

Abstract
Angiopoietins are ligands for the endothelial cell tyrosine kinase receptor Tie-2. Ang-1, the major physiological activator of Tie-2, promotes blood vessel maturation and stability. Ang-2 counteracts this effect by competitively inhibiting the binding of Ang-1 to Tie-2. Using a combined RNase protection/semiquantitative reverse transcriptase-polymerase chain reaction approach, we demonstrate that hypoxia up-regulates Ang-2 mRNA levels by up to 3.3-fold in two human endothelial cell lines. In bovine microvascular endothelial (BME) cells, the flavoprotein oxidoreductase inhibitor diphenylene iodonium (DPI) and the related compound iodonium diphenyl mimic induction of Ang-2 but not vascular endothelial growth factor (VEGF) by hypoxia; in combination with hypoxia, DPI further increases Ang-2 expression but has no effect on the induction of VEGF by hypoxia. Neither Ang-2 or VEGF was increased by cyanide or rotenone, suggesting that failure in mitochondrial electron transport is not involved in the oxygen-sensing system that controls their expression. In ischemic rat dorsal skin flaps or in the brain of rats maintained for 12 hours under conditions of hypoxia, Ang-2 mRNA was up-regulated 7.5- or 17.6- fold, respectively. VEGF was concomitantly increased, whereas expression of Ang-1, Tie-2, and the related receptor Tie-1 was unaltered. In situ hybridization localized Ang-2 mRNA to endothelial cells in hypoxic skin. These findings 1) show that up-regulation of Ang-2 by hypoxia occurs widely in endothelial cells in vitro and in vivo; 2) suggest that induction of Ang-2, but not VEGF, by hypoxia in BME cells is controlled by a flavoprotein oxidoreductase that is sensitive to iodonium compounds; and 3) point to Ang-2 and VEGF as independently regulated and selective effectors of hypoxia-induced vascular sprouting.
Angiogenesis, the sprouting of new capillary blood vessels from preexisting microvasculature, is an absolute requirement for the development and maintenance of tissues and organs and is an important component in a number of pathological settings, including diabetic retinopathy, rheumatoid arthritis, and tumor growth. During angiogenesis, endothelial cells loose their intimate contacts with adjacent pericytes, degrade their underlying basement membrane, and migrate into the surrounding stroma as cell cords (sprouting phase). Later these cords form a lumen, deposit a new basement membrane, develop cell-cell junctions, and recruit perivascular cells (maturation phase), thus resulting in new functional vessels. 1,2
As might have been predicted, the search for angiogenic regulators has resulted in the discovery of a number of molecules, including polypeptide growth factors (vascular endothelial growth factors, angiopoietins, fibroblast growth factors, transforming growth factors-α and -β, platelet-derived growth factor-BB, tumor necrosis factor-α, hepatocyte growth factor), chemokines (platelet factor 4), enzymes (angiogenin, platelet-derived endothelial cell growth factor/thymidine phosphorylase), extracellular matrix components/coagulation factors or fragments thereof (thrombospondin, endostatin, 29 kd-fibronectin fragment, angiostatin), adhesion molecules (integrins αvβ3 and α5β1), prostaglandins, adipocyte lipids, and copper ions (rev. by 3–5). However, although all of these molecules modulate angiogenesis in the experimental setting, a role for a limited number has been clearly demonstrated in the endogenous regulation of this process.
Among these factors, the vascular endothelial growth factor (VEGF) and angiopoietin (Ang) families are the only two with target specificity for endothelial cells. VEGF, the prototype member of an expanding family of angiogenic polypeptides, binds to the endothelial cell receptors VEGFR-1/flt1 and VEGFR-2/KDR/Flk-1, the expression of which is restricted to endothelial cells and progenitors of the hematopoietic/endothelial cell lineage. 6 The relevance of the VEGF/VEGF receptor system to both developmental and pathological angiogenesis has been clearly demonstrated. 6 In cultured endothelial cells, VEGF stimulates proliferation, extracellular proteolytic enzyme production, as well as the formation of capillary-like structures in reconstituted collagen matrices. 7-12 Taken together, these results suggest that VEGF plays a key role in the sprouting phase of angiogenesis.
The discovery of the angiopoietin family has contributed significantly to our understanding of the process of blood vessel maturation. The first angiopoietin described, angiopoietin-1 (Ang-1), is an agonist of the endothelial cell tyrosine kinase receptor Tie-2, which is prominently expressed during development in the myocardium, and later throughout the embryo in the mesenchyme surrounding the developing vasculature. 13,14 Analysis of Ang-1 function in vivo through targeted inactivation of the Ang-1 locus in the mouse resulted in embryonic lethality at E12.5. 14 This was due to impaired development of myocardium, defective remodeling of the primitive vascular plexus into large and small vessels, as well as the complete lack of perivascular cells. 14 Similar vascular defects had previously been reported in mice lacking Tie-2. 15 Taken together, these findings demonstrated that, when compared to the VEGF/VEGF receptor system, the Ang-1/Tie-2 system plays a role in the later stages of angiogenesis.
Angiopoietin-2 (Ang-2) shares ∼60% amino acid identity with Ang-1 and binds to Tie-2 with similar affinity. However, Ang-2 does not induce Tie-2 autophosphorylation, but instead blocks Tie-2 autophosphorylation induced by Ang-1, thus behaving like an Ang-1 antagonist. 16 Thus overexpression of Ang-2 in the developing mouse embryo mimicked the phenotypes of Ang-1 or Tie-2 inactivation. 16 During embryogenesis and adult life, Ang-2 expression occurs almost exclusively at sites of vascular remodeling, where it begins in endothelial cells of the sprouting microvasculature and where VEGF is concomitantly expressed. 16-18 However, Ang-2 expression is also pronounced at sites of vascular regression, where VEGF mRNA is almost undetectable. 16,18 Taken together, these findings have led to the proposal that by virtue of its capacity to counteract blood vessel maturation/stability, the function of Ang-2 may be context-dependent. When acting in the absence of angiogenic inducers (such as VEGF), Ang-2 induces endothelial cell apoptosis with consequent vascular regression. When acting in concert with VEGF, Ang-2 may facilitate endothelial cell migration and proliferation, thus serving as a permissive angiogenic signal. 19,16
Hypoxia is a fundamental angiogenic stimulus that is believed to play a key role in a number of settings in which angiogenesis is an important component, including embryonic development and tumor growth. It is believed that the effects of hypoxia are initiated at the cellular level by an as yet uncharacterized heme protein oxygen sensor that in turn activates hypoxia inducible factor-1, a heterodimeric basic helix-loop-helix transcription factor composed of a constitutive β subunit (HIF-1β/ARNT) and a regulatory α subunit (HIF-1α) (reviewed in refs. 20-22 ). Functional HIF-1 binding sites have been described in the majority of genes regulated by hypoxia, including VEGF, erythropoietin (Epo), tyrosine hydroxylase, glucose transporter-1 (GLUT-1), most glycolytic enzymes, and inducible nitric oxide synthase (iNOS), 20,22 suggesting that HIF-1 is a central component of the organism’s response to hypoxia. However, the recent finding that both HIF subunits exist as gene families suggests that a precise tailoring of the hypoxic response might be achieved through differences in HIFα subunit activation and/or target gene specificity.
We 23 and others 24 have previously reported that hypoxia increases Ang-2 expression in bovine endothelial cells but not in smooth muscle cells, 23 which is consistent with the pattern of Ang-2 expression observed in vivo. This finding strongly suggested that, in addition to VEGF, Ang-2 might be a crucial effector of hypoxia-induced neovascularization. In the present study, we wished to assess whether hypoxia also induces Ang-2 expression in human endothelial cells and whether induction of Ang-2 by hypoxia occurs in vivo. Finally, we wished to compare the pharmacological features of hypoxia-inducible Ang-2 expression with those of VEGF.
Materials and Methods
Cell Culture
Simian virus 40 large T-antigen-transformed human dermal microvascular endothelial (HMEC-1) cells 25 (kindly provided by Drs. E. W. Ades and T. Lawley) were cultured in endothelial cell basal medium (EBM) 131 medium (Clonetics) supplemented with 10% fetal calf serum (FCS) (Life Technologies), 10 ng/ml of recombinant human epidermal growth factor (EGF) (Boehringer Mannheim), and 1 μg/ml of hydrocortisone. Human umbilical vein endothelial (HUVE) cells (kindly provided by Dr. N. Maggiano) and bovine microvascular endothelial (BME) cells from adrenal cortex 26 (kindly provided by Drs. M. B. Furie and S. C. Silverstein) were cultured as described. 12
Twenty-four to 72 hours after the last medium change, confluent monolayers of HUVE, HMEC-1, or BME cells were incubated for the times indicated in an airtight Plexiglas container in which atmospheric air was replaced with a 95% N2/5% CO2 gas mixture. In experiments aimed at characterizing Ang-2 and VEGF regulation in BME cells, confluent monolayers were incubated for 15 hours in the presence of the above-mentioned hypoxic conditions, or in the presence of the following treatments: 100 μmol/L cobalt chloride hexahydrate (catalog no. C-8661; Sigma); 130 μmol/L desferrioxamine mesylate (DFO) (catalog no. D-9533; Sigma); 1 mmol/L potassium cyanide (KCN) (catalog no. 60180; Fluka); 5 μg/ml of cycloheximide (CHX) (catalog no. C-4218; Sigma), alone or in combination with hypoxia (CHX was added to the cells 30 minutes before the hypoxic incubation was started); 5 μmol/L diphenylene iodonium chloride (DPI) (catalog no. D-2926; Sigma), alone or in combination with hypoxia; 50 μmol/L iodonium diphenyl (IDP) (catalog no. 43088; Fluka); or 100 nmol/L rotenone (catalog no. R-8875; Sigma). Stock solutions were prepared in H2O, except for DPI (EtOH 50% in H2O), IDP (EtOH 50% in H2O), and rotenone (dimethyl sulfoxide, DMSO), and diluted to the indicated concentrations in cell culture medium. Equivalent volumes of EtOH or DMSO were tested alone in parallel cultures for possible effects on Ang-2 mRNA levels. Where indicated, cells were washed twice with serum-free medium and incubated for 15 hours in serum-free medium under normoxic or hypoxic conditions.
Ischemic Dorsal Skin Flap Model
Female Wistar rats weighing 250–300 g were anesthetized with intraperitoneal sodium pentobarbital, 35 mg/kg. Caudally-based 9 × 3 cm dorsal skin/panniculus carnosus flaps 27,28 were raised with the two constant sacral axial vessels systematically cut. The caudal border of the flaps was marked 1 cm below the posterior iliac crests. The flaps were resutured back onto their original bed without further manipulation. Postoperatively the rats were fed ad libitum and housed in individual cages. Flaps were examined daily after surgery. Rats were sacrificed using an intracardiac pentobarbital overdose of 200 mg/kg. Immediately after sacrifice, sections II and III of the flaps (Figure 6) ▶ were divided into longitudinal strips; edges of the flap were excluded. Flap segments were snap frozen in liquid nitrogen-cooled isopentane. RNA was extracted from frozen tissues.
Figure 6.

Rat ischemic dorsal skin flap model. A caudally based 9 × 3 cm dorsal skin/panniculus carnosus flap was raised in rats, with the two constant sacral axial vessels systematically cut. Under these conditions, the skin flaps develop tissue hypoxia and necrosis in their distal part (III) in a predictable manner and with low SD. Ang-2 mRNA levels were studied in both the middle (II) and the distal (III) part over a time period of 7 days after intervention.
Systemic Hypoxia in Rats
Three- to six-month-old male Sprague-Dawley rats were kept for 12 hours in an airtight Plexiglas container in which atmospheric air was replaced with a continuous flow of a 6% oxygen/94% nitrogen gas mixture. Under these conditions, animals appeared to feed normally. Age-matched animals were used as controls. At the end of the incubation, rats were killed by cervical dislocation, and the whole brain was removed, immediately frozen in liquid nitrogen, and stored at −80°C until RNA extraction.
RNA Purification
Total cellular RNA was purified from cultured cells or rat tissues using Trizol reagent (Life Technologies).
RNase Protection Assay
Ten micrograms of total RNA from cultured endothelial cells or rat skin was analyzed. In the case of hypoxic brain, 30 μg of total RNA was used. Rat and bovine Ang-2 cRNA probes were synthesized from the partial cDNA fragments previously described, 23 with the modification that the rat Ang-2 fragment was digested at the internal PvuII site, thus resulting in a shorter cRNA probe (∼320 bases). The human Ang-2 probe was generated by reverse transcriptase-polymerase chain reaction (RT-PCR) from total RNA from HUVE cells exposed to the above-mentioned hypoxic conditions for 15 hours. Briefly, total cellular RNA was reverse transcribed using oligo-dT15 (Boehringer Mannheim) and Superscript II reverse transcriptase (RT) (Life Technologies), according to the manufacturer’s instructions. The oligonucleotide primer sequences used were as follows: forward: 5′- GCGTTGATTTTCAGAGGACTTGG; reverse: 5′-GCGAATAGCCTGAGCCTTTCCA. PCR cycles were as follows: 94°C, 2 minutes (1×); 94°C, 1 minute, 60°C, 1 minute, 72°C, 1 minute (30×); 72°C, 3 minutes. The unique ∼450-bp product was cloned into pGEM-TE (Promega) and sequenced in its entirety. The 393-bp rat VEGF164 cDNA probe, spanning the common and alternatively spliced regions of rat VEGF mRNA, was kindly provided by Dr. B. Berse. Equal amounts of acidic ribosomal phosphoprotein P0 cRNA probe (obtained by RT-PCR from the corresponding species as previously described) 29 were included in all samples as an internal control. RNase protection was performed as described. 30
Semiquantitative RT-PCR
Two micrograms (5 μg in the case of experiments using rat brains) of total RNA was reverse transcribed using oligo-dT15 (Boehringer Mannheim) and Superscript II RT (Life Technologies). Where indicated, RT was omitted. For each RT product, 1/20 of the final reaction volume, or an equivalent volume of H2O, was amplified in parallel PCR reactions, using, as indicated in each case, the above-mentioned primers for human Ang-2, a pair of specific primers for human VEGF (forward: 5′-GGAGCCTCGCCTTGCTGCTCTACC; reverse: 5′-CCGAAACCCTGAGGGAGGCTCC; located outside of the alternatively spliced region), 31 a pair of specific primers for bovine VEGF (forward: 5′-CCGGAATTCCAGGAGTACCCAGATGAG; reverse: 5′-CGCGGATCCGGCTCACCGCCTCGGCTTGTC, containing an artificial EcoRI or BamHI site at the 5′ end, respectively, and located outside of the alternatively spliced region), 31 a pair of partially degenerate primers for Ang-1 or Ang-2 previously described, 23 a pair of partially degenerate primers for Tie-1 (forward: 5′-GATGTAGACAGGCC(A/G/T/C)GA(A/G)GA(A/G); reverse: 5′-CTCAAAGGT(A/G/T)AT(A/G)TC(C/T)TCCCA), a pair of partially degenerate primers for Tie-2 (forward: 5′-GGCAA(A/G)AATGAAGA(C/T)CA(A/G)CA(C/T); reverse: 5′-TCTTGAAA(C/T)TTGAT(A/G)TC(A/G)TTCCA), a pair of specific primers for mouse and rat VEGF, 32 or, finally, a pair of partially degenerate primers for the acidic ribosomal phosphoprotein P0 29 that efficiently amplify the corresponding cDNA of bovine, human, and rat origin. For all of the reactions, preliminary experiments were performed to determine the number of PCR cycles at which saturation occurred, and the experiments mentioned were carried out with a number of cycles that precedes saturation. The specificity of the PCR reactions using degenerate primers for Ang-1, Ang-2, and P0 has previously been confirmed by sequencing. 23,29 For reactions with degenerate Tie-1 and Tie-2 primers, human Ang-2 primers, and bovine VEGF primers (only for the isoform corresponding in size to the 164 aa isoform), identity of the RT-PCR product was confirmed by cloning into pGEM-TE (Promega) or pBluescriptKS (Stratagene), entire sequencing, and RNase protection analysis using the appropriate total cellular RNA from the corresponding species. Sequences of rat Tie-1 and Tie-2 partial cDNAs were unreported and have been deposited in GenBank with accession numbers AF030377 and AF030423, respectively.
PCR cycles were as follows: Human Ang-2 primers (HUVE and HMEC-1 cells): 94°C, 2 minutes (1×); 94°C, 1 minute, 60°C, 1 minute, 72°C, 45 sec (25×); 72°C, 3 minutes. Human VEGF primers (HUVE and HMEC-1 cells): 94°C, 2 minutes (1×); 94°C, 1 minute, 65°C, 1 minute, 72°C, 1 minute (23×); 72°C, 3 minutes. Bovine VEGF primers (BME cells): 94°C, 2 minutes (1×); 94°C, 1 minute, 62°C, 1 minute, 72°C, 1 minute (23×); 72°C, 3 minutes. Degenerate Ang-1, Ang-2, Tie-1, and Tie-2 primers (rat brain): 94°C, 2 minutes (1×); 94°C, 30 seconds, 55°C, 1 minute, 72°C, 45 seconds (25×; 28× in the case of Ang-2); 72°C, 3 minutes. Mouse/rat VEGF primers (rat brain): 94°C, 2 minutes (1×); 94°C, 30 seconds, 60°C, 1 minute, 72°C, 45 seconds (23×); 72°C, 3 minutes. Degenerate P0 primers (HUVE, HMEC-1, and BME cells; rat brain): 94°C, 2 minutes (1×); 94°C, 30 seconds, 60°C, 1 minute, 72°C, 45 seconds (16×; 20× in the case of rat brain); 72°C, 3 minutes. Five microcuries of 32P-labeled dCTP were added to each sample to visualize PCR products by autoradiography. Equal volumes of PCR products were electrophoresed in 6% acrylamide gels.
In Situ Hybridization
A 35S-UTP-labeled antisense rat Ang-2 cRNA probe of ∼320 bases was generated by transcription of a PvuII-digested rat Ang-2 cDNA fragment, 23 whereas 35S-UTP-labeled antisense rat VEGF cRNA probe was synthesized from the above-mentioned rat VEGF cDNA fragment. Both probes were used for in situ hybridization on paraffin sections of 4% paraformaldehyde perfusion-fixed rat skinflap fragments, using a previously published procedure. 33
Results
We and others have previously reported that hypoxia increases Ang-2 mRNA 23,24 and protein 24 in bovine endothelial cells. This phenomenon was apparently endothelial cell-specific, in that it was not observed in smooth muscle cells (SMCs). 23 To investigate whether expression of Ang-2 is inducible by hypoxia in other endothelial cell lines, confluent cultures of HUVE or transformed HMEC-1 cells were incubated for 15 hours under hypoxic conditions. Total RNA was reverse transcribed with oligo-dT, and Ang-2 mRNA levels were studied by semiquantitative RT-PCR, using the same partially degenerated oligonucleotide primers that we previously designed to clone bovine and rat Ang-2 partial cDNAs. 23 Alternatively, specific primers corresponding to the same regions of human Ang-2 were used. Parallel reactions on the same RT products assessed 1) the levels of VEGF mRNAs as an internal control for the hypoxic response and 2) the levels of the acidic ribosomal phosphoprotein P0 mRNA as an internal control of the amount of total cDNA used in each amplification reaction.
After 25 PCR cycles using specific primers for human Ang-2, a unique band of ∼450 bp, thus corresponding in size to the RT-PCR product expected from human Ang-2 cDNA (456 bp), was detectable in normoxic HUVE cells, and to a much lesser extent in normoxic HMEC-1 cells (Figure 1) ▶ . When normalized with respect to the P0 signal, the intensity of the putative Ang-2 band was increased by 3.3- or 2.5- fold by hypoxia in HUVE or HMEC-1 cells, respectively. Identical results were obtained using the partially degenerate primers for Ang-2 mentioned above (data not shown). Massive induction of VEGF expression occurred in both cell lines in response to hypoxia (Figure 1) ▶ , which is consistent with previous results. 34 The Ang-2, VEGF, and P0 bands were undetectable when RT was omitted (Figure 1) ▶ , demonstrating that they were not amplified from contaminating genomic DNA.
Figure 1.

Hypoxia increases Ang-2 mRNA levels in HUVE and HMEC-1 cells. Two micrograms of total RNA from confluent monolayers of HUVE or HMEC-1 cells incubated for 15 hours under normoxic conditions (Control) or in the presence of a gas mixture containing 95% N2/5% CO2 (Hypoxia) was analyzed for Ang-2 and VEGF expression (or acidic ribosomal phosphoprotein P0 expression as an internal control) by semiquantitative RT-PCR. Where indicated, RT was omitted. When normalized with respect to the P0 signal, hypoxia increased both Ang-2 and VEGF expression in both cell types.
To confirm that the ∼450-bp RT-PCR product from HUVE and HMEC-1 cells was in fact Ang-2, the RT-PCR product amplified from hypoxic HUVE cells was cloned and sequenced in its entirety. Nucleotide sequence analysis revealed 100% identity with the human Ang-2 cDNA. 16 This fragment was therefore used in RNase protection assays to more precisely characterize the induction of Ang-2 expression by hypoxia in HUVECs. Confluent monolayers of HUVE cells were incubated under hypoxic conditions for time periods of 3, 8, or 15 hours. Total RNAs were analyzed by RNase protection, using a 32P-labeled cRNA probe transcribed from the above-mentioned human Ang-2 cDNA fragment. As an internal control, an equal amount of 32P-labeled cRNA from a human P0 partial cDNA was included in all samples. When normalized with respect to the P0 signal, Ang-2 mRNA was up-regulated by hypoxia in HUVE cells in a time-dependent manner, by 2.6-fold after 8 hours, and further increasing by up to 3.3-fold after 15 hours (Figure 2) ▶ , which confirms and extends the results shown in Figure 1 ▶ . Similar results were obtained in independent Northern blot experiments using the same probe (data not shown). Ang-2 mRNA levels at a later time point (48 hours) could not be assessed, because cells died after ∼36 hours of exposure to hypoxia.
Figure 2.

Kinetics of induction of Ang-2 by hypoxia in HUVE cells. Purified 32P-labeled human Ang-2 cRNA probe (Ang-2 pr.) or human P0 cRNA probe (P0 pr.) was hybridized to hybridization mix (Ang-2 pr. + h.m., P0 pr. + h.m.), yeast tRNA (tRNA), or 10 μg of total RNA from confluent monolayers of HUVE cells incubated for the indicated time periods under conditions of normoxia or hypoxia (a gas mixture of 95% N2/5% CO2). The graph shows values for Ang-2 mRNA levels obtained by scanning densitometry normalized with respect to the P0 signal.
Taken together with previous results for bovine endothelial cells, 23,24 these findings showed that up-regulation of Ang-2 expression by hypoxia is a general property of vascular endothelial cells. These findings prompted us to explore the mechanisms underlying this phenomenon in greater detail.
Hypoxia-inducible expression of typical hypoxia-responsive genes such as VEGF and erythropoietin (Epo) has well-known pharmacological features. For instance, it is mimicked in normoxic cells by transition metals such as cobalt, nickel, and manganese, and by the iron chelator DFO, but not by poisons of mitochondrial electron transport, such as cyanide or azide (reviewed in ref. 20 ). DPI, a potent inhibitor of flavoproteins, blocks hypoxia-induced VEGF and Epo expression in HepG2 and Hep3B cells, whereas it has no effect on normoxic levels of either gene. 35 A similar effect has been reported for the DPI-related molecule IDP. 35 Because cultured endothelial cells express both VEGF and Ang-2, we wished to compare the pharmacological features of hypoxia-inducible Ang-2 and VEGF expression in the same cell type.
To this end, confluent monolayers of BME cells cultured in serum-containing medium were incubated for 15 hours in the presence of hypoxia, 100 μmol/L cobalt chloride, 130 μmol/L DFO, 1 mmol/L potassium cyanide (KCN), 5 μg/ml of CHX alone or in combination with hypoxia, 5 μmol/L DPI alone or in combination with hypoxia, or were exposed to hypoxia in the absence of serum. Ang-2 mRNA levels were studied by RNase protection, using the bovine Ang-2 cDNA fragment previously described, 23 whereas VEGF mRNA levels were studied in the same samples by means of semiquantitative RT-PCR, because basal levels of VEGF mRNA were barely detectable by RNase protection in BME cells (data not shown). PCR primers for bovine VEGF were chosen outside of the alternatively spliced region, thus allowing us to identify VEGF164 and VEGF120 as the two VEGF mRNA isoforms expressed in equal proportion by BME cells (Figure 4) ▶ ; an additional band corresponding in size to VEGF188 was detectable in bovine adult lung (Figure 4) ▶ . The specificity of the PCR reaction was confirmed by cloning and sequencing of the VEGF164 isoform (data not shown). In both RNase protection and semiquantitative RT-PCR analysis, modulations of Ang-2 or VEGF expression were normalized with respect to the P0 signal. When compared to controls, hypoxia induced both Ang-2 and VEGF in BME cells (Figures 3 and 4) ▶ ▶ . However, whereas the amplitude of VEGF induction was dramatic (20.6- or 14.7-fold for VEGF164 or VEGF120, respectively), that of Ang-2 was relatively modest (2.9-fold), which is consistent with previous results. 23,24 Cobalt chloride and DFO induced a weak and similar increase in VEGF164 and VEGF120 (1.9- and 1.8-fold, respectively, in the case of cobalt chloride, and 3.0- or 2.6-fold, respectively, in the case of DFO) (Figure 4) ▶ , but did not alter Ang-2 mRNA levels (Figure 3) ▶ , perhaps because of the low amplitude of Ang-2 inducibility by hypoxia. KCN had no effect on VEGF or Ang-2 expression (Figures 3 and 4) ▶ ▶ , suggesting that hypoxia-inducible Ang-2 expression, similar to that of VEGF, is not a consequence of failure in mitochondrial electron transport.
Figure 4.

Pharmacological characterization of the regulation of VEGF mRNA expression in BME cells as assessed by semiquantitative RT-PCR. Two micrograms of total RNA from the same samples shown in Figure 3 ▶ or from adult bovine lung (Lung) was analyzed for VEGF mRNA levels (or P0 mRNA levels as an internal control) by semiquantitative RT-PCR. Where indicated, RT was omitted. The graph shows values obtained by scanning densitometry (normalized with respect to the P0 signal) ± SD from two independent experiments. An arbitrary value of 1 was assigned to controls.
Figure 3.

Pharmacological characterization of the regulation of Ang-2 mRNA expression in BME cells as assessed by RNase protection. Purified 32P-labeled bovine Ang-2 cRNA probe (Ang-2 pr.) or bovine P0 cRNA probe (P0 pr.) was hybridized to hybridization mix (Ang-2 pr. + h.m., P0 pr. + h.m.), yeast tRNA (tRNA), or 10 μg of total RNA from confluent monolayers of BME cells incubated for 15 hours under conditions of normoxia (Control), in the presence of a gas mixture containing 95% N2/5% CO2 (Hypoxia), 100 μM cobalt chloride (CoCl2), 130 μmol/L desferrioxamine (DFO), 1 mmol/L KCN (KCN), 5 μg/ml of cycloheximide (CHX), alone or in combination with hypoxia (CHX + Hyp.), or 5 μmol/L diphenylene iodonium (DPI), alone or in combination with hypoxia (DPI + Hyp.). Where indicated, cells were incubated in serum-free medium. The graph shows values obtained by scanning densitometry (normalized with respect to the P0 signal) ± SD from two independent experiments. An arbitrary value of 1 was assigned to controls.
Experiments using CHX, DPI, and serum-free medium revealed the most interesting differences between Ang-2 and VEGF. CHX very strongly increased VEGF164 and VEGF120 expression when tested alone (53.3- or 11.0-fold, respectively) or in combination with hypoxia (212.3- or 88.8- fold, respectively) (Figure 4) ▶ . Because CHX inhibits activity of hypoxia-inducible factor (HIF)−1 (reviewed in ref. 20 ), the effect of CHX on VEGF expression most probably reflects increased stability of VEGF mRNA, which, interestingly, is more marked in the case of the 164-aa isoform (Figure 4) ▶ . In contrast, CHX reduced the basal level of Ang-2 mRNA by 50% and did not alter the extent of Ang-2 inducibility by hypoxia (3.4-fold) (Figure 3) ▶ , showing that ongoing protein synthesis is required for the maintenance of basal Ang-2 mRNA levels but not for hypoxia-inducible expression of Ang-2.
DPI is an inhibitor of flavoproteins that does not alter Epo or VEGF mRNA levels under normoxic conditions and that blocks hypoxia-inducible VEGF and Epo expression in HepG2 and Hep3B cells. 35 When assessed in BME cells, DPI increased Ang-2 mRNA levels in normoxic cells to an extent similar to that of hypoxia (3.3-fold) (Figure 3) ▶ and further slightly increased Ang-2 mRNA levels when tested in combination with hypoxia (3.8-fold) (Figure 3) ▶ . Intriguingly, DPI, which did not alter the basal levels of VEGF mRNA in BME cells (Figure 4) ▶ , did not affect hypoxia-inducible VEGF expression (Figure 4) ▶ , suggesting that the response to DPI might vary, depending on the cell type considered.
Because most hypoxia-inducible genes need to integrate diverse regulatory signals to efficiently responding to hypoxia, we wished to assess whether serum had any effect on hypoxia-inducible Ang-2 expression. When cultured in the absence of serum for 15 hours, normoxic BME cells showed a basal level of Ang-2 mRNA that was ∼2.0-fold higher than that observed in the same cells cultured in the presence of serum (Figure 3) ▶ . Under hypoxic conditions, the level of Ang-2 mRNA was equivalent in the presence or absence of serum (Figure 3) ▶ . Similar results were obtained by semiquantitative RT-PCR in independent experiments (data not shown). Thus, in the absence of serum, induction of Ang-2 by hypoxia was reduced by 40%, and this was apparently due to the fact that the absence of serum per se increases the levels of Ang-2 mRNA, rather than to the fact that hypoxic-inducible expression of Ang-2 is serum dependent. In contrast, the absence of serum decreased the expression of VEGF164 and VEGF120 by 30% and 50%, respectively (Figure 4 ▶ and data not shown). Serum-free medium did not affect hypoxic inducibility of either VEGF isoform (Figure 4) ▶ .
Finally, prompted by the positive effect of DPI on Ang-2 mRNA levels, we wished to assess the role of two additional flavoprotein inhibitors in the regulation of Ang-2 expression in normoxic BME cells. When analyzed by RNase protection and normalized with respect to the internal control P0, IDP also stimulated BME cell Ang-2 mRNA levels by 4.8- or 3.1- fold, respectively, in two independent experiments (Figure 5) ▶ . In the same experiments, DPI induced Ang-2 mRNA levels by 4.5- or 3.9-fold, respectively (Figure 5) ▶ . In contrast, rotenone, a specific inhibitor of complex I in the mitochondrial respiratory chain, had no effect (Figure 5) ▶ .
Figure 5.

Effect of iodonium diphenyl and rotenone on the regulation of Ang-2 mRNA levels in BME cells as assessed by RNase protection. Purified 32P-labeled bovine Ang-2 cRNA probe (Ang-2 pr.) or bovine P0 cRNA probe was hybridized to hybridization mix (Ang-2 pr. + h.m., P0 pr. + h.m.), yeast tRNA (tRNA), 10 μg of total RNA from BME cells incubated for 15 hours in the presence of 95% N2/5% CO2 (BME/Hypoxia), culture medium alone (Control), DPI (5 μmol/L), iodonium diphenyl (IDP) (50 μmol/L), rotenone (100 nmol/L), or an equivalent volume of EtOH 50% (EtOH) or DMSO. Samples from cells treated with DPI, IDP, rotenone, EtOH, and DMSO are from two experiments.
Taken together, these findings show both similarities and differences between Ang-2 and VEGF regulation. First, experiments using KCN and rotenone suggested that, similar to VEGF and Epo, 20 hypoxic induction of Ang-2 is not mediated by sensing of failure in electron mitochondrial transport. However, a DPI- and IDP-sensitive flavoprotein oxidoreductase seems to be involved in the mechanism of oxygen sensing that controls Ang-2 but not VEGF expression in BME cells. Second, hypoxia-inducible expression of either VEGF or Ang-2 in BME cells does not require ongoing protein synthesis. However, opposing effects were observed with CHX alone, in that it decreased basal levels of Ang-2 expression and strongly increased basal levels of VEGF expression. Third, hypoxia-inducible expression of both Ang-2 and VEGF in BME cells occurred independently of the presence of serum. When considered alone, serum had opposite effects on the regulation of VEGF or Ang-2 in BME cells, in that it was required to maintain the expression of VEGF, and, intriguingly, repressed the expression of Ang-2.
We next wished to assess Ang-2 expression in response to tissue hypoxia in vivo. The first model analyzed was a caudally based rat dorsal skin/panniculus carnosus flap (Figure 6) ▶ that predictably develops acute tissue hypoxia and massive necrosis in its distal (cranially localized) portion. 27,28 Ang-2 mRNA levels were studied by RNase protection over a time period of 7 days after intervention, using a rat Ang-2 partial cDNA 23 as a probe and a rat P0 cDNA fragment as an internal control. Interestingly, a low but detectable level of Ang-2 mRNA could be observed in the skin of control rats (Figure 7) ▶ . When assessed in the distal part of the skin flap, where tissue hypoxia is pronounced and tissue necrosis reaches a maximum after 3 days ( refs. 27, 28, and data not shown), Ang-2 mRNA levels were increased by 3.7- fold after 12 hours, further increased by up to 7.5- fold after 2 days, and gradually returned to control levels after 1 week (Figures 7 and 8) ▶ ▶ . In the middle part of the flap, where tissue hypoxia is insufficient to promote necrosis, Ang-2 mRNA was induced to a lesser extent (Figure 8 ▶ and data not shown). In situ hybridization of the middle and distal part of the skin flaps at day 2 showed selective expression of Ang-2 mRNA in endothelial cells of small and medium-sized blood vessels (Figure 9, A and A ▶ ′, and data not shown). Analysis of VEGF expression revealed an expression profile that was similar in terms of kinetics, but heterogeneous in nature, in that it occurred in endothelial cells, stromal cells (most likely macrophages), basal keratinocytes, hair follicles, and brown adipocytes (Figure 9, B and B ▶ ′, and our unpublished results). This was most likely due to the strong and persistent tissue ischemia that occurs in the distal part of the skin flap. Both Ang2 and VEGF were undetectable by in situ hybridization in the skin of control animals, or when the corresponding sense probes were used (Figure 9, C and C ▶ ′, and data not shown). Thus, in the setting of tissue ischemia, in contrast to VEGF, the expression of which is widespread, Ang-2 is specifically induced in vascular endothelial cells. In accord with previous reports, 16-18,24 these results suggest that, in ischemic tissues, Ang-2 might contribute to hypoxia-induced neovascularization and/or to tissue necrosis through induction of vascular regression. However, in the specific case of the model presented here, it should be pointed out that although neovascularization accompanied the formation of granulation tissue at the edges of the flap (data not shown), over the time period of 7–10 days neovascularization in the skin flap in itself was limited. This is most likely due to the extensive necrosis observed in this model.
Figure 7.

RNase protection analysis of Ang-2 mRNA levels in rat ischemic dorsal skin flaps. Purified 32P-labeled rat Ang-2 cRNA probe (Ang-2 pr.) or rat P0 cRNA probe was hybridized to hybridization mix (Ang-2 pr. + h.m., P0 pr. + h.m.), yeast tRNA (tRNA), or 10 μg of total RNA from 12.5-day rat embryo + placenta (E/P12.5), rat dorsal skin (Control), or the distal part of dorsal skin flaps (see Figure 6 ▶ ) from two different animals per time point. Ang-2 mRNA levels were quantitated as detailed in the legend to Figure 8 ▶ .
Figure 8.
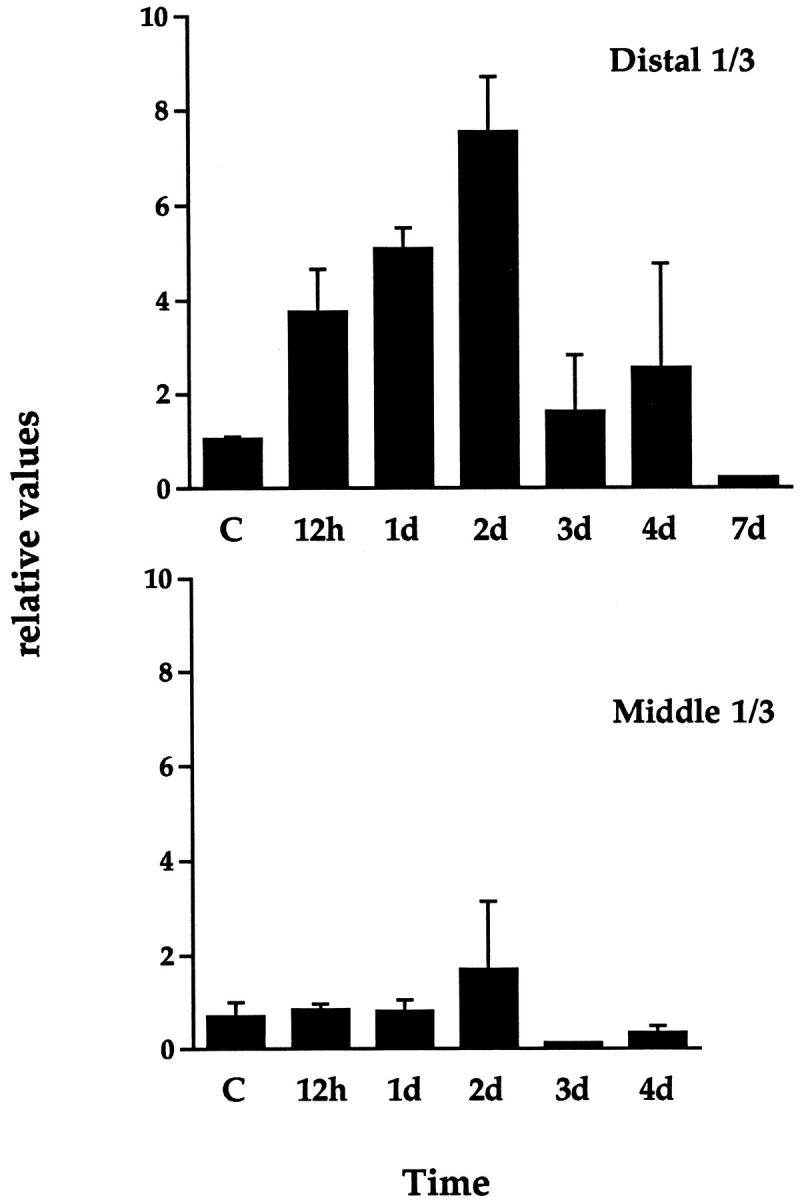
Quantitation of Ang-2 mRNA levels in rat ischemic dorsal skin flaps as assessed by RNase protection analysis. Samples were harvested from the distal or middle third (regions III + II in Figure 6 ▶ ) at various times after surgical intervention. For each time point, the graph shows values for Ang-2 mRNA levels obtained by scanning densitometry (normalized with respect to the P0 signal) ± SEM from two different animals. Ang-2 mRNA levels were increased in the distal third (region III) in a time-dependent manner, reaching maximum after 2 days and returning to baseline levels after 1 week. A weak increase in Ang-2 mRNA levels was also observed in the middle third (region II).
Figure 9.
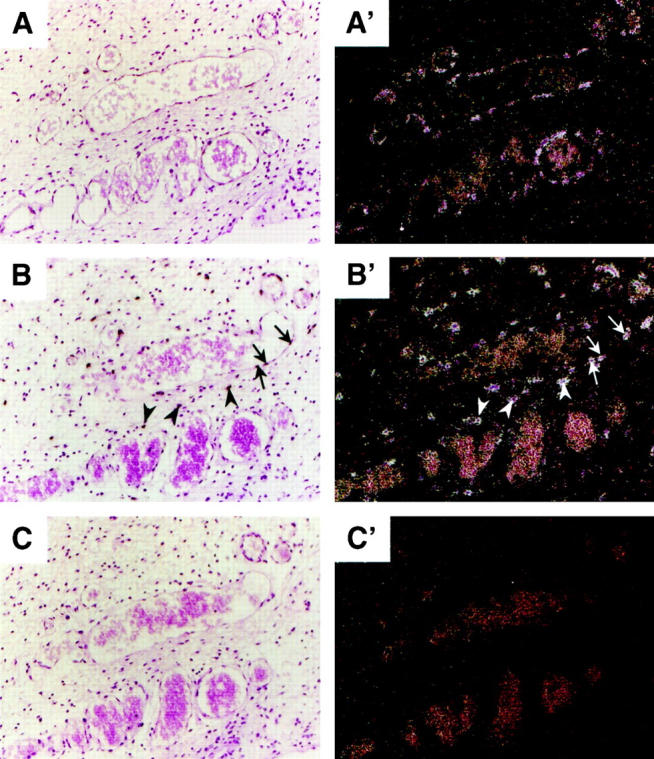
In situ hybridization for Ang-2 and VEGF in rat ischemic skin flaps. Animals were perfusion-fixed with 4% paraformaldehyde, and skin flap tissue was paraffin-embedded. Consecutive sections of the distal third (region III in Figure 6 ▶ ), harvested 2 days after surgical intervention, were analyzed by in situ hybridization using 35S-UTP labeled antisense (A, A′) or sense (C, C′) rat Ang-2 or antisense VEGF (B, B′) cRNA probes. A′, B′, and C′ are dark-field views of A, B, and C, respectively. Ang-2 mRNA was confined to vascular endothelial cells of small and medium vessels (A, A′), whereas VEGF mRNA was expressed by both stromal cells (arrowheads) and endothelial cells (arrows).
Ang-2 mRNA levels were next assessed in the brains of rats exposed to systemic hypoxia, a model in which angiogenesis is known to occur as part of the adaptive response to hypoxia. 36 As a first approach, Ang-2 mRNA levels were studied by semiquantitative RT-PCR, using the partially degenerate oligonucleotide primers mentioned above and previously described 23 in conjunction with a parallel reaction for P0 as an internal control. After 12 hours of hypoxia (6% oxygen), in two independent experiments, Ang-2 mRNA levels were increased in the brain by 7.4- or 7.9- fold, respectively (Figure 10) ▶ . A strong increase in VEGF expression was also observed (4.7- or 2.9-fold for VEGF164 and 15.5- or 5.6-fold for VEGF120) (Figure 10) ▶ , which is consistent with previous results. 37-39 A semiquantitative RT-PCR screening of various organs of mice subjected to the same hypoxic conditions revealed a similar increase in Ang-2 and VEGF in the brain and to some extent of Ang-2 in the liver, but no significant up-regulation of either gene in the lung, spleen, or kidney, despite strong and selective induction of Epo mRNA in the latter organ (data not shown). In contrast to Ang-2 and VEGF, the levels of Ang-1, Tie-1, and Tie-2 mRNAs were unaffected or slightly decreased in the brain of hypoxic rats (Figure 10) ▶ , which is consistent with our previous findings in cultured cells. 23 The specificity of the PCR reactions for Ang-1, Ang-2, Tie-1, and Tie-2 was confirmed by cloning and sequencing of the respective RT-PCR products ( ref. 23 ; see also Materials and Methods).
Figure 10.

Semiquantitative RT-PCR analysis of Ang-1, Ang-2, Tie-1, Tie-2, and VEGF mRNA levels in the brains of rats maintained for 12 hours under hypoxic conditions. Five micrograms of total RNA from 12.5-day rat embryo + placenta (E/P12.5) or from the brains of rats maintained for 12 hours under normoxic (Control) or hypoxic (Hypoxia) conditions (6% oxygen) in two independent experiments was analyzed for Ang-1, Ang-2, Tie-1, Tie-2, or VEGF mRNA expression (using P0 mRNA as an internal control) by semiquantitative RT-PCR. Where indicated, RT was omitted.
Up-regulation of Ang-2 and VEGF mRNAs in the hypoxic rat brain was confirmed by RNase protection analysis. When analyzed by this technique and normalized with respect to the P0 signal, the same RNA samples of Figure 10 ▶ revealed an increase in Ang-2 mRNA of 17.6- or 17.0-fold, respectively, and an increase in VEGF164 of 3.0- or 5.3- fold, respectively (Figure 11) ▶ .
Figure 11.

RNase protection analysis of Ang-2 and VEGF164 mRNA levels in the brains of hypoxic rats. Purified 32P-labeled rat Ang-2 or VEGF164 cRNA probes (pr.) or rat P0 cRNA probe (P0 pr.) was hybridized to hybridization mix (pr. + h.m., P0 pr. + h.m.), yeast tRNA (tRNA), 30 μg of total RNA from 12.5-day rat embryo + placenta (E/P12.5), or 30-μg aliquots of the brain RNA samples shown in Figure 10 ▶ .
Thus selective up-regulation of VEGF and Ang-2 occurs in the brain of rats and mice exposed to systemic hypoxia at a time point that precedes the formation of new capillaries, suggesting a crucial role for these two molecules in the promotion of vascular sprouting in this model.
Discussion
Genetic studies have shown that Ang-1, the major physiological agonist of the endothelial cell tyrosine kinase receptor Tie-2, serves important functions during embryonic angiogenesis. Although not required for vasculogenesis, Ang-1 is essential for remodeling of the primary vascular plexus, branching of new blood vessels from the latter, and maturation of the vessel wall through the recruitment of perivascular cells. 14 The finding that Ang-1 is constitutively expressed in most adult organs, 16 where Tie-2 is also constitutively phosphorylated, 40 has led to the hypothesis that, besides being essential for blood vessel maturation during embryonic development, Ang-1 could also be a key molecule for the maintenance of blood vessel stability in the adult. 19 It has been proposed that Ang-2, the Ang-1 antagonist that binds to Tie-2 and blocks Ang-1-induced Tie-2 phosphorylation, could be an important pro-angiogenic factor, in that by interrupting the stabilizing Ang-1 signal it might promote a process of vessel wall disassembly that would allow endothelial cells to respond to angiogenic inducers. 19,16,23 Consistent with this view, an increasing number of reports have shown that expression of Ang-2 occurs almost exclusively at physiological and pathological sites of vascular remodeling, where it appears to begin at the tip of the developing vessel sprouts. 16-18,24,41 In addition, we have previously reported that expression of Ang-2 is increased in bovine microvascular endothelial cells by a number of angiogenic inducers, including VEGF and bFGF, 23 thus providing a possible mechanism for the induction of Ang-2 in vivo and suggesting that induction of an autocrine Ang-2 loop resulting in Ang-1 inactivation may represent part of the mechanism by which angiogenic inducers act.
Perhaps most importantly, we 23 and others 24 also found that endothelial cell expression of Ang-2 is increased by hypoxia. Hypoxia is a fundamental angiogenic stimulus that plays a key role during a variety of physiological and pathological settings, including embryonic development and tumor growth. Although intensely studied, the mechanisms by which hypoxia induces angiogenesis are only partially known. Hypoxia induces up-regulation of VEGF expression in a variety of in vivo and in vitro systems, a phenomenon that is initiated at the level of a yet uncharacterized cellular oxygen sensor and effected intracellularly through the activation of the heterodimeric basic helix-loop-helix PAS transcription factor HIF-1 (reviewed in refs. 6, 21, and 22). However, although VEGF induction is certainly a relevant mechanism in hypoxia’s angiogenic effect, it is certainly not the only one, because a large body of experimental evidence clearly shows that VEGF is necessary but not sufficient for angiogenesis to occur, and that the angiogenic effects of VEGF or other inducers are largely determined by a local, complex, and probably tissue-specific balance between different positive and negative angiogenic regulators. 3 A search for other hypoxia-inducible angiogenic molecules has revealed a role for PDGF-B 42 and VEGF receptor-1/flt. 43 However, neither of these molecules appears to play a role in the promotion of vascular sprouting, because PDGF-B is involved in blood vessel maturation and VEGFR-1 seems to act as a dominant negative regulator of VEGF’s effects on the vascular endothelium and progenitors of the hematopoietic/endothelial lineage. 44-47 Regulation of other angiogenic molecules by hypoxia has led to conflicting and unclear results: although Tie-1 has been reported to be induced by hypoxia in one line of cultured endothelial cells, 48 we found that hypoxia does not affect Tie-1 expression in BME cells, 23 and is even slightly decreased by hypoxia in the rat brain model (this study). An equivalent argument concerning Tie-2 can be raised, because, although the activity of the transfected Tie-2 promoter has been shown to be inducible by the HIF-1α-related transcription factor HIF-2α, 49 we have not observed up-regulation of Tie-2 by hypoxia, neither in BME cells 23 nor in the rat brain model (this study). Thus the observation that Ang-2 is consistently increased by hypoxia in a number of different endothelial cell lines ( refs. 23 and 24 and this study), as well as in vivo ( ref. 24 and this study), strongly points to the VEGF/Ang-2 pair as a selective and key effector of sprouting angiogenesis in response to hypoxia.
Despite these similarities, it is important to point out that VEGF and Ang-2 appear to be regulated by hypoxia in different ways. First, in vitro experiments have shown that up-regulation of Ang-2 by hypoxia is specific for endothelial cells, as it did not occur in SMC cells in which a massive induction of VEGF expression was observed. 23 These results are consistent with the finding that in hypoxic tumors 17 and the ischemic retina 24 Ang-2 is specifically expressed by endothelial cells of the sprouting microvasculature. The reasons for this specificity might be due to the fact that autocrine expression of Ang-2 by microvascular endothelial cells, by tight restriction of the inactivation of the Ang-1 signal to the sprouting microvasculature, allows nonangiogenic vessels to remain unresponsive to angiogenic stimuli. Second, the pharmacological characterization of Ang-2 and VEGF expression presented in this study, which was performed in the same cell type, gives some preliminary and interesting insights into the mechanisms of their regulation by hypoxia. Similar to VEGF and most other typical hypoxia-responsive genes, 20 Ang-2 mRNA expression was not induced by KCN or rotenone, suggesting that this is not due to sensing of failure in mitochondrial electron transport. Experiments using CHX showed that in BME cells, neither VEGF induction nor Ang-2 induction by hypoxia was dependent on ongoing protein synthesis, yet the effect of CHX alone on these genes was the opposite, in that it greatly increased VEGF expression and reduced basal levels of Ang-2 expression. Third, neither VEGF induction nor Ang-2 induction by hypoxia was dependent on the presence of serum, yet when studied alone, the absence of serum again revealed opposite effects on Ang-2 or VEGF regulation: it increased basal levels of Ang-2 but reduced those of VEGF. One likely explanation for the latter finding is that the serum effect on either gene is due to transforming growth factor-β, because this molecule is known to up-regulate VEGF expression in a number of cells, including BME cells ( ref. 50 and our unpublished results), and to down-regulate basal levels of Ang-2 mRNA in the same cells. 23
However, the most intriguing difference between Ang-2 and VEGF regulation and the more pertinent finding with respect to the specific mechanism of hypoxic regulation of either gene, came from experiments using DPI. Currently, the most accepted paradigm of oxygen sensing and signal transduction involves a still unidentified heme protein oxygen sensor that binds atmospheric oxygen and continuously reduces it to H2O, thus generating an oxidized environment intracellularly that keeps HIF-1 in an inactive state. Hypoxia would stop this electron flow, thus creating a reduced intracellular environment that results in HIF-1 activation. 20 It is important to point out that this model is still largely speculative and does not easily accommodate all of the existing evidence on oxygen sensing and signal transduction. One of the most conflicting pieces of evidence comes from experiments using DPI. DPI is an inhibitor of flavoprotein oxidoreductases, which are molecules that are often linked to heme proteins in various types of electron transport systems and have been proposed by various authors to act as oxygen sensors. Somewhat surprisingly, experiments assessing the effects of DPI (and its related compound IDP) on the human hepatoma cell lines HepG2 and HepB3, the human fibrosarcoma cell line HT1080, and the human trophoblastic cell line BeWo have shown that these compounds have no or little effect on normoxic levels of hypoxia-responsive genes such as Epo, VEGF, lactate dehydrogenase-A (LDH-A), glucose transporter-1 (GLUT-1), or placental growth factor (PLGF), but potently inhibit the hypoxic regulation of all of these genes. This effect occurred irrespective of whether the hypoxic response was induction (Epo, VEGF, LDH-A, GLUT-1) or inhibition (PLGF) of gene expression. 35 This suggested that iodonium compounds operate specifically in the pathway of oxygen sensing by interfering with the activity of an unidentified flavoprotein oxidoreductase. These results, which are in opposition to what might be expected on the basis of the capacity of iodonium compounds to block flavoprotein-mediated electron flow, have been accommodated by some authors into the above-mentioned model of oxygen sensing, with the proposal that oxygen may provide an alternative electron acceptor, thereby diverting electron flow from a pathway that maintains transcription factors in a reduced and active form. 21,35
Our results using iodonium compounds reveal two intriguing findings. First, in contrast to VEGF and the other above-mentioned hypoxia-regulated genes (with the exception of PLGF, the basal levels of which were very slightly decreased by both DPI and hypoxia), 35 iodonium compounds mimic hypoxia-inducible expression of Ang-2 in BME cells, and DPI further increases the effect of hypoxia on Ang-2 expression. This finding, together with the fact that hypoxia-inducible Ang-2 expression in bovine endothelial cells could not be blocked by neutralizing antibodies against VEGF, 24 rules out the possibility that hypoxia-inducible expression of Ang-2 in endothelial cells is due to an autocrine effect of endogenous VEGF and points to a direct effect of hypoxia on appropriate responsive elements in the Ang-2 gene. Second, in contrast to findings in HepG2 cells, DPI had no effect on the induction of VEGF by hypoxia in BME cells. Although the fragmentary nature of current knowledge of the mechanisms of oxygen sensing does not allow us to fully interpret these findings, they nonetheless offer the following intriguing hypotheses: first, that hypoxic regulation of Ang-2, but not VEGF, in BME cells is under the control of an oxidoreductase flavoprotein that is sensitive to iodonium compounds; second, that the mechanism of oxygen sensing in endothelial cells may have different properties when compared to other cell types. It is tempting to speculate that different cell types have different mechanisms of oxygen sensing, perhaps due to the existence of a family of oxygen sensors (in association with cell-specific components), whose precise distribution and configuration would depend on the cell type considered, its localization in the body with respect to blood, its metabolic requirements and specific pattern of gene expression in response to hypoxia.
Is hypoxia a physiological stimulator of Ang-2 expression in the microvasculature in vivo? Although the constant presence of blood in vessels may argue against this idea, four pieces of evidence suggest that this may indeed be the case. First, endothelial cells of the microvasculature are likely to be subjected to hypoxic conditions in situations of exceptionally high tissue metabolism and/or reduced blood flow, as would result, for example, from the formation of microthrombi or in situations of vascular stasis that may occur in tumors and other pathological settings. Second, HIF-1α protein has been detected in the microvascular endothelium of hypoxic ferret lungs. 51 HIF-1α is continuously synthesized within cells and degraded by the proteasome, and protein stabilization is a hallmark of the cellular response to hypoxia. 22 Third, expression of VEGFR-1, which has a functional HIF-1-binding site within its promoter region, 43 is induced in the microvasculature of mice exposed to systemic hypoxia. 38 Fourth, the HIF-1α-related transcription factor, HIF-2α, the expression of which is restricted almost exclusively to endothelial cells, is activated by conditions of intermediate hypoxia, whereas HIF-1α is not. 52 This is interesting in view of the fact that when compared to other cell types, endothelial cells, by being constantly exposed to the circulating blood, may need to sense and to respond to conditions of less severe hypoxia. Taken together, these findings suggest that endothelial cells do sense hypoxia in vivo and respond to hypoxic conditions with an appropriate and specific program of gene expression, in which induction of the Ang-2 gene is prominent.
Acknowledgments
We thank Dr. P. Maisonpierre for helpful discussions, Dr. N. Maggiano for the HUVE cells; Dr. E.W. Ades and T. Lawley for the HMEC-1 cells; Drs. M.B. Furie and S.C. Silverstein for the BME cells; Dr. B. Berse for the rat VEGF probe; M. Quayzin, Joao Oliveira, J. Mandelbaum, and B. Richter for excellent technical assistance; and P.-A. Ruttimann and J.-P. Gerber for photographic work.
Footnotes
Address reprint requests to Dr.Michael S. Pepper, Department of Morphology, University Medical Center, 1 rue Michel Servet, 1211 Geneva 4, Switzerland. E-mail: michael.pepper@medecine.unige.ch.
Supported by grants from the Swiss National Science Foundation (3100-043364.95 and 3200-052957.97), the Ligue Genevoise contre le Cancer, the Fondation Carlos et Elise de Reuter, and the Foundation Suisse de Cardiologie.
Dr. Mandriota’s present address is The Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford OX3 7BN, United Kingdom.
References
- 1.Folkman J: Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995, 1:27-31 [DOI] [PubMed] [Google Scholar]
- 2.Pepper MS: Manipulating angiogenesis. From basic science to the bedside. Arterioscler Thromb Vasc Biol 1997, 17:605-619 [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86:353-364 [DOI] [PubMed] [Google Scholar]
- 4.Pepper MS, Mandriota SJ, Vassalli JD, Orci L, Montesano R: Angiogenesis regulating cytokines: activities and interactions. Curr Top Microbiol Immunol 1996, 213:31-67 [DOI] [PubMed] [Google Scholar]
- 5.Davis S, Yancopoulos GD: The angiopoietins: yin and yang in angiogenesis. Curr Top Microbiol Immunol 1999, 237:173-185 [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N: Vascular endothelial growth factor: molecular and biological aspects. Curr Top Microbiol Immunol 1999, 237:1-30 [DOI] [PubMed] [Google Scholar]
- 7.Ferrara N, Henzel WJ: Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989, 161:851-858 [DOI] [PubMed] [Google Scholar]
- 8.Pepper MS, Ferrara N, Orci L, Montesano R: Vascular endothelial growth factor (VEGF) induces plasminogen activators and plasminogen activator inhibitor-1 in microvascular endothelial cells. Biochem Biophys Res Commun 1991, 181:902-906 [DOI] [PubMed] [Google Scholar]
- 9.Pepper MS, Ferrara N, Orci L, Montesano R: Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun 1992, 189:824-831 [DOI] [PubMed] [Google Scholar]
- 10.Unemori EN, Ferrara N, Bauer EA, Amento EP: Vascular endothelial growth factor induces interstitial collagenase expression in human endothelial cells. J Cell Physiol 1992, 153:557-562 [DOI] [PubMed] [Google Scholar]
- 11.Nicosia RF, Nicosia SV, Smith M: Vascular endothelial growth factor, platelet-derived growth factor, and insulin-like growth factor-1 promote rat aortic angiogenesis in vitro. Am J Pathol 1994, 145:1023-1029 [PMC free article] [PubMed] [Google Scholar]
- 12.Mandriota SJ, Seghezzi G, Vassalli JD, Ferrara N, Wasi S, Mazzieri R, Mignatti P, Pepper MS: Vascular endothelial growth factor increases urokinase receptor expression in vascular endothelial cells. J Biol Chem 1995, 270:9709-9716 [DOI] [PubMed] [Google Scholar]
- 13.Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD: Isolation of angiopoietin-1, a ligand for the Tie2 receptor, by secretion-trap expression cloning. Cell 1996, 87:1161-1169 [DOI] [PubMed] [Google Scholar]
- 14.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD: Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 1996, 87:1171-1180 [DOI] [PubMed] [Google Scholar]
- 15.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y: Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 1995, 376:70-74 [DOI] [PubMed] [Google Scholar]
- 16.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD: Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277:55-60 [DOI] [PubMed] [Google Scholar]
- 17.Stratmann A, Risau W, Plate KH: Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol 1998, 153:1459-1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ: Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284:1994-1998 [DOI] [PubMed] [Google Scholar]
- 19.Hanahan D: Signaling vascular morphogenesis and maintenance. Science 1997, 277:48-50 [DOI] [PubMed] [Google Scholar]
- 20.Bunn HF, Poyton RO: Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev 1996, 76:839-885 [DOI] [PubMed] [Google Scholar]
- 21.Ratcliffe PJ, O’Rourke JF, Maxwell PH, Pugh CW: Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol 1998, 201:1153-1162 [DOI] [PubMed] [Google Scholar]
- 22.Semenza GL: Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev 1998, 8:588-594 [DOI] [PubMed] [Google Scholar]
- 23.Mandriota SJ, Pepper MS: Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ Res 1998, 83:852-859 [DOI] [PubMed] [Google Scholar]
- 24.Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y: Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem 1999, 274:15732-15739 [DOI] [PubMed] [Google Scholar]
- 25.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ: HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol 1992, 99:683-690 [DOI] [PubMed] [Google Scholar]
- 26.Furie MB, Cramer EB, Naprstek BL, Silverstein SC: Cultured endothelial cell monolayers that restrict the transendothelial passage of macromolecules and electrical current. J Cell Biol 1984, 98:1033-1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McFarlane RM, DeYoung MD, Henry RA: The design of a pedicle flap in the rat to study necrosis and its prevention. Plast Reconstr Surg 1965, 35:177-182 [DOI] [PubMed] [Google Scholar]
- 28.Khouri RK, Angel MF, Edstrom LE: Standardizing the dorsal rat flap. Surg Forum 1986, 37:590-591 [Google Scholar]
- 29.Pepper MS, Mandriota SJ: Regulation of vascular endothelial growth factor receptor-2 (Flk-1) expression in vascular endothelial cells. Exp Cell Res 1998, 241:414-425 [DOI] [PubMed] [Google Scholar]
- 30.Pepper MS, Soriano JV, Menoud PA, Sappino AP, Orci L, Montesano R: Modulation of hepatocyte growth factor and c-met in the rat mammary gland during pregnancy, lactation, and involution. Exp Cell Res 1995, 219:204-210 [DOI] [PubMed] [Google Scholar]
- 31.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N: Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246:1306-1309 [DOI] [PubMed] [Google Scholar]
- 32.Gorden DL, Mandriota SJ, Montesano R, Orci L, Pepper MS: Vascular endothelial growth factor is increased in devascularized rat islets of Langerhans in vitro. Transplantation 1997, 63:436-443 [DOI] [PubMed] [Google Scholar]
- 33.Rømer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Danø K: Impaired wound healing in plasminogen-deficient mice. Nat Med 1996, 2:287-292 [DOI] [PubMed] [Google Scholar]
- 34.Shweiki D, Itin A, Soffer D, Keshet E: Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359:843-848 [DOI] [PubMed] [Google Scholar]
- 35.Gleadle JM, Ebert BL, Ratcliffe PJ: Diphenylene iodonium inhibits the induction of erythropoietin and other mammalian genes by hypoxia. Implications for the mechanism of oxygen sensing. Eur J Biochem 1995, 234:92-99 [DOI] [PubMed] [Google Scholar]
- 36.Boero JA, Ascher J, Arregui A, Rovainen C, Woolsey TA: Increased brain capillaries in chronic hypoxia. J Appl Physiol 1999, 86:1211-1219 [DOI] [PubMed] [Google Scholar]
- 37.Kalaria RN, Cohen DL, Premkumar DR, Nag S, LaManna JC, Lust WD: Vascular endothelial growth factor in Alzheimer’s disease and experimental cerebral ischemia. Brain Res Mol Brain Res 1998, 62:101-105 [DOI] [PubMed] [Google Scholar]
- 38.Marti HH, Risau W: Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci USA 1998, 95:15809-15814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu F, Severinghaus JW: Rat brain VEGF expression in alveolar hypoxia: possible role in high-altitude cerebral edema. J Appl Physiol 1998, 85:53-57 [DOI] [PubMed] [Google Scholar]
- 40.Wong AL, Haroon ZA, Werner S, Dewhirst MW, Greenberg CS, Peters KG: Tie2 expression and phosphorylation in angiogenic and quiescent adult tissues. Circ Res 1997, 81:567-574 [DOI] [PubMed] [Google Scholar]
- 41.Bunone G, Vigneri P, Mariani L, Buto S, Collini P, Pilotti S, Pierotti MA, Bongarzone I: Expression of angiogenesis stimulators and inhibitors in human thyroid tumors and correlation with clinical-pathologic features. Am J Pathol 1999, 155:1967-1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kourembanas S, Hannan RL, Faller DV: Oxygen tension regulates the expression of the platelet-derived growth factor-B chain gene in human endothelial cells. J Clin Invest 1990, 86:670-674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerber HP, Condorelli F, Park J, Ferrara N: Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem 1997, 272:23659-23667 [DOI] [PubMed] [Google Scholar]
- 44.Folkman J, D’Amore PA: Blood vessel formation: what is its molecular basis? Cell 1996, 87:1153-1155 [DOI] [PubMed] [Google Scholar]
- 45.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M: Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci USA 1998, 95:9349-9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fong GH, Rossant J, Gertsenstein M, Breitman ML: Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376:66-70 [DOI] [PubMed] [Google Scholar]
- 47.Fong GH, Zhang L, Bryce DM, Peng J: Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development 1999, 126:3015-3025 [DOI] [PubMed] [Google Scholar]
- 48.McCarthy MJ, Crowther M, Bell PR, Brindle NP: The endothelial receptor tyrosine kinase tie-1 is upregulated by hypoxia and vascular endothelial growth factor. FEBS Lett 1998, 423:334-338 [DOI] [PubMed] [Google Scholar]
- 49.Tian H, McKnight SL, Russell DW: Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 1997, 11:72-82 [DOI] [PubMed] [Google Scholar]
- 50.Pertovaara L, Kaipainen A, Mustonen T, Orpana A, Ferrara N, Saksela O, Alitalo K: Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells. J Biol Chem 1994, 269:6271-6274 [PubMed] [Google Scholar]
- 51.Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL: Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol 1998, 275:L818-L826 [DOI] [PubMed] [Google Scholar]
- 52.Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH: Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood 1998, 92:2260-2268 [PubMed] [Google Scholar]