia, Severe Ductal Dysplasia and Invasion in Each Case and Heterogeneity Index of Severe Ductal Dysplasia
| Allele | Ordinary epithelium | Hyperplasia | Severe ductal dysplasia | Invasion | Tumor suppressor gene | ||
|---|---|---|---|---|---|---|---|
| Nonpapillary | Papillary | Heterogeneity Index† | |||||
| 9p | 0 /4* | 1 /9 | 1 /6 | 90% (9/10) | 11% (1/9) | 100% (10/10) | p16 |
| 17p | 0 /4 | 0 /9 | 0 /6 | 80% (8/10) | 13% (1/8) | 80% (8/10) | p53.MKK4 |
| 18q | 0 /4 | 0 /7 | 0 /5 | 88% (7/8) | 50% (4/8) | 88% (7/8) | DCC, DPC4 |
| 6q | 0 /2 | 0 /7 | 0 /5 | 63% (5/8) | 40% (2/5) | 50% (4/8) | |
| 13q | 0 /3 | 0 /6 | 0 /6 | 14% (1/7) | 0% (0/1) | 29% (2/7) | RB, BRCA2 |
| 3p | 0 /3 | 1 /8 | 1 /5 | 11% (1/9) | 100% (1/1) | 33% (3/9) | |
| 8q | 0 /2 | 0 /5 | 0 /4 | 0% (0/6) | — | 17% (1/6) | |
| 10q | 0 /2 | 1 /4 | 0 /4 | 40% (2/5) | 100% (2/2) | 0% (0/5) | |
| 5q | 0 /1 | 0 /4 | 1 /3 | 0% (0/4) | — | 25% (1/4) | APC |
| 4q | 0 /1 | 0 /3 | 1 /3 | 33% (1/3) | 0% (0/1) | 33% (1/3) | |
| LOH (−)‡ | 4 /4 | 7 /9 | 5 /6 | 0% (0/10) | 0% (0/10) |
*Numerators indicate the number of cases with LOH (including heterogeneous LOH), and denominators indicate the number of cases with tested and informative markers at the designated locus.
†Calculated as the number of cases with heterogeneous patterns of allelic loss of the chromosomal arms in severe ductal dysplasia divided by the total number of cases with at least one dysplastic foci showing LOH.
‡LOH(−), none of the chromosomal loci tested showed allelic loss.
When individually microdissected foci were separately scored, all severe ductal dysplasia and invasive foci showed LOH at more than one locus. Allelic loss of 9p was most frequently observed (95% of severe ductal dysplasia, 96% of invasion), followed by 17p (83% of severe ductal dysplasia, 89% of invasion), 18q (74% of severe ductal dysplasia, 90% of invasion), and 6q (66% of severe ductal dysplasia, 29% of invasion). 17% of non-pap HP and 33% of pap HP showed LOH. Only one focus of pap HP showed multiple LOH (Table 3) ▶ .
Table 3.
Summary of LOH Pattern in Each Case
| Case | p53 protein expression* | LOH pattern | Lesion | Number of foci | ||||
|---|---|---|---|---|---|---|---|---|
| Ordinary epithelium | Hyperplasia | Severe ductal dysplasia | Invasion | |||||
| Nonpapillary | Papillary | |||||||
| G2 | − | LOH (−) | T6, T12 | 1 | 1 | |||
| +++ | 9p, 17p, 6q−α, 18q−α | T1, T2, T3, T7, T9 | 5 | |||||
| +++ | 9p, 17p, 6q−α, 18q−α, 3p−α | T4 | 1 | |||||
| +++ | 9p, 17p, 6q−α, 18q−α, 3p−α, 13q | T11 | 1 | |||||
| +++ | 9p, 17p, 6q−α, 18q−α, 3p−β | T5 | 1 | |||||
| +++ | 9p, 17p, 6q−β, 18q−β | T10 | 1 | |||||
| +++ | 9p, 17p, 6q−β, 18q−β, 10q | T8, T15 | 2 | |||||
| G4 | − | LOH(−) | T4, T5, T6, T8, T7, T13, T14 | 3 | 1 | 3 | ||
| +++ | 6q, 9p, 17p | T9 | 1 | |||||
| +++ | 6q, 9p, 17p, 18q | T1, T2, T3, T10, T11, T12 | 6 | |||||
| +++ | 6q−, 9p, 17p, 18q, 8q | T16 | 1 | |||||
| G5 | − | LOH(−) | T15, T8 | 1 | 1 | |||
| +++ | 18q | T1 | 1 | |||||
| +++ | 18q, 9p | T16, T17 | 2 | |||||
| +++ | 18q, 9p, 17p | T2, T3, T4, T5, T6, T12, T13, T14 | 8 | |||||
| +++ | 18q, 9p, 13q | T7 | 1 | |||||
| +++ | 18q, 9p, 13q, 17p | T18 | 1 | |||||
| G8 | − | LOH (−) | T13, T14, T12 | 2 | 1 | |||
| − | 9p | T8 | 1 | |||||
| − | 9p, 18q | T7 | 1 | |||||
| − | 9p, 18q, 3p−β | T1, T2 | 2 | |||||
| − | 9p, 3p−α | T6 | 1 | |||||
| G21 | − | LOH (−) | T5, T10, T11 | 1 | 2 | |||
| +++ | 9p | T9 | 1 | |||||
| +++ | 9p, 13q | T6 | 1 | |||||
| +++ | 9p, 13q, 18q−α | T7, T1, T2 | 1 | 2 | ||||
| +++ | 9p, 13q, 18q−β | T3, T8 | 2 | |||||
| G24 | − | LOH (−) | T15 | 1 | ||||
| − | 17p, 9p | T13, T14 | 2 | |||||
| − | 17p, 9p, 6q−α | T1, T2, T3, T4, T6, T7, T8, T9, T10, T11, T12 | 11 | |||||
| − | 17p, 9p, 6q−α, 3p−α | T16 | 1 | |||||
| − | 17p, 9p, 6q−β | T17 | 1 | |||||
| G25 | − | LOH (−) | T9, T5 | 1 | 1 | |||
| +++ | 9p, 17p | T3, T4, T8, T1, T2, T7 | 3 | 3 | ||||
| +++ | 9p, 17p, 5q | T6 | 1 | |||||
| G30 | − | 9p | T1 | 1 | ||||
| +++ | 9p, 17p, 18q−α, 4q | T4, T2, T3, T11 | 1 | 3 | ||||
| + | 9p, 17p, 18q−α, 4q, 6q, 10q−α | T8 | 1 | |||||
| − | 10q−β | T7 | 1 | |||||
| − | 3p | T6, T5, T9 | 1 | 2 | ||||
| − | 4q, 5q | T10 | 1 | |||||
| G31 | − | LOH (−) | T1, T7, T8 | 3 | ||||
| +++ | 17p | T2, T5, T3 | 2 | 1 | ||||
| +++ | 17p, 18q, 9p, 6q, | T4 | 1 | |||||
| G33 | − | LOH (−) | T13, T18, T24 | 3 | ||||
| +++ | 9p, 17p, 18q−α | T9, T10, T12, T14, T15, T17, T22, T23 | 8 | |||||
| +++ | 9p, 17p, 6q | T1, T2, T3, T4, T6, T8, T16, T19 | 8 | |||||
| +++ | 9p, 17p, 6q, 18q−β | T5, T7, T20 | 3 |
*, p53 protein expression; −, negative; +, focal; ++, moderate; +++, diffuse.
T + number (bold), microdissected invasive foci.
LOH (−): no chromosomes exhibited loss of heterogeneity.
Patterns of Allelic Loss within a Tumor
Heterogeneity with respect to allelic loss among different neoplastic foci of severe ductal dysplasia was found in 80% (8 out of 10: G2, G4, G5, G8, G21, G24, G30, and G33) of the cases examined. Heterogeneity was also observed among invasive areas in 57.1% (four of seven cases in which multiple invasive areas were dissected: G2, G24, G25, and G31) (Table 3) ▶ . For severe ductal dysplasia, the heterogeneity index was calculated as the number of cases with heterogeneous patterns of allelic loss of the chromosomal arms in severe ductal dysplasia divided by the total number of cases with at least one dysplastic foci showing LOH (Table 2) ▶ . A low heterogeneity index suggests that LOH of a particular chromosomal arm occurred relatively early in the dysplastic foci. LOH of chromosomal arms with a high heterogeneity index is considered a late event in the intraductal neoplastic process. Both 9p (11%) and 17p (13%) showed low heterogeneity indexes, thus, these chromosomal losses were considered to have occurred early. LOH of 18q (heterogeneity index, 50%) and 6q (heterogeneity index, 40%) were still assumed to have occurred early in the dysplasia, but may have followed 9p and 17p LOH as shown for G2 and G33 below.
Two examples of heterogeneous patterns of LOH from HP to invasive carcinoma through severe ductal dysplasia are illustrated for G2 (Figure 1 ▶ for distribution of the foci microdissected and representative histology, Figure 2 ▶ for representative gels and deduced sequence of genetic alterations) and G33 (Figure 3 ▶ for distribution of the foci microdissected and representative histology, and Figure 4 ▶ for representative gels and deduced sequence of genetic alterations). When both alleles of the same chromosome were independently lost from different foci, LOHα and LOHβ were used for the designation. To clarify the heterogeneous patterns of LOH, gels of the two different markers on the same chromosomal arms are shown for 6q, 18q, and 3p.
Figure 1.
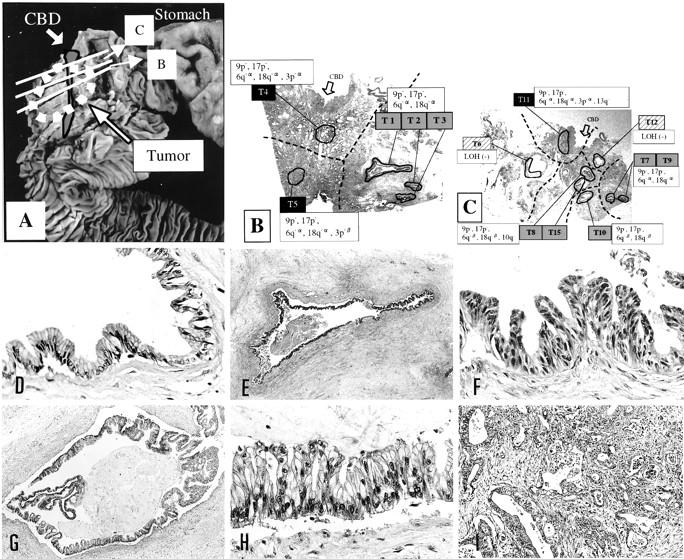
Representative foci microdissected from case G2 and spatial distribution of heterogeneous genetic patterns. A: Gross photo of the specimen showing the histology sections taken. B and C: Low power views of the section B and section C showing the microdissected foci and the distribution of heterogeneous LOH. T6,T12, hyperplasia; T1-T3, T7-T10, T15, foci of severe ductal dysplasia; T4, T5, T11, invasive foci; dotted lines divide areas with divergent genetic changes. D–I: Representative histology. D: T12, pap HP. Original magnification, ×100. E and F: T1, severe ductal dysplasia. Original magnifications: E, ×2.5; F, ×100. G and H: T15, severe ductal dysplasia. Original magnifications: G, ×10; H, ×100. I: T5, invasive focus. Original magnification, ×50.
Figure 2.

Representative gels and proposed genetic pathways for case G2. Gel for D9S1749 shows homogeneous LOH at 9p for all dysplastic and invasive foci (T1–T5, T7–T11, T15). Hyperplastic foci (T6 and T12) retained all alleles tested. Subsequent and divergent LOH at 18q is shown with two different markers (D18S487 and D18S55) to clarify the true nature of the heterogeneous patterns and to exclude spurious PCR reactions. Arrowheads indicate loss of the lower allele (18q-β: T8, T10, T15). Other foci except hyperplastic foci showed loss of the upper allele (18q-α). Further divergence of LOH at 3p (D3S1293 and D3S1286) in invasive foci (T4, T5, and T11) is shown by arrows. *, Normal alleles; N, normal control DNA; T#, microdissected foci.
Figure 3.
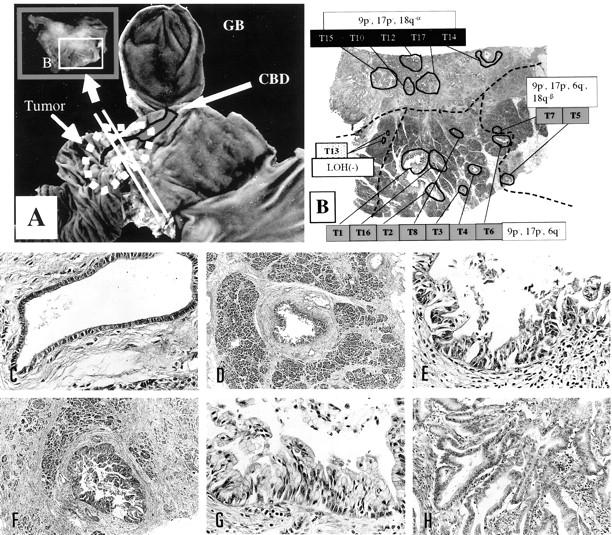
Representative foci microdissected for case G33 and spatial distribution of heterogeneous genetic changes. A: Gross photo of the specimen showing the plane of the histology section taken. B: Low power view of the section showing the microdissected foci and the distribution of heterogeneous LOH. T13 , ordinary epithelium; T1-T8, T16 , foci of severe ductal dysplasia; T10, T12, T14, T15, T17 , invasive foci; dotted lines divide areas with the divergent genetic changes. C–I: Representative histology. C: T13, ordinary epithelium. Original magnification, ×100. D and E: T4, severe ductal dysplasia. Original magnifications: D, ×10; E, ×100. F and G: T5, severe ductal dysplasia. Original magnifications: F, ×16; G, ×100 (G). H: T12, invasive focus. Original magnification, ×50.
Figure 4.

Representative gels and deduced genetic pathways for case G33. Gel for D17S786 shows homogeneous LOH at 17p for all dysplastic and invasive foci (T2, T4, T5, T7, T10, T12, and T14). Ordinary epithelium (T13 and T24) retained all alleles tested. Divergent LOH at 18q and 6q is shown with two different markers (D18S487 and D18S46 for 18q, and D6S264 and D6S473 for 6q) to clarify the true nature of the heterogeneous patterns and to exclude spurious PCR reactions. *, Normal alleles; arrows, divergent patterns of allelic losses detected at 18q (D18S487, and D18S46) in some of the dysplastic foci (18q-β at T5, T7, T20) and all of the invasive foci (18q-α); arrowheads, LOH at 6q detected only in some of the dysplastic foci (T2, T4, T5, and T7); N, normal control DNA; T#, microdissected foci.
In case G2, LOH was not seen in HP (T6 and T12), LOH at 9p and 17p was seen in all severe ductal dysplasia and invasive foci, suggesting that these genetic changes occurred early and that the neoplastic process was clonal. Two subsequent divergent patterns of LOH were observed in severe ductal dysplasia. The LOHα of 6q and 18q was seen not only in severe ductal dysplasia (T1, T2, T3, T7, and T9) but also in invasion (T4, T5, and T11). In contrast, the LOH β of 6q, 18q was seen only in severe ductal dysplasia (T8, T10, and T15). In the three invasive foci microdissected (T4, T5, and T11), further divergent and progressive genetic changes were detected at 3p and 13q. Summarizing case G2, the clonal neoplastic process started with early and homogeneous LOH at 9p and 17p, with subsequent genetic divergence resulting in different allelic loss patterns of 6q, 18q, 10q, 3p, and 13q, some of which were seen in some dysplastic and invasive foci (Figure 2 ▶ , right).
For case G33, LOH of 9p and 17p was detected in all microdissected dysplastic and invasive foci, indicating that these clonal chromosomal alterations occurred early in the intraductal stage. The neoplastic clone subsequently diverged into two genetic pathways; one pathway developed into invasive foci with 18q-α and the other subclone with 6q− and 18q-β was exclusively seen in intraductal foci.
Spatial Distribution of Genetic Heterogeneity
As shown in Figure 1, B and C ▶ , for G2 and in Figure 3B ▶ for G33, foci with identical genetic changes were clustered in a certain area in all cases that showed heterogeneous LOH. Genetically heterogeneous but closely related foci were clustered and located nearby. Thus, all genetic progression and heterogeneity occurred in spatially continuous manners within the ducts. The clustering of foci with heterogeneous genetic changes also excludes the possibility of PCR artifacts, which most likely produce a random distribution of heterogeneity.
Concurrent Allelic Loss of Chromosomal Regions
Concurrent alterations of multiple loci (more than two) of LOH were more common in severe ductal dysplasia (94% of the foci) and invasive foci (96% of the foci) than in HP (3%). Concurrent alterations of these three loci of LOH (9p, 17p, and 18q) in severe ductal dysplasia and invasive foci were evaluated. Coexistence of 9p−, 17p−, and 18q− occurred in 63% of invasive foci and 42% of severe ductal dysplasia. In the remaining foci, coexistence of 9p− and 17p− was observed in 22% of invasion and 38% of severe ductal dysplasia. Coexistence of 9p− and 18q− was observed in 7% of invasion and 12% of severe ductal dysplasia. Coexistence of 17p− and 18q− without 9p− was never observed in either invasion or severe ductal dysplasia.
p53 Immunohistochemistry
Eight of ten cases were strongly stained for p53 in both severe ductal dysplasia and invasive foci (Table 3 ▶ ; G2, G4, G5, G21, G25, G30, G31, and G33). In seven cases (G2, G4, G5, G25, G30, G31, and G33), both p53 positivity and 17p LOH were detected throughout the dysplastic intraductal and invasive foci, indicating early inactivation of p53.
Discussion
Carcinomas, in general, have been postulated to progress by the sequential accumulation of multiple genetic changes from early neoplastic lesions to invasive foci. During this early noninvasive stage of cancer, subclones with a growth advantage expand and gradually acquire an aggressive character. When multiple preinvasive pathological foci are individually microdissected and analyzed for genetic alterations, this temporal development of a clonal neoplastic process becomes evident. By microdissecting DNA from paraffin-embedded tissue sections, sequential LOH changes by Vogelstein et al 45 and K-ras gene mutation by Shibata et al 46 were shown in benign adenoma to carcinoma sequences of the colon and rectum. Fujii et al 43,44 described genetic divergence, as well as genetic progression in the clonal evolution of breast cancer by studying allelic loss of ductal in situ carcinoma and invasive components of breast cancers. Similarly, they reported clonal progression and heterogeneity in intraductal papillary-mucinous neoplasia of the pancreas. 23
In the present study, by extensive sampling of intraductal lesions with microdissection and LOH analysis of multiple chromosomal loci, we successfully demonstrated the dynamic and successive accumulation of genetic alterations in early pancreatic intraductal neoplastic foci for the first time.
All foci of severe ductal dysplasia and invasive foci showed LOH at more than one chromosomal locus. For each case, LOH on 9p, 17p, and 18q was frequently observed in invasive areas (9p−, 100%; 17p−, 80%; and 18q−, 88%), similar to previous reports. 9,21,22 These high frequencies of LOH on 9p, 17p, and 18q were also detected in individually microdissected severe ductal dysplasia foci (9p− in 95%, 17p− in 83%, and 18q− in 74% of total foci dissected) in the present study. These allelic losses shared by most of the dysplastic foci and invasive foci strongly suggest that the clonal neoplastic process begins early in intraductal dysplastic epithelium.
Heterogeneity of the LOH pattern among dissected severe ductal dysplasia and invasive foci, as shown in Figures 1, 2, 3, and 4 ▶ ▶ ▶ was seen in 57% of invasion and in 80% of severe ductal dysplasia. (Table 3) ▶ . In all cases, however, some of the allelic losses identified in the in situ cancers were generally conserved in the synchronous infiltrating tumors, supporting the concept that infiltrating tumors are clonally derived from noninvasive intraductal lesions. Heterogeneity was either genetic progression, genetic diversion, or a combination of both. In the genetic progression, there was a linear and gradual accumulation of LOH from hyperplastic foci to severe ductal dysplasia to invasive foci. In clonal diversion, the clone with early genetic changes will diverge, and multiple subclones will expand within the duct. Only some of these divergent subclones may develop invasive tumors as shown in G2 and G33. Many pancreas cancers probably evolve by a combination of these two heterogeneous pathways.
Spatial distributions of genetic heterogeneity were not random but were strongly indicative of the pathways for clonal neoplastic progression. Genetically identical foci were all clustered in a certain area and closely related foci were clustered nearby as shown in Figure 1, B and C, and 3B ▶ ▶ . Thus, genetic progression and diversion was assumed to occur when the clonal neoplastic process continuously spreads within the duct. At some point, in some foci, the clone assumes invasive character.
Chromosomal losses common to all tumor foci most likely preceded the chromosomal losses observed only in the tumor foci of more advanced stages. The 9p loss, especially, was detected very early (in HP foci in two cases), and in many cases tended to be clonally homogeneous in multiple dissected foci. Thus, 9p loss and possibly inactivation of p16 occur very early in the intraductal lesions and most likely in the transition from hyperplastic to early severe ductal dysplasia stage. 17p LOH is also relatively homogeneous throughout the intraductal foci and thus occurs early. Successive loss of 18q and 6q follows, still in the intraductal stage. Loss of other chromosomal loci (3p, 4q, 5q, 8p, 10q, and 13q) was less frequent and much more limited in the foci dissected. Thus, these additional losses occur later in the severe ductal dysplasia stage when the neoplastic subclones diverge and expand.
In previous studies of invasive pancreatic ductal adenocarcinoma, using frozen tissues, cell lines, or xenografts, nonrandom LOH on many chromosomes has been reported. 21-25 Seymour et al 21 described allelic loss of chromosome 18q (86%) and 17p (80%) by analyzing fresh-frozen tissues from seven cases of surgically-resected exocrine adenocarcinomas of the pancreas. Hahn et al 22 used xenografts taken from 18 human pancreatic ductal carcinomas and described frequent allelic loss (>60%) for 1p (69%), 9p (89%), 17p (100%), and 18q (89%), in addition, moderately frequent allelic loss (40 to 60%) was seen at 3p (44%), 6p (50%), 6q (53%), 8p (56%), 10q (50%), 12q (56%), 13q (50%), 18p (44%), 21q (56%), and 22q (59%). Mahlamäki et al 24 also described a high frequency of LOH for 18q (100%) and 9p (91%) by comparative genomic hybridization.
The specific genetic alterations in adenocarcinomas of the pancreas include activation of the K-ras oncogene in 80 to 100% of cancers 13,47-49 and inactivation of p16 (on chromosome 9p) 13-17,38 , p53 (chromosome 17p), 9-13 and DPC4 (chromosome 18q). 13,18-20 Rozenblum et al 13 described the frequencies of tumor suppressor gene inactivation for p16, p53, and DPC4 as 82% (33 of 40), 76% (31 of 41), and 53% (20 of 38) of tumors, respectively, by direct sequencing from xenograft materials. All were accompanied by LOH. Most of these studies, however, have focused on invasive lesions and limited their sampling of in situ foci.
Pancreatic intraductal lesions found around invasive carcinoma have been reported to be precursors to invasive pancreas cancers in histopathological studies. 26-31 These lesions have been labeled as nonpapillary hyperplasia (mucous cell hypertrophy), papillary-hyperplasia and severe ductal dysplasia (atypical hyperplasia, carcinoma in situ). 30,31,33 Of these, mucous cell hyperplasia has been reported to be the most important early precursor lesion. Severe cellular atypia of ductal epithelium, with or without papillary projection, has been defined as ”severe ductal dysplasia“ (atypical hyperplasia). Recently, cases with severe ductal dysplasia have been reported to have developed invasive cancer 17 months and 10 years later. 50
Few molecular genetic studies of such intraductal pancreatic lesions have been reported. K-ras mutation has been shown to be the most important precursor of pancreatic ductal carcinoma. 3-6 Recently, however, K-ras mutation has been observed in mucous cell hyperplasia in chronic pancreatitis and other nonneoplastic lesions. 7,8 Moskaluk et al 6 described an alteration of the p16 gene in a subset of pancreatic intraductal neoplasia that contains a mutation of K-ras, and they found K-ras and p16 mutations to be important high-risk precursors of invasive disease. Hruban et al 32 suggested the possibility that duct lesions are the precursors to infiltrating carcinoma of the pancreas based on both morphological and molecular analyses. The present findings further support the previous studies and confirmed that the clonal neoplastic process starts early in the intraductal stage with early involvement of 9p loss (consistent with alteration of p16), 17p loss and progressive accumulation of other genetic changes during the preinvasive stage.
The p16 alteration is observed in 85% of xenografts or cell lines from pancreatic carcinoma. 13,16,38 Schutte et al 38 described three patterns in xenografts or cell lines of pancreatic carcinoma: homozygous deletions in 48% (24 out of 50), intragenetic mutations in 36% (18 out of 50), and wild type in 16% (8 out of 50) where methylation of the CpG island occurred in 39% (7 out of 18). Loss of 9p (primers INFA and D9S 171, which were located near the p16 locus) was observed in 98% of all cases, including homozygous deletion and wild type. The present finding of 9p loss is also consistent with these previous studies and further supports the early involvement of p16 in the intraductal stage of the pancreatic neoplasms.
Concurrent alterations of three tumor suppressor genes in tumors were described by Rozenblum et al. 13 p53, p16, and DPC4 inactivation were observed in 39.5% (15 out of 38 cases of pancreatic ductal carcinoma), p53 and p16 inactivation in 26.3% (10 out of 38 cases), and p16 and DPC4 inactivation in 13.2% (5 out of 38 cases). However, p53 and DPC4 inactivation without p16 alteration was never observed. In the present study, not only foci of invasion but also foci of severe ductal dysplasia frequently showed coexistent loss of 9p, 17p, and 18q. LOH of 9p, 17p, and 18q, 9p and 17p, and 9p and 18q were frequently detected in both invasive and severe ductal dysplasia foci, whereas 17p and 18q without 9p loss was never observed. Thus, a combination of these three genetic alterations is characteristic of pancreatic ductal carcinoma and tends to be acquired very early in the intraductal stage; 9p loss seems to be an obligatory step for later 17p and 18q loss.
In conclusion, pancreatic ductal carcinomas develop from hyperplasia through severe ductal dysplasia to invasive foci by a progressive and often divergent accumulation of genetic changes, which reflect the spectrum of intraductal morphological alterations around invasive foci. LOH of 17p and 9p is observed at a high frequency in both invasive and severe ductal dysplasic foci, and 9p loss may be the earliest event in the transition from hyperplasia to early dysplasia. Other chromosomal alterations including 18q and 6q follow later in the intraductal dysplastic and invasive stage and confer subclones with a further growth advantage.
Footnotes
Address reprint requests to Hiroaki Fujii, Department of Pathology II, Juntendo University School of Medicine, 2–1-1, Hongo, Bunkyo-ku, Tokyo 113-8421, Japan. E-mail: hfujii@med.juntendo.ac.jp.
Supported in part by a research grant from the Ministry of Education, Science and Culture of Japan.
References
- 1.Parkin DM, Läärä E, Muir CS: Estimates of the worldwide frequency of sixteen major cancers in 1980. Int J Cancer 1988, 41:184-197 [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto M, Ohashi O, Saitoh Y: Japan pancreatic cancer registry: current status. Pancreas 1998, 3:238-242 [DOI] [PubMed] [Google Scholar]
- 3.Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern SE: Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res 1994, 54:3568-3573 [PubMed] [Google Scholar]
- 4.Tada M, Omata M, Ohta M: Ras gene mutations in intraductal papillary neoplasms of the pancreas. Analysis in five cases. Cancer 1991, 67:634-637 [DOI] [PubMed] [Google Scholar]
- 5.Matsubayashi H, Watanabe H, Nishikura K, Ajioka Y, Kijima H, Saito T: Determination of pancreatic ductal carcinoma histogenesis by analysis of mucous quality and K-ras mutation. Cancer 1998, 82:651-660 [DOI] [PubMed] [Google Scholar]
- 6.Moskaluk CA, Hruban RH, Kern SE: p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res 1997, 57:2140-2143 [PubMed] [Google Scholar]
- 7.Yanagisawa A, Ohtake K, Ohashi K, Hori M, Kitagawa T, Sugano H, Kato Y: Frequent c-Ki-ras oncogene activation in mucous cell hyperplasias of pancreas suffering from chronic inflammation. Cancer Res 1993, 53:953-956 [PubMed] [Google Scholar]
- 8.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R, Kishi K, Omata M: Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology 1996, 110:227-231 [DOI] [PubMed] [Google Scholar]
- 9.Hahn SA, Kern SE: Molecular genetics of exocrine pancreatic neoplasms. Surg Clin North Am 1995, 75:857-869 [DOI] [PubMed] [Google Scholar]
- 10.Ruggeri B, Zhang ST, Caamato J, DiRado M, Flynn SD, Klein-Szanto AJP: Human pancreatic carcinomas and cell lines reveal frequent and multiple alterations in the p53 and Rb-1 tumor-suppressor genes. Oncogene 1992, 7:1503-1511 [PubMed] [Google Scholar]
- 11.Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C, Hirohashi S: Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol 1993, 142:1534-1543 [PMC free article] [PubMed] [Google Scholar]
- 12.Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, Kern SE: p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994, 54:3025-3033 [PubMed] [Google Scholar]
- 13.Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE: Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 1997, 57:1731-1734 [PubMed] [Google Scholar]
- 14.Bartsch D, Shevlin DW, Tung WS, Kisker O, Wells SA, Jr, Goodfellow PJ: Frequent mutations of CDKN2 in primary pancreatic adenocarcinomas. Genes Chromosom Cancer 1995, 14:189-195 [DOI] [PubMed] [Google Scholar]
- 15.Huang L, Goodrow TL, Zhang SY, Klein-Szanto AJP, Chang H, Ruggeri BA: Deletion and mutation analyses of the p16/MTS-1 tumor suppressor gene in human ductal pancreatic cancer reveals a higher frequency of abnormalities in tumor-derived cell lines than in primary ductal adenocarcinomas. Cancer Res 1996, 56:1137-1141 [PubMed] [Google Scholar]
- 16.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE: Frequent somatic mutations and homozygous deletions of the p16(MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994, 8:27-32 [DOI] [PubMed] [Google Scholar]
- 17.Naumann M, Savitskaia N, Eilert C, Schramm A, Kalthoff H, Schmiegel W: Frequent codeletion of p16/MTS1 and p15/MTS2 and genetic alterations in p16/MTS1 in pancreatic tumors. Gastroenterology 1996, 110:1215-1224 [DOI] [PubMed] [Google Scholar]
- 18.Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H, Sidransky D, Casero RA, Jr, Meltzer PS, Hahn SA, Kern SE: DPC4 gene in various tumor types. Cancer Res 1996, 56:2527-2530 [PubMed] [Google Scholar]
- 19.Hahn SA, Schutte M, Hoque ATMS, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271:350–353 [DOI] [PubMed]
- 20.Hahn SA, Hoque ATMS, Moskaluk CA, da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, Kern SE: Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res 1996, 56:490–494 [PubMed]
- 21.Seymour AB, Hruban RH, Redston M, Caldas C, Powell SM, Kinzler KW, Yeo CJ, Kern SE: Allelotype of pancreatic adenocarcinoma. Cancer Res 1994, 54:2761-2764 [PubMed] [Google Scholar]
- 22.Hahn SA, Seymour AB, Hoque ATMS, Schutte M, da Costa LT, Redston MS, Caldas C, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res 1995, 55:4670-4675 [PubMed] [Google Scholar]
- 23.Fujii H, Inagaki M, Kasai S, Miyokawa N, Tokusashi Y, Gabrielson E, Hruban RH: Genetic progression and heterogeneity in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol 1997, 151:1447-1454 [PMC free article] [PubMed] [Google Scholar]
- 24.Mahlamäki EH, Höglund M, Gorunova L, Karhu R, Dawiskiba S, Andrén-Sandberg Å, Kallioniemi OP, Johansson B: Comparative genomic hybridization reveals frequent gains of 20q, 8q, 11q, 12p, and 17q, and losses of 18q, 9p, and 15q in pancreatic cancer. Genes Chromosom Cancer 1997, 20:383–391 [DOI] [PubMed]
- 25.Sugio K, Molberg K, Albores-Saavedra J, Virmani AK, Kishimoto Y, Gazdar AF: K-ras mutations and allelic loss at 5q and 18q in the development of human pancreatic cancers. Int J Pancreatol 1997, 21:205-217 [DOI] [PubMed] [Google Scholar]
- 26.Cubilla AL, Fitzgerald PJ: Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res 1976, 36:2690-2698 [PubMed] [Google Scholar]
- 27.Sommers SC, Murphy SA, Warren S: Pancreatic duct hyperplasia and cancer. Gastroenterology 1954, 27:629-640 [PubMed] [Google Scholar]
- 28.Kozuka S, Sassa R, Taki T, Masamoto K, Nagasawa S, Saga S, Hasegawa K, Takeuchi M: Relation of pancreatic duct hyperplasia to carcinoma. Cancer 1979, 43:1418-1428 [DOI] [PubMed] [Google Scholar]
- 29.Pour PM, Sayed S, Sayed G: Hyperplastic, preneoplastic and neoplastic lesions found in 83 human pancreases. Am J Clin Pathol 1982, 77:137-152 [DOI] [PubMed] [Google Scholar]
- 30.Kasuya K, Watanabe H, Ajioka Y, Ohashi Y, Takei K, Yoshida M: The classification of pancreatic ductal epithelium according to atypia. Tan to Sui 1993, 14:491–499 (in Japanese)
- 31.Solcia E, Capella C, Klöppel G: Tumors of the pancreas. ed 3 Rosai J eds. Atlas of Tumor Pathology, 1995, :pp 215-236 Armed Forces Institute of Pathology, Washington DC [Google Scholar]
- 32.Hruban RH, Wilentz RE, Goggins M, Offerhaus GJA, Yeo CJ, Kern SE: Review. Pathology of incipient pancreatic cancer. Ann Oncol 1999, 10(Suppl 4):S9-S11 [PubMed] [Google Scholar]
- 33.American Joint Committee on Cancer: Exocrine Pancreas. Cancer Staging Manual, ed 5, Edited by ID Fleming, JS Cooper, DE Henson, RVP Hutter, BJ Kennedy, GP Murphy, B O’Sullivan, LH Sobin, JW Yarbro. Philadelphia, Lippincott-Raven, 1997, pp 121–126
- 34.Teng DHF, Perry WL, III, Hogan JK, Baumgard M, Bell R, Berry S, Davis T, Frank D, Frye C, Hattier T, Hu R, Jammulapati S, Janecki T, Leavitt A, Mitchell JT, Pero R, Sexton D, Schroeder M, Su PH, Swedlund B, Kyriakis JM, Avruch J, Bartel P, Wong AKC, Oliphant A, Thomas A, Skolnick MH, Tavtigian SV: Human mitogen-activated protein kinase kinase 4 as a candidate tumor suppressor. Cancer Res 1997, 57:4177-4182 [PubMed] [Google Scholar]
- 35.Su GH, Hilgers W, Shekher MC, Tang DJ, Yeo CJ, Hruban RH, Kern SE: Alterations in pancreatic, biliary, and breast carcinomas support: MKK4 as a genetically targeted tumor suppressor gene. Cancer Res 1998, 58:2339-2342 [PubMed] [Google Scholar]
- 36.Horii A, Nakatsuru S, Miyoshi Y, Ichii S, Nagase H, Ando H, Yanagisawa A, Tsuchiya E, Kato Y, Nakamura Y: Frequent somatic mutations of the APC gene in human pancreatic cancer. Cancer Res 1992, 52:6696-6698 [PubMed] [Google Scholar]
- 37.McKie AB, Filipe MI, Lemoine NR: Abnormalities affecting the APC and MCC tumor suppressor gene loci on chromosome 5q occur frequently in gastric cancer but not in pancreatic cancer. Int J Cancer 1993, 55:598-603 [DOI] [PubMed] [Google Scholar]
- 38.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, Baylin SB, Kern SE, Herman JG: Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997, 57:3126-3130 [PubMed] [Google Scholar]
- 39.Moskaluk CA, Kern SE: Technical advance. Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997, 150:1547-1552 [PMC free article] [PubMed] [Google Scholar]
- 40.Williams C, Pontén F, Moberg C, Söderkvist P, Uhlén M, Pontén J, Sitbon G, Lundeberg J: Technical advance. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol 1999, 155:1467-1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ben-Ezra J, Johnson DA, Rossi J, Cook N, Wu A: Effect of fixation on the amplification of nucleic acids from paraffin-embedded material by the polymerase chain reaction. J Histochem Cytochem 1991, 39:351-354 [DOI] [PubMed] [Google Scholar]
- 42.Pääbo S, Irwin DM, Wilson AC: DNA damage promotes jumping between templates during enzymatic amplification. J Biol Chem 1990, 265:4718-4721 [PubMed] [Google Scholar]
- 43.Fujii H, Marsh C, Cairns P, Sidransky D, Gabrielson E: Genetic divergence in the clonal evolution of breast cancer. Cancer Res 1996, 56:1493-1497 [PubMed] [Google Scholar]
- 44.Fujii H, Szumel R, Marsh C, Zhou W, Gabrielson E: Genetic progression, histological grade, and allelic loss in ductal carcinoma in situ of the breast. Cancer Res 1996, 56:5260-5265 [PubMed] [Google Scholar]
- 45.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AMM, Bos JL: Genetic alterations during colorectal-tumor development. N Engl J Med 1988, 319:525-532 [DOI] [PubMed] [Google Scholar]
- 46.Shibata D, Schaeffer J, Li ZH, Capella G, Perucho M: Genetic heterogeneity of the c-K-ras locus in colorectal adenomas but not in adenocarcinomas. J Natl Cancer Inst 1993, 85:1058-1063 [DOI] [PubMed] [Google Scholar]
- 47.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M: Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53:549-554 [DOI] [PubMed] [Google Scholar]
- 48.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL: KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988, 16:7773-7782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mariyama M, Kishi K, Nakamura K, Obata H, Nishimura S: Frequency and types of point mutation at the 12th codon of the c-Ki-ras gene found in pancreatic cancers from Japanese patients. Jpn J Cancer Res 1989, 80:622-626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH: Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am J Surg Pathol 1998, 22:163-169 [DOI] [PubMed] [Google Scholar]