

Abstract
Endometrial hyperplasia is regarded as a precursor lesion of endometrioid adenocarcinomas of the endometrium. The genetic events involved in the multistep process from normal endometrial glandular tissue to invasive endometrial carcinomas are primarily unknown. We chose endometrial hyperplasia as a model for identifying chromosomal aberrations occurring during carcinogenesis. Comparative genomic hybridization (CGH) was performed on 47 formalin-fixed, paraffin-embedded specimens of endometrial hyperplasia using the microdissection technique to increase the number of tumor cells in the samples and reduce contamination from normal cells. CGH analysis revealed that 24 out of 47 (51%) samples had detectable chromosomal imbalances, whereas 23 (49%) were in a genetically balanced state. The incidence of aberrant CGH profiles tended to parallel dysplasia grade, ranging from 22% aberrant profiles in simple hyperplasia to 67% in complex hyperplasia with atypia. The most frequent imbalances were 1p, 16p, and 20q underrepresentations and 4q overrepresentations. Copy number changes in 1p were more frequent in atypical complex hyperplasia than in complex lesions without atypical cells or simple lesions (42% versus 20% and 0%). Our results show that endometrial hyperplasia reveals recurrent chromosomal imbalances which tend to increase with the presence of atypical cells. The most frequent aberrations in endometrial cancer, 1q and 8q overrepresentations, are not present or are rare in its precursor lesions. This analysis provides evidence that tumorigenesis proceeds through the accumulation of a series of genetic alterations and suggests a stepwise mode of tumorigenesis.
Endometrial hyperplasia is a pathological condition of the endometrium that is known to be a precursor lesion of endometrioid adenocarcinoma. The risk of developing invasive endometrial cancer depends on the presence or absence of atypical cells, which are graded according to a histological classification including simple hyperplasia, complex hyperplasia, and complex hyperplasia with cytological atypia. 1 The risk of a subsequent endometrial cancer increases from ∼1% in simple hyperplasia to up to 45% in atypical complex lesions. 2,3
A series of genetic aberrations is required for tumor initiation and progression. 4 The phenotypes of endometrial hyperplasia possess different malignant potential and can be defined as different histomorphological stages during the carcinogenesis of endometrioid adenocarcinomas of the endometrium. They are an appropriate model for studying the sequences of genetic alterations during tumorigenesis. So far, the genetic events involved in the multistep process in endometrial cancer are primarily unknown. Microsatellite instability (MI) is one of the genetic alterations observed in ∼25% of sporadic endometrial cancers. 5 Furthermore, mutations and inactivations of various oncogenes and tumor suppressor genes, eg, c-erb-2, c-myc, PTEN, and TP53 have been reported in endometrial cancer. 6,7 However, with the exception of PTEN alterations, which occur in ∼40% of sporadic endometrial carcinomas, the frequencies of these alterations were low, and it has been suggested that mutations in other genes may play key roles in endometrial cancer. DNA replication error-positive (RER+) phenotypes were also detected in a small subset of cases of atypical endometrial hyperplasia, which progress to RER+ endometrial carcinomas. 8 Recently, Levine et al 9 and Maxwell et al 10 reported that somatic PTEN mutations occur in ∼20 to 27% of cases of endometrial hyperplasia and might be an early event in endometrial carcinogenesis.
For the analysis of genetic imbalances within entire tumor genomes and the identification of gross genetic target regions, comparative genomic hybridization (CGH) is a powerful tool. 11 With this method, information about gains and losses of DNA sequences throughout the genome is easily obtained without the need for mitotic cells and time-consuming tissue culture. It has also been successfully applied to microdissected archival paraffin-embedded tissue by several investigators. 12
We applied CGH to 47 paraffin-embedded specimens of endometrial hyperplasia using the microdissection technique to establish a correlation between the microscopic phenotype and the genotype of defined stages of endometrial carcinogenesis. It was the aim of our study to gain insights into genetic changes in precursor lesions of endometrial adenocarcinomas to verify whether the multistep model of Fearon and Vogelstein 13 is also applicable to adenocarcinomas of the endometrium.
Materials and Methods
Tissue Samples
Forty-seven formalin-fixed, paraffin-embedded specimens of endometrial hyperplasia were obtained from the archives of the Departments of Pathology at the Universities of Kiel and Mainz. Ten serial 10-μm sections were cut from the tissue blocks. The first and the last section were stained with hematoxylin and eosin (H&E) for histological analysis and the endometrial lesions were classified using standard World Health Organization criteria. 14 Areas of unequivocal endometrial hyperplasia were identified on the first section, which was stained with H&E. The corresponding areas were labeled on the subsequent serial sections for tissue microdissection and DNA extraction. Evaluation of the last section, which was again stained with H&E, assured that the correct area of the tissue section was indeed chosen for further analysis.
DNA Extraction
DNA was extracted as described by Speicher et al 15 with minor modifications. Briefly, after dewaxing (3 × 10 minutes in xylol and 2 × 5 minutes in methanol), cells were dissected from labeled areas of the eight unstained serial sections and collected in sterile tubes. The tissues were incubated overnight in 450 μl of DNA extraction buffer (75 mmol/L NaCl, 2.5 mmol/L ethylenediaminetetraacetic acid, pH 8.0, 0.5% Tween 20, 0.1 μg/μl proteinase K) at 55°C. The mixture was boiled for 10 minutes to inactivate the proteinase K and chilled on ice. After incubation with RNase A (20 μg/ml) for one hour at 37°C, the DNA was extracted by phenol/chloroform and ethanol precipitation. There was enough DNA of sufficient quality so there was no need to perform degenerate oligonucleotide-priomed polymerase chain reaction amplification. Reference DNA was isolated from peripheral blood lymphocytes of normal males according to standard procedures.
Comparative Genomic Hybridization
CGH analysis was done as described by Arnold et al 16 with minor modifications. Briefly, reference metaphase spreads were prepared following standard procedures from peripheral blood lymphocytes of a healthy male donor. Preparations were stored in 70% ethanol at 4°C until use. To minimize background signals and to increase accessibility of the DNA probes to the target chromosomes RNase A and pepsin treatment of the slides before denaturation was performed. Genomic DNA from a healthy male donor (reference DNA) was labeled in a standard nick translation assay with digoxigenin-11dUTP, and test DNA was labeled with biotin-16dUTP. Equal amounts of labeled reference and test DNA including 30 μg of Cot-1 DNA (Life Technologies, Inc., Rockville, MD) were hybridized to normal metaphase spreads. After incubation for 3 days at 37°C in a humidified chamber, the slides were washed two times in 1× phosphate buffered detergent (PBD; Appligene Oncor, Illkirch, France), once in 2× standard saline citrate (0.15 mol/L NaCl, 0.015 mol/L Na-citrate), pH 7.0, at 70°C without shaking and once in 1× PBD at room temperature for 5 minutes each. Slides were stained with 5 μg/ml of streptavidin-fluorescein isothiocyanate (FITC) and 1 μg/ml of antidigoxigenin rhodamine (both from Boehringer Mannheim, Mannheim, Germany) diluted in 1× PBD at 37°C for 45 minutes. Slides were then washed three times in 1× PBD at room temperature for 2 minutes and mounted in an antifade solution (Vectashield, Vector Laboratories, Burlingame, CA) containing 4′,6-diamidino-2-phenylindole as a counterstain.
Digital Image Analysis
Images of the hybridized metaphases were obtained and evaluated with a digital imaging system (MetaSystems GmbH, Sandhausen, Germany), connected to a Zeiss fluorescence microscope (Jena, Germany). Images were captured using an integrated uncooled black and white charge-coupled device camera. Subsequently, the three color components, green (fluorescein isothiocyanate) for the tumor DNA, red (rhodamine) for the normal reference DNA, and blue (4′,6-diamidino-2-phenylindole) for the DNA counterstain were digitized. The automatic exposure control of the imaging system made sure that the full dynamic range of the system was used for each color component. A real-color image was displayed on the monitor. A detailed description of the evaluation process is given by Arnold et al. 16 The average number of copy alterations (ANCA) were calculated by dividing the total number of copy alterations per chromosome arm presented in a karyogram by the number of analyzed lesions.
Results
Table 1 ▶ summarizes the pertinent histological data and the CGH results. Twenty-four out of 47 (51%) samples revealed imbalanced CGH profiles. Both the incidence of aberrant CGH profiles and the ANCA tended to parallel the histological subtypes. Two out of nine specimens of simple hyperplasia (22%), 10 out of 20 of complex hyperplasia (50%), and 12 out of 18 complex atypical lesions (67%) had detectable chromosomal imbalances. The ANCA values increased with increasing cellular atypia. Simple hyperplasia revealed 0.3, complex hyperplasia 1.85, and atypical complex hyperplasia 2.55 ANCA. In lesions showing simple hyperplasia, a total of three random chromosomal imbalances were found (Table 1) ▶ .
Table 1.
Chromosomal Imbalances Detected by CGH and Pertinent Clinical Data in 47 Cases of Endometrial Hyperplasia
| No. | Case | Age | Histology type | Chromosomal imbalances |
|---|---|---|---|---|
| 1 | 97 /21253 | 56 | SH | |
| 2 | 95 /50685 | 44 | SH | |
| 3 | 96 /50859 | 55 | SH | |
| 4 | 96 /41619 | 52 | SH | |
| 5 | 95 /50062 | 51 | SH | |
| 6 | 97 /22157 | 50 | SH | dim(2p16) enh(5q14) |
| 7 | 95 /50211 | 60 | SH | dim(7q21q22) |
| 8 | 97 /2897 | 30 | SH | |
| 9 | 95 /50738 | 41 | SH | |
| 10 | 95 /50748 | 79 | CH | |
| 11 | 97 /32244 | 78 | CH | |
| 12 | 96 /50664 | 67 | CH | |
| 13 | 97 /51150 | 60 | CH | |
| 14 | 95 /50419 | 55 | CH | |
| 15 | 97 /12983 | 59 | CH | |
| 16 | 97 /16057 | 75 | CH | |
| 17 | 96 /28026 | 55 | CH | dim(3q26.1q26.3; 9p23) enh(9p12q13; 17qter; 22cenqter) |
| 18 | 96 /35144 | 61 | CH | enh(1p33; 1p36.2p36.1) |
| 19 | 95 /50140 | 44 | CH | dim(4q22q24) enh(16p13.1) |
| 20 | 96 /36872 | 60 | CH | dim(1p32p31; 4q24q26) enh(1q32q42) |
| 21 | 96 /50649 | 59 | CH | |
| 22 | 96 /50649 | 59 | CH | dim(4q34qter) |
| 23 | 97 /26703 | 78 | CH | enh(1pterp31; 16p12; 19q13.1q13.3; 22q12qter) |
| 24 | 97 /29728 | 55 | CH | |
| 25 | 97 /31942 | 36 | CH | |
| 26 | 97 /6751 | 73 | CH | dim(1pterp35; 2q36qter; 9q33qter; 16pterp12; 16q23qter; 17q22qter; 20pterp12; 20q13.1qter) enh(4p12; 4q33q22; 5q21; 6q12q22) |
| 27 | 96 /7441 | 62 | CH | dim(16pterp13.1; 20q12q13.2) enh(4p13p12) |
| 28 | 96 /21497 | 58 | CH | enh(4q31.3; 15q21) |
| 29 | 98 /20598 | 40 | CH | dim(20q13.1qter) |
| 30 | 95 /50108 | 63 | CAH | |
| 31 | 97 /4680 | 61 | CAH | |
| 32 | 96 /27168 | 43 | CAH | |
| 33 | 96 /50343 | 39 | CAH | |
| 34 | 98 /14959 | 70 | CAH | dim(20p11; 20q12–13.2) enh(3p12; 3q13.1q13.2; 4q22; 5p13p12; 6p12cen) |
| 35 | 97 /17600 | 74 | CAH | dim(22q12qter) |
| 36 | 96 /24958 | 48 | CAH | |
| 37 | 96 /8363 | 46 | CAH | |
| 38 | 97 /12660 | 55 | CAH | dim(16p13.1cen; 16q23qter) enh(1cenqter) |
| 39 | 96 /11646 | 66 | CAH | dim(1pterp36.1; 9q34qter; 16q22qter) enh(2q32; 12p12p11.2; 12q14q21) |
| 40 | 98 /16692 | 37 | CAH | dim(1p31cen) |
| 41 | 97 /10256 | 52 | CAH | enh(17q12) |
| 42 | 96 /16674 | 66 | CAH | dim(10q25q26; 19q13.1q13.2; 20q12q13.2) enh(6q16; 6q12q14) |
| 43 | 97 /555 | 72 | CAH | dim(1pterp36.1) enh(1q32) |
| 44 | 97 /12661 | 75 | CAH | enh(6p12p11.2; 21q21) |
| 45 | 96 /12985 | 63 | CAH | enh(2p23; 9pterp21) |
| 46 | 97 /5168 | 61 | CAH | dim(1pterp35; 16p13.2p12; 22q12qter) enh(4q12q13; 6q16) |
| 47 | 98 /15147 | 69 | CAH | dim(1pterp33; 9q33qter; 12q24.1qter; 16p13.1p12; 16q21q23; 17q21; 20q12q13.2) enh(3p12; 4p14p12; 4q25q31.1; 5q21; 6q22) |
SH, simple hyperplasia; CH, complex hyperplasia; CAH, complex atypical hyperplasia.
In contrast to simple hyperplasia, complex hyperplasia with and without atypical cells showed recurrent alterations. Among all 22 specimens of complex hyperplasia with aberrant CGH profiles, the most common aberrations were 1p underrepresentation (7 out of 22; 32%), 20q− (6 out of 22; 27%) as well as 4q+, and 16p− (5 out of 22; 23%). Moreover, 1p underrepresentations were twice as frequent in atypical hyperplasia (5 out of 12; 42%) as in complex lesions without atypia (2 out of 10; 20%). Figure 1 ▶ shows an example of a CGH analysis with a specimen of complex hyperplasia (case 27) revealing an underrepresentation of 16p and 20q. In Figure 2 ▶ the recurrent chromosomal imbalances were mapped. They showed smallest regions of overlap at 1pterp36, 16p13.1, and 20q13.1q13.2. So far, no smallest regions of overlap could be designated on 4q, because the observed regions of amplification were scattered throughout the whole chromosome arm.
Figure 1.
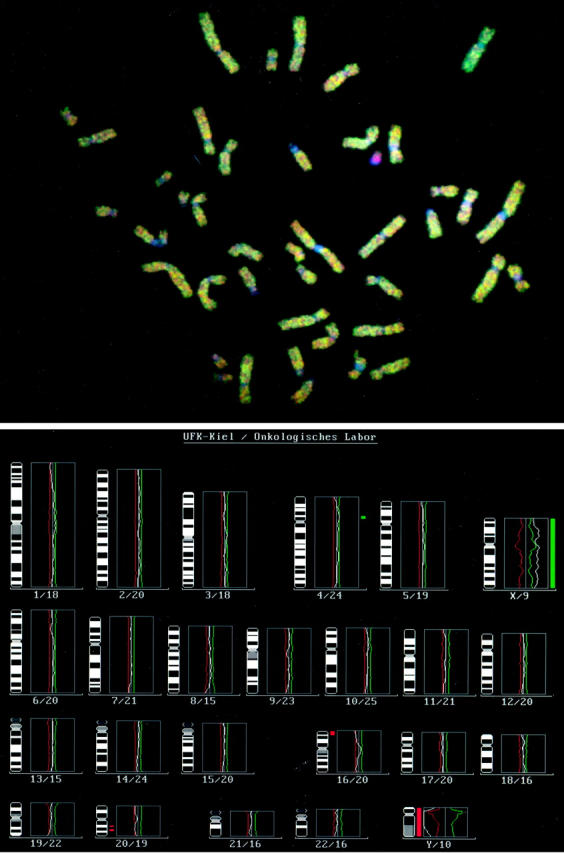
Images of normal metaphase chromosomes from a male donor hybridized to a complex hyperplasia (case 27) DNA (green) and normal male DNA (red). The metaphase plate (top) and the mean green to red ratio profiles from 13 metaphases (bottom) are shown. The central vertical line represents the baseline ratio value (1.0) typical for a balanced status of chromosome material, whereas the red and green lines represent thresholds for chromosomal losses and gains. The numbers below reflect the chromosome number and the number of chromosomes that are used for the profiling analysis. This profile represents the following aberrations indicated by red and green bars: Gain of chromosomal material 4p13p12 and loss of chromosomal material 16pterp13.1 and 20q12q13.2. As an internal control, the Y-chromosome appears red, because it is not present in the tumor sample. The X-chromosome lies over the threshold and is regarded to be present in two copies.
Figure 2.

Partial karyogram showing the localization of recurrent genetic alterations in complex endometrial hyperplasia. Vertical lines on the left side of a chromosome indicate the extent of loss of genetic material.
Discussion
The aim of this study was to determine the prevalence of chromosomal imbalances in endometrial hyperplasia to verify Fearon and Vogelstein’s 13 model of cancerogenesis for endometrioid adenocarcinoma of the endometrium. To study the chromosomal imbalances during the initiation and progression of endometrioid adenocarcinomas of the endometrium, microdissected, formalin-fixed, paraffin-embedded specimens showing endometrial hyperplasia of morphologically defined stages were screened by means of a molecular cytogenetic method known as CGH.
To our knowledge, this is the first study indicating that endometrial hyperplasia reveals recurrent chromosomal changes. A total of 24 hyperplastic lesions (51%) had detectable DNA copy changes. The incidence of aberrant CGH profiles rose with increasing architectural complexity and cellular atypia. Only 2 out of 9 specimens of simple hyperplasia (22%) had detectable chromosomal imbalances, whereas specimens with complex hyperplasia revealed DNA copy number changes in 50% of cases (10 out of 20) and complex atypical lesions in 67% (12 out of 18). Our findings support the concept that accumulating genetic alterations increase genomic instability, which is phenotypically associated with an increasing risk of developing an invasive carcinoma. This idea is further underlined by the observation that the ANCA also increases with increasing cellular atypia. Simple hyperplasia showing a low risk for the development of endometrial cancer revealed an ANCA value of 0.3. Hyperplasia with architectural abnormalities, however, which is associated with a 3 to 17% risk of endometrial cancer, 2,3 had an ANCA value of 1.85. The presence of atypical cells in complex atypical hyperplasia with a 29 to 45% risk of an invasive cancer, 2,3 resulted in ANCA values of 2.55. ANCA values in endometrioid adenocarcinomas of the endometrium are even higher, rising to 4.6. 17 Earlier CGH studies on precursor and invasive lesions of cervical 18 and colon cancer 19,20 are in line with our results, suggesting that the ANCA index correlates with disease progression and might be a valid indicator of genomic instability and malignant potential.
Among those complex lesions with DNA copy number changes (n = 22), the most consistent alterations were decreased fluorescence values at 1p, 20q, and 16p, which occurred in 23 to 32% (5 to 7 out of 22) of the samples. Increased DNA copy number changes were less common. Mostly, they were observed on 4q in 23% (5 out of 22) of the samples with CGH imbalances. These consistent aberrations were only observed in complex hyperplasia. Simple hyperplasia showed only three random alterations in two cases. The partial karyogram of common chromosomal gains and losses in endometrial hyperplasia (Figure 2) ▶ indicates the smallest regions of overlap mapping the regions of interest to chromosome 1pterp36, 20q13.1q13.2, and 16p13.1. These findings suggest that genes involved in the initiation and progression of complex hyperplasia may be located within these chromosomal regions. A possible candidate gene deleted at 1p36 might be the cell division cycle 2-like 1 protein (CDC2L1). This well-conserved protein kinase p58 is supposed to be a negative regulator of normal cell cycle progression. 21 This cell division control-related gene may also be implicated in the pathogenesis of other tumors that have deletions in the region of 1p36, such as ductal carcinoma of the breast, endocrine neoplasia, and malignant melanoma. 22 Recently, the epithelial membrane protein 2 was mapped to 16p13.2 by Liehr et al. 23 It has been suggested that it is involved in cell proliferation and cell-cell interactions. 24 In addition, the deleted regions on 16p and 20q contain two ubiquitin-conjugating enzymes (UBE2I and UBE2V1). This family of proteins is involved in essential cellular processes such as DNA repair, cell cycle control, and stress responses. 25,26 An additional gene of interest at 20q13 is the cellular apoptosis susceptibility (CAS) gene, the human homologue of the yeast chromosome segregation gene CSE1. 27 The CAS protein, which has a dual function in mammalian cells, may be involved in both apoptosis and cell proliferation processes. Interestingly, CAS is associated with the mitotic spindle and has been suggested to be part of the cell cycle checkpoint that assures correct chromosome distribution to daughter cells. CAS depletion may lead to aberrant chromosome segregation during mitosis in both yeast and mammalian cells. 27 Alterations of these candidate genes in endometrial cells may provide growth advantages and increase genomic instability.
The pattern of chromosomal changes in invasive endometrial cancer is totally different from that in endometrial hyperplasia. So far, CGH analysis of 47 endometrioid adenocarcinomas of the endometrium has been reported. 18,28,29 Aberrant CGH profiles were found in 27 carcinomas, mainly comprising cases without MI. 1q and 8q overrepresentations were found to be the most consistent alterations, occurring in 20 out of 27 (74%) and 8 out of 27 (62%) of abnormal cases, respectively. In contrast to endometrial hyperplasia, decreased fluorescence intensities were less frequent in endometrial carcinomas. Moreover, 1q amplifications were less common in endometrial hyperplasia and were detected in only three of 47 hyperplastic lesions in the present study. 8q overrepresentations, the second most common alteration found in invasive endometrial cancer, were totally lacking in its precursor lesions.
The comparison of chromosomal gains and losses in simple, complex, and complex atypical hyperplasia suggests a sequence of genetic events (Figure 3 ▶ ). Progression from simple to complex hyperplasia involves loss of genetic material at 16p and 20q, which did not occur in any of the specimens of simple hyperplasia, in contrast to 23 to 27% of complex hyperplasia. Deletions in 1p seem to be associated with the presence of atypical cells. They are not detected in simple hyperplasia, are rare in complex lesions (2 out of 20; 10%), and occur in 42% (5 out of 12) of specimens of complex atypical hyperplasia with aberrant CGH profiles. The combination of our data with the results of CGH analysis of invasive endometrial cancer in the literature 18,28,29 suggests that the gain of chromosome arms 1q and 8q, which is rare or lacking in the precursor lesions, may define the transition from complex atypical hyperplasia to invasive endometrioid adenocarcinoma of the endometrium.
Figure 3.

Model of the stepwise accumulation of genetic alterations in the carcinogenesis of endometrioid adenocarcinomas of the endometrium. The photographs represent defined phenotypes of endometrial hyperplasia, starting with simple hyperplasia at the right side and ending with an endometrioid adenocarcinoma at the left side. The ANCA values increase with the presence of architectural abnormalities and cellular atypia. 20q− and 16p− are most likely early events connected with architectural changes, whereas 1p− occurs later and is associated with the presence of atypical cells. 1q+ and 8q+ seem to be responsible for the transition from complex atypical hyperplasia to invasive endometrial cancer.
It should be noted that a subset of both lesions showing complex hyperplasia (16 out of 38; 42%) and carcinomas (20 out of 47; 43%) has no detectable aberrations. Because we used a microdissection technique for tissue selection, major contamination with normal cells is unlikely. Various point mutations in proto-oncogenes or tumor suppressor genes caused by repeat instability because of mismatch repair deficiency are a more likely causative mechanism in these tumors. It is well known that MI occurs in a subset of endometrial carcinomas 5 and lesions showing atypical endometrial hyperplasia. 8 Additionally, it has been shown that MI positive endometrial carcinomas and colon carcinomas are primarily devoid of chromosomal changes that are detectable by CGH. 28,30
In summary, endometrial hyperplasia reveals consistent chromosomal abnormalities, including 16p, 20q, and 1p underrepresentations. Targeted molecular investigations of these regions may reveal genes that play an important role in the initiation and progression of endometrial tumors that are probably MI-negative. In the present study, aberrant CGH profiles tended to parallel cellular complexity and atypia and might be a useful marker for a phenotype with an increased malignant potential.
Acknowledgments
We are very grateful to Doris Karow for her excellent technical assistance.
Footnotes
Address reprint requests to Priv.-Doz. Dr. Marion Kiechle, Oncological Laboratory, Department of Gynecology and Obstetrics, University of Kiel Michaelisstrasse 16, D-24105 Kiel, Germany. E-mail: mkiechle@email.uni-kiel.de.
Supported by a grant from the Interdisciplinary Center of Clinical Research of the University of Kiel (IZKF).
The research was performed in the oncological laboratory of the Department of Gynecology and Obstetrics, University of Kiel Medical School, Michaelisstrasse 16, D-24105 Kiel, Germany.
References
- 1.Kurman RJ, Mazur MT: Benign diseases of the endometrium. Kurman RJ eds. Blaustein’s Pathology of the Female Genital Tract. 1994, :pp 367-410 Springer, New York-Berlin-Heidelberg [Google Scholar]
- 2.Kurman RJ, Kamisnki PF, Norris HJ: The behavior of endometrial hyperplasia. A long term study of “untreated” hyperplasias in 170 patients. Cancer 1985, 56:403-412 [DOI] [PubMed] [Google Scholar]
- 3.Baak JPA, Wisse-Brekelmans ECM, Fleege JC, van der Putten HW, Bezemer PD: Assessment of the risk on endometrial cancer in hyperplasia by means of morphological and morphometrical features. Pathol Res Pract 1992, 188:856-859 [DOI] [PubMed] [Google Scholar]
- 4.Nowell PC: The clonal evolution of tumor cell populations. Science 1976, 194:23-28 [DOI] [PubMed] [Google Scholar]
- 5.Gurin CC, Federici MG, Kang L, Boyd J: Causes and consequences of microsatellite instability in endometrial cancer. Cancer Res 1999, 59:462-466 [PubMed] [Google Scholar]
- 6.Burton JL, Wells M: Recent advances in the histopathology and molecular pathology of carcinoma of the endometrium. Histopathology 1998, 33:297-303 [DOI] [PubMed] [Google Scholar]
- 7.Berchuk A, Boyd J: Molecular basis of endometrial cancer. Cancer 1995, 76:2034-2040 [DOI] [PubMed] [Google Scholar]
- 8.Mutter GL, Boynton KA, Faquin WC, Ruiz RE, Jovanovic AS: Allelotype mapping of unstable microsatellites establishes direct lineage continuity between endometrial precancers and cancer. Cancer Res 1996, 56:4483-4486 [PubMed] [Google Scholar]
- 9.Levine RL, Cargile CB, Blazes MS, van Rees B, Kurman RJ, Ellenson LH: PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res 1998, 58:3254-3258 [PubMed] [Google Scholar]
- 10.Maxwell GL, Risinger JI, Gumbs C, Shaw H, Bentley RC, Barrett JC, Berchuk A, Futreal PA: Mutation of the PTEN tumor suppressor gene in endometrial hyperplasias. Cancer Res 1998, 58:2500-2503 [PubMed] [Google Scholar]
- 11.Kallioniemi A, Kallioniemi OP, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258:818-821 [DOI] [PubMed] [Google Scholar]
- 12.James L, Varley J: Preparation, labeling and detection of DNA from archival tissue sections suitable for comparative genomic hybridization. Chromosom Res 1996, 4:163-164 [DOI] [PubMed] [Google Scholar]
- 13.Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 14.Scully RE, Bonfiglio TA, Kurman RJ, Silverberg SG, Wilkinson EJ: WHO international histological classification of tumors. Histological Typing of Female Genital Tract Tumors. New York-Berlin-Heidelberg Springer, 1994, pp 13–14
- 15.Speicher MR, du Manoir S, Schröck E, Holtgreve-Grez H, Schoell B, Lengauer C, Cremer T, Ried T: Molecular cytogenetic analysis of formalin fixed, paraffin embedded solid tumors by comparative genomic hybridization after universal DNA-amplification. Hum Mol Genet 1993, 2:1907-1914 [DOI] [PubMed] [Google Scholar]
- 16.Arnold N, Hägele L, Walz L, Schempp W, Pfisterer J, Bauknecht T, Kiechle M: Overrepresentation of 3q and 8q material and loss of 18q material are recurrent findings in advanced ovarian cancer. Genes Chromosom Cancer 1996, 16:46-54 [DOI] [PubMed] [Google Scholar]
- 17.Pere H, Tapper J, Wahlström T, Knuutila S, Butzow R: Distinct chromosomal imbalances in uterine serous and endometrioid carcinomas. Cancer Res 1998, 58:892-895 [PubMed] [Google Scholar]
- 18.Heselmeyer K, Schröck E, du Manoir S, Blegen H, Shah K, Steinbeck R, Auer G, Ried T: Gain of chromosome 3q defines the transition from severe dysplasia to invasive carcinoma of the uterine cervix. Proc Natl Acad Sci USA 1996, 93:479-484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ried T, Knutzen R, Steinbeck R, Blegen H, Schröck E, Heselmeyer K, du Manoir S, Auer G: Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosom Cancer 1996, 15:234-245 [DOI] [PubMed] [Google Scholar]
- 20.Ried T, Heselmeyer-Haddad K, Blegen H, Schrock E, Auer G: Genomic changes defining the genesis, progression, and malignancy potential in solid human tumors: a phenotype/genotype correlation. Genes Chromosom Cancer 1999, 25:195-204 [DOI] [PubMed] [Google Scholar]
- 21.Bunnell B, Heath LS, Adams DE, Lahti JM, Kidd VJ: Elevated expression of a p58 protein kinase leads to changes in the CHO cell cycle. Proc Nat Acad Sci USA 1990, 87:7467-7471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eipers PG, Barnoski BL, Han J, Carroll AJ, Kidd VJ: Localization of the expressed human p58 protein kinase chromosomal gene to chromosome 1p36 and a highly related sequence to chromosome 15. Genomics 1991, 11:621-629 [DOI] [PubMed] [Google Scholar]
- 23.Liehr T, Kuhlenbaumer G, Wulf P, Taylor V, Suter U, Van Broeckhoven C, Lupski JR, Claussen U, Rautenstrauss B: Regional localization of the human epithelial membrane protein genes 1, 2, and 3 (EMP1, EMP2, EMP3) to 12p12.3, 16p13.2, and 19q13.3. Genomics 1999, 58:106–108 [DOI] [PubMed]
- 24.Ben-Porath I, Benvenisty N: Characterization of a tumor-associated gene, a member of a novel family of genes encoding glycoproteins. Gene 1996, 183:69-75 [DOI] [PubMed] [Google Scholar]
- 25.Wang ZJ, Qiu QQ, Seufert W, Taguchi T, Testa JR, Whitmore SA, Callen DF, Welsh D, Shenk T, Deuel TF: Molecular cloning of the cDNA and chromosome localization of the gene for human ubiquitin-conjugating enzyme 9. J Biol Chem 1996, 271:24811-24816 [DOI] [PubMed] [Google Scholar]
- 26.Rothofsky ML, Lin SL: Croc-1 encodes a protein which mediates transcriptional activation of the human FOS promoter. Gene 1997, 195:141-149 [DOI] [PubMed] [Google Scholar]
- 27.Brinkmann U: CAS, the human homologue of the yeast chromosome-segregation gene CSE1, in proliferation, apoptosis, and cancer. Am J Hum Genet 1998, 62:509-513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonoda G, du Manoir S, Godwin AK, Bell DW, Liu Z, Hogan M, Yakushiji M, Testa JR: Detection of DNA gains and losses in primary endometrial carcinomas by comparative genomic hybridization. Genes Chromosom Cancer 1997, 18:115-125 [DOI] [PubMed] [Google Scholar]
- 29.Suzuki A, Fukushige S, Nagase S, Ohuchi N, Satomi S, Horii A: Frequent gains on chromosome arms 1q and/or 8q in human endometrial cancer. Hum Genet 1997, 100:629-636 [DOI] [PubMed] [Google Scholar]
- 30.Schlegel J, Stumm G, Scherthan H, Bocker T, Zirngibl H, Rüschoff J, Hofstädter F: Comparative genomic in situ hybridization of colon carcinomas with replication error. Cancer Res 1995, 55:6002-6005 [PubMed] [Google Scholar]