

Abstract
The activation of the death receptors, tumor necrosis factor-receptor-1 (TNF-R1) or CD95, is a hallmark of inflammatory or viral liver disease. In different murine in vivo models, we found that livers depleted of γ-glutamyl-cysteinyl-glycine (GSH) by endogenous enzymatic conjugation after phorone treatment were resistant against death receptor-elicited injury as assessed by transaminase release and histopathology. In apoptotic models initiated by engagement of CD95, or by injection of TNF or lipopolysaccharide into galactosamine-sensitized mice, hepatic caspase-3-like proteases were not activated in the GSH-depleted state. Under GSH depletion, also caspase-independent, TNF-R1-mediated injury (high-dose actinomycin D or α-amanitin), as well as necrotic hepatotoxicity (high-dose lipopolysaccharide) were entirely blocked. In the T-cell-dependent model of concanavalin A-induced hepatotoxicity, GSH depletion resulted in a suppression of interferon-γ release, delay of systemic TNF release, hepatic nuclear factor-κB activation, and an abrogation of sinusoidal endothelial cell detachment as assessed by electron microscopy. When GSH depletion was initiated 3 hours after concanavalin A injection, ie, after the peak of early pro-inflammatory cytokines, livers were still protected. We conclude that sufficient hepatic GSH levels are a prerequisite for the execution of death receptor-mediated hepatocyte demise.
Apoptosis is an active and highly regulated form of cell death responsible for the cellular default demise of the hepatocyte. This process is thus in charge of tissue homeostasis and maintenance of vital function of the liver. 1-3 Accordingly, a dysregulation of apoptosis might underlay several pathophysiological disturbances of the liver, eg, hepatitis of viral or autoimmune origin, alcoholic hepatitis, acute hyperinflammatory liver failure, primary biliary cirrhosis, and toxic liver injury. 3,4 In these cases, a causal involvement of cytokines such as tumor necrosis factor (TNF) or CD95 ligand (CD95L) in the initiation of hepatocyte cell death has been described. 1,2,5,6 A better understanding of the detailed modulation of cytokine-mediated destruction of hepatic tissue is therefore the basis for future therapeutic intervention strategies in liver disease.
The tripeptide glutathione (GSH, γ-glutamyl-cysteinyl-glycine) represents the major intracellular nonprotein thiol. GSH has a central role in sulfhydryl homeostasis, serves as the major cytosolic antioxidant, and provides defense against xenobiotics as a phase II conjugate substrate. 7,8 Numerous central cellular functions are controlled by the GSH/glutathione disulfide system, eg, key enzymes of metabolism, cell growth, gene transcription, and apoptosis. 9-11 Cells therefore tightly regulate synthesis, utilization, and export of glutathione. Its intracellular concentrations are maintained within the millimolar range under normal conditions. 12 GSH is synthesized by the consecutive ATP-dependent enzymes, γ-glutamylcysteine-synthase and GSH synthase, and glutathione is primarily maintained in its reduced form by glutathione disulfide reductase using NADPH as cosubstrate. 12
The total intracellular GSH concentration varies considerably, especially in the liver, and hepatic GSH can dramatically decrease as a result of drug metabolism, 13 after oxidative stress, 10 or because of inherited deficiencies in GSH synthesis. 10 Thus, low GSH levels are observed during sepsis, acetaminophen intoxication, chronic alcohol consumption, and in acute Wilson’s disease. 10,13-17 Moreover, hepatic GSH is subject to pronounced circadian alterations. 18 A common experimental approach to create a sustained GSH-deficiency is the inhibition of glutathione synthesis by buthionine-sulfoximine, which affects all organs and requires repetitive treatment of animals. 12 In contrast, α,β-unsaturated carbonyl compounds such as diethyl maleate or phorone preferentially deplete hepatic GSH via enzymatic conjugation by GSH-transferases, followed by biliary excretion of these conjugates. 19 GSH-depleting compounds do not induce redox-stress in the liver per se. 20 However, depletion of GSH predisposes cells to oxidative injury, with the consequence that the liver toxicity of many xenobiotics in man and in animal models is greatly enhanced under this condition. Vice versa, a pharmacological enhancement of hepatic GSH renders the liver less vulnerable and protects against many direct hepatotoxins. 7,21,22
Increasing evidence argues for a dichotomal role of GSH with respect to cellular damage. In some paradigms of cell death where the primary event is apoptosis, a protective (ie, anti-apoptotic), and not an aggravating, effect of GSH depletion was reported. To date, NO-induced apoptosis of macrophages, 23 CD95-mediated apoptosis of T cells, 24 and cytokine-mediated hepatocyte apoptosis in vivo 25,26 were found to depend on a sufficient intracellular GSH level of the respective cells. In these studies, the redox sensitivity of apoptosis-executing caspases, ie, aspartate-specific cysteine proteases, 27 was hypothesized to be responsible for the observed protection because of decreased GSH levels. However, elevated intracellular GSH levels can also abrogate apoptosis in various cell lines, and GSH export has been reported to be an integral part of death receptor-mediated apoptosis in some experimental systems. 11,28,29
Concerning in vivo studies, protection by GSH depletion was reported in models of apoptotic, caspase-dependent liver injury. 25,26 On the contrary, in animal models of primary necrotic hepatotoxicity, this condition always resulted in an enhanced toxicity. 7,22 It seemed therefore appropriate to comparatively study the influence of GSH depletion on apoptotic and necrotic liver injury models. As a common feature, they all depend on the activation of death receptors, ie, either tumor necrosis factor-receptor-1 (TNF-R1) or CD95. 6,30,31
Therefore, the following models of acute inflammatory liver injury were studied: 1) in galactosamine-sensitized mice, injection of recombinant TNF (GalN/TNF), or low-dose lipopolysaccharide (GalN/LPS), or injection of activating anti-CD95 antibody (αCD95) in naive mice. These regimens commonly induce hepatocyte apoptosis via the activation of caspases. 2,6,30,32,33 2) High-dose treatment of naive mice with two hepatotoxins of fungal origin, actinomycin-D (ActD) or α-amanitin, ie, with inhibitors of transcription that strongly sensitize hepatocytes toward endogenously produced TNF and induce hepatocyte apoptosis. 34 3) Injection of naive mice with concanavalin A (Con A), a plant lectin that activates T cells and thereby induces selective liver injury. In this model, necrotic and apoptotic hepatocyte demise without caspase activation was described. 6,35-37 4) Injection of high-dose LPS (endotoxic shock model). Here, the mode of hepatocyte cell death is regarded to be necrotic, despite its dependence on TNF. 33,38
We compared these models with regard to their GSH dependence, the mode of cell death, and the activation of caspases. In the LPS shock models and the Con A models, we also examined the possible role of immunosuppression because cellular GSH levels are known to influence the immune response. 9,39,40 When GSH was depleted, the onset of liver injury was blocked in all models investigated at the target cell level, ie, the hepatocyte. Additionally, we found that in the Con A model, the structure of sinusoidal endothelial cells was preserved in the GSH-depleted state.
Materials and Methods
Materials
Phorone was obtained from Aldrich (Steinheim, Germany), benzyloxycarbonyl-Val-Ala-Asp-(OMe)-fluoromethylketone (z-VAD-fmk) from Bachem (Heidelberg, Germany), N-acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (DEVD-afc), Pefablock from Biomol (Hamburg, Germany), galactosamine (GalN) from Roth (Karlsruhe, Germany), 1-cis-chloro-2,4-dinitrobenzene and Epon from Fluka (Buchs, Switzerland), LPS (Salmonella abortus equi) from Metalon (Wusterhausen, Germany), and acetaminophen from EGA (Steinheim, Germany). Interferon-γ (IFN-γ) and recombinant murine TNF were kindly provided by Dr. G. A. Adolf (Bender & Co., Vienna, Austria). All other reagents and recombinant enzymes not further specified were purchased from Sigma (Deisenhofen, Germany).
Animal Experiments and Sampling of Material
Specific pathogen-free male BALB/c mice (∼25 g, from the in-house animal breeding station of the University of Konstanz) were maintained under controlled conditions (22°C and 55% humidity, constant day/night cycle of 12 hours) and fed a standard laboratory chow. All animals received humane care in concordance with the National Institutes of Health guidelines as well as with the legal requirements in Germany. Mice were starved overnight before the onset of experiments, which generally started at 8 am.
Phorone (250 mg/kg) and 1-cis-chloro-2,4-dinitrobenzene (100 mg/kg) were dissolved in 300 μl of vegetable oil and injected intraperitoneally, either before challenge with GalN/TNF, GalN/LPS, acetaminophen, αCD95, or Con A, or delayed 1 hour after challenge to avoid interference of the solvent with LPS, ActD, or α-amanitin at the site of injection. l-buthionin-S,R-sulfoximin (buthionine-sulfoximine, 890 mg/kg), LPS (various doses as indicated), galactosamine (GalN, 700 mg/kg), α-amanitin (3 mg/kg), and ActD (2 mg/kg) were administered intraperitoneally in 300 μl of endotoxin-free saline. Activating anti-CD95 antibody (αCD95, 2 μg per animal), recombinant murine TNF (5 to 10 μg/kg), and IFN-γ (50 μg/kg) were injected intravenously in a volume of 300 μl of saline supplemented with 0.1% human serum albumin. Con A (25 or 50 mg/kg) was given intravenously in 300 μl of endotoxin-free saline.
At the time points indicated, mice were euthanized by intravenous injection of 150 mg/kg of pentobarbital plus 0.8 mg/kg of heparin. Blood was withdrawn by cardiac puncture and centrifuged (5 minutes, 14,000 × g, 4°C) to obtain plasma, and the extent of liver damage was assessed by measuring plasma alanine aminotransferase activity with an EPOS 5060 analyzer (Netheler & Hinz, Hamburg, Germany) according to the method of Bergmeyer. 41 Blood samples for the cytokine determinations were obtained either from the tail veins using heparinized syringes, or by cardiac puncture as described above, centrifuged (5 minutes, 14,000 × g, 4°C) and stored at −80°C. To determine further organ parameters, livers were perfused for 10 seconds with a cold perfusion buffer containing 50 mmol/L phosphate, 120 mmol/K NaCl, 10 mmol/L ethylenediaminetetraacetic acid (EDTA), pH 7.4, and subsequently excised. A slice of the large anterior lobe was frozen in liquid nitrogen and stored at −80°C until the measurement of caspase-3-like activity or preparation of nuclear extracts for the nuclear factor-κB (NF-κB) electrophoretic mobility gel shift assay, or was freeze-clamped with liquid nitrogen precooled pliers, and stored at −80°C for the determination of total glutathione (GSx) (GSx = GSH + 2 × glutathione disulfide), that was quantified according to the enzymatic cycling method of Tietze as described in detail. 25,42 For histology, liver specimens were immediately cut into 1-mm-thick slices and fixed in 2.5% glutaraldehyde in phosphate buffer (0.1 mol/L, pH 7.4) for electron microscopy, or in 4% buffered formalin solution for light microscopy. Additionally, spleen cells were isolated by grinding the spleen through a steel grid (pore diameter, 100 μm) into 5 ml of RPMI 1640 medium. To determine GSx, the cells were centrifuged (5 minutes, 14,000 × g, 4°C) and lysed with 1% sulfosalicylic acid.
Cytokine Determination
All enzyme-linked immunosorbent assays were performed on flat-bottomed high-binding polystyrene microtiter plates (Greiner, Nürtingen, Germany). Antibody pairs (specific rat anti-murine mAb) were purchased from Pharmingen (San Diego, CA), except for the TNF enzyme-linked immunosorbent assay (capture: polyclonal ovine anti-mouse TNF antibody, in-house preparation, IgG fraction, 20 mg/ml; detection antibody: polyclonal anti-mouse TNF antibody from Endogen, Boston, MA). Streptavidin-peroxidase was obtained from Jackson Immuno Research (West Grove, PA), and the TMB liquid substrate system was from Sigma (Deisenhofen, Germany). Interleukin (IL)-1β was determined using a commercially available enzyme-linked immunosorbent assay kit (Endogen). The detection limits of the assays were 10 pg/ml for TNF and IFN-γ, 30 pg/ml for IL-2, 10 pg/ml for IL-4, and 15 pg/ml for IL-1β.
Measurement of Caspase-3-Like Activity
Cytosolic extracts from liver tissue were prepared by Dounce homogenization in hypotonic extraction buffer (25 mmol/L HEPES, pH 7.5; 5 mmol/L MgCl2; 1 mmol/L EGTA; 1 mmol/L Pefablock; and pepstatin, leupeptin, and aprotinin, 1 μg/ml each), subsequently centrifuged (15 minutes, 14,000 × g, 4°C) and stored at −80°C. The fluorometric DEVD-afc cleavage assay was carried out on microtiter plates (Greiner, Nürtingen, Germany) according to the method originally described by Thornberry. 43 Cytosolic extracts (10 μl, ∼1 mg/ml protein) were diluted 1:10 with substrate buffer (55 μmol/L of fluorogenic substrate DEVD-afc in 50 mmol/L HEPES, pH 7.4, 1% sucrose, 0.1% CHAPS, 10 mmol/L dithiothreitol. Blanks contained 10 μl of extraction buffer and 90 μl of substrate buffer. Generation of free 7-amino-4-trifluoromethylcoumarin (afc) at 37°C was kinetically determined by fluorescence measurement (excitation, 385 nm; emission, 505 nm) using the fluorometer plate reader Victor 2 (Wallac Instruments, Turku, Finland). Protein concentrations of the corresponding samples were estimated with the Pierce Assay (Pierce, Rockford, IL), and the activity was calculated using serially diluted standards (0 to 5 μmol/L of afc). Control experiments confirmed that the activity was linear with time and with protein concentration under the conditions described above.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared from frozen liver sections using a modification of a method by Schreiber et al. 44 Briefly, tissue samples were homogenized in 3 ml of ice-cold hypotonic buffer A (10 mmol/L HEPES pH 7.9, 10 mmol/L KCl, 0.1 mmol/L EDTA, 0.1 mmol/L EGTA, 1 mmol/L dithiothreitol, 0.5 mmol/L Pefablock) with a Dounce homogenizer. The homogenate was incubated for 10 minutes on ice and centrifuged (10 minutes, 1,000 × g, 4°C). The cell pellet was suspended in 1.4 ml of ice-cold buffer A, and 90 μl 10% solution of Nonidet P-40 solution was added followed by 10 seconds of vigorous vortexing. The suspension was incubated on ice for 10 minutes and centrifuged (30 seconds, 12,000 × g, 4°C). The supernatant was removed and the nuclear pellet was extracted with 200 μl of hypertonic buffer B (20 mmol/L HEPES, pH 7.9, 0.4 mol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 1 mmol/L Pefablock) by shaking at 4°C for 30 minutes. The extract was centrifuged (10 minutes, 12,000 × g, 4°C), and the supernatant was stored at −80°C. A double-stranded oligonucleotide probe containing a consensus binding-sequence for NF-κB (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) (Promega, Heidelberg, Germany) was 5′-end-labeled with γ[hyph]32P-ATP (3000 Ci/mmol, Amersham, Braunschweig, Germany) using T4 polynucleotide kinase (Promega, Heidelberg, Germany). Ten μg of nuclear protein were incubated at room temperature in a 15 μl reaction volume containing 10 mmol/L Tris-HCl pH 7.5, 5 × 10 4 cpm radiolabeled oligonucleotide probe, 2 μg poly(dIdC), 4% glycerol, 1 mmol/L MgCl2, 0.5 mmol/L EDTA, 50 mmol/L NaCl, and 0.5 mmol/L dithiothreitol for 20 minutes. Nucleoprotein-oligonucleotide complexes were resolved by electrophoresis in a 4.5% nondenaturing polyacrylamide gel in 0.25× Tris borate-EDTA at 100 V. The gel was autoradiographed with an intensifying screen at −80°C overnight. The specificity of the DNA-protein complex was confirmed by competition with a 100-fold excess of unlabeled NF-κB sequence (5′-GAT CGA ACT GAC CGC CCG CGG CCC GT-3′, Promega, Heidelberg, Germany).
Light and Electron Microscopy
For light microscopy, liver samples were fixed in 4% buffered formalin and embedded in paraffin. Five-micrometer sections were cut and stained with hematoxylin and eosin. For transmission electron microscopy, the liver samples were stored in 2.5% glutaraldehyde in phosphate buffer (0.1 mol/L, pH 7.4) for 2 to 3 days before further processing. Specimens were postfixed with osmium tetroxide, dehydrated in graded alcohol, and embedded in Epon. Ultrathin sections (60 to 80 nm) were cut on a Reichert ultramicrotome (Leica, Glattbrugg, Switzerland) and contrasted with uranyl acetate and lead citrate. Stained sections were reviewed in a Phillips (Dietikon, Switzerland) CM 10 electron microscope operating at 60 KV.
Statistics
All data are given as means ± SD. Statistical differences were determined by one-way analysis of variance (ANOVA) followed by Dunnett multiple comparison test of the control versus other groups. P < 0.05 was considered significant.
Results
Inhibition of Apoptotic Caspase-Dependent Liver Injury by GSH Depletion
We investigated the effect of the GSH depletor phorone in various models of acute, inflammatory liver injury that involve caspase activation. The enzymatic activity of caspase-3-like proteases, ie, caspase-3 and caspase-7 in the liver, 45 was used as a quantitative biochemical parameter for the detection of caspase-dependent hepatocyte apoptosis in vivo. 25,45-47 As shown in Table 1 ▶ , the caspase-3-like activity in liver tissue from control animals was under the detection limit of the assay, whereas in both TNF-R1-mediated models, a greatly increased caspase-3-like activity was observed 6 hours after treatment of mice with GalN/LPS or GalN/TNF, respectively. This pivotal event in TNF-R1-signaling was entirely abrogated (Table 1) ▶ in animals that were pretreated with a dose of phorone which depletes hepatic GSH stores by 90% within 30 minutes. 25
Table 1.
Prevention of Hepatotoxicity and Hepatic Caspase-3-Like Activity by Phorone Treatment in Different Models of Cytokine-Mediated Liver Injury
| Treatment | Phorone | ALT plasma activity [U/L] ± SD | DEVD-afc cleavage [μU/mg] ± SD |
|---|---|---|---|
| Control | − | 15 ± 5 (3) | ≤20 (3) |
| + | 15 ± 5 (3) | ≤20 (3) | |
| Acetaminophen | − | 230 ± 200 (3) | ≤20 (3) |
| + | 3,790 ± 1,290 (3) | ≤20 (3) | |
| GalN/LPS | − | 6,930 ± 1,070 (3) | 480 ± 20 (3) |
| + | 20 ± 10 (3) | ≤20 (3) | |
| GalN/TNF | − | 9,680 ± 4,300 (3) | 420 ± 20 (3) |
| + | 760 ± 670 (3) | 60 ± 20 (3) | |
| αCD95 | − | 9,950 ± 2,900 (12) | 120 ± 20 (3) |
| + | 600 ± 700 (12) | ≤20 (3) | |
| ActD | − | 9,800 ± 880 (3) | ≤20 (3) |
| + | 550 ± 400 (3) | ≤20 (3) | |
| α-Amanitin | − | 3,850 ± 1,800 (3) | ≤20 (3) |
| + | 340 ± 40 (3) | ≤20 (3) | |
| Con A | − | 5,950 ± 3,350 (12) | ≤20 (3) |
| + | 350 ± 550 (12) | ≤20 (3) | |
| LPS | − | 2,560 ± 1,140 (6) | ≤20 (3) |
| + | 100 ± 20 (6) | ≤20 (3) |
Mice were injected with acetaminophen (175 mg/kg), GalN/LPS (700 mg/kg; 5 μg/kg), GalN/TNF (700 mg/kg; 2 μg/kg), αCD95 (2 μg/animal), ActD (2 mg/kg), α-amanitin (3 mg/kg), Con A (25 mg/kg), or LPS (10 mg/kg), and additionally treated with phorone (250 mg/kg) as indicated at various time points (acetaminophen; GalN/TNF; αCD95; Con A: t = −30 min; GalN/LPS: t = +1 hour; ActD; α-amanitin: t = +2 hours). Mice were sacrificed for the evaluation of plasma ALT levels at 8 hours (acetaminophen; GalN/TNF; GalN/LPS; αCD95; Con A) or 20 hours (ActD; α-amanitin; LPS), and for the determination of caspase-3-like activity at 4 (αCD95), 6 (GalN/TNF; GalN/LPS), 8 (control, acetaminophen, Con A), or 20 hours (ActD; α-amanitin; LPS). The numbers of animals per group are indicated in parentheses.
Likewise, the histological examination of liver specimens from mice injected with GalN/LPS (8 hours) revealed massive hepatocyte apoptosis, as judged by the frequent appearance of nuclear fragmentation and hyperchromatic nuclear membranes (Figure 1A) ▶ as described. 32,33 At this late time point, necrotic foci, mild neutrophil infiltration, erythrocyte agglutination, and a complete destruction of the sinusoidal structure of the liver were also observed. In phorone-treated animals, signs of liver injury elicited by GalN/LPS were absent (Figure 1B) ▶ . Accordingly, the alanine aminotransferase (ALT) release was at control levels in mice that had received phorone in addition to GalN/LPS or GalN/TNF (Table 1) ▶ . In the GalN/LPS model, the systemic release of TNF was increased threefold in the phorone-treated group compared to controls (without figure), which might be specific for GSH deficiency. 48 A similar hepatoprotection on GSH depletion was observed in the CD95 model (Table 1) ▶ , 25 and, as a further control, the hepatotoxicity of acetaminophen was increased 16-fold in phorone-treated mice (Table 1) ▶ . 7,22
Figure 1.
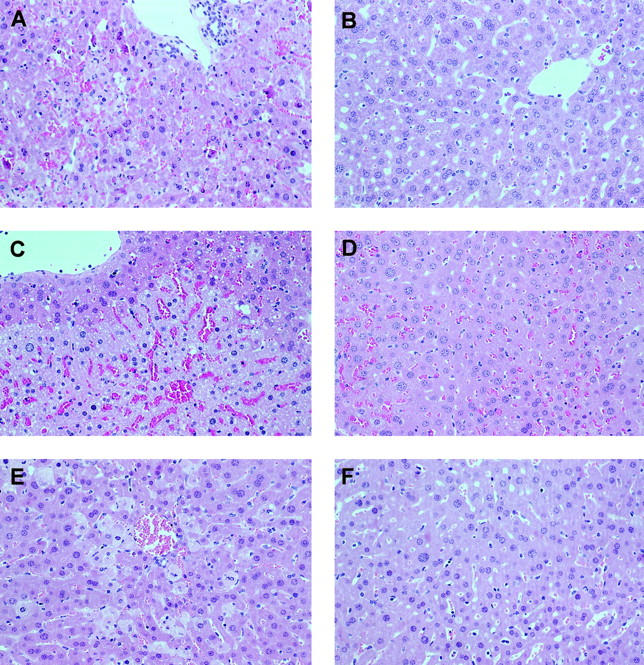
Histopathology of apoptotic and necrotic liver damage (left panel) and prevention by phorone treatment (right panel) in mice. Mice received GalN/LPS (A, 700 mg/kg; 5 μg/kg, 8 hours), and one group additionally was treated with phorone (B, 250 mg/kg, t = +1 hour); Con A (C, 25 mg/kg, 8 hours), one group treated with phorone (D, 250 mg/kg, t = +3 hours); high-dose LPS (E, 10 mg/kg, 20 hours), one group treated with phorone (F, 250 mg/kg, t = +1 hour). Original magnification, ×200.
Recently, it was reported that GSH efflux is an important event in apoptosis, also in cell death triggered by CD95, 28,49 and lowering of the hepatic GSH level provoked a profound hepatoprotection in the models described here. Therefore, we were further interested whether hepatic GSH stores are affected by massive hepatocyte apoptosis. In a separate time course experiment, mice received 2 μg per animal of the activating anti-CD95 antibody (αCD95) Jo-2, and liver samples for the determination of total glutathione (GSx) were taken by freeze-clamp technique at different times. The activity of caspase-3-like proteases became detectable at 3 hours (70 ± 10 μU/mg DEVD-afc cleavage, n = 3 per group) and peaked at 4 hours (210 ± 50 μU/mg, DEVD-afc cleavage, n = 3 per group), whereas the liver enzyme release was determined at 4 hours. Even after 8 hours, when the ALT release was exceedingly high (4,250 ± 950 U/L ALT), the concentration of total hepatic glutathione (GSx = GSH + 2 × glutathione disulfide) remained primarily at control levels (34.6 ± 3.4 nmol GSx per mg protein at 0 hours, 32.0 ± 2.7 nmol GSx per mg protein 4 hours after αCD95; 35.4 ± 1.6 nmol GSx/mg protein 8 hours after αCD95; n = 3 per group). These findings demonstrate that death receptor-dependent, caspase-mediated hepatocyte apoptosis in vivo depends on a sufficient availability of intracellular GSH, and that, in contrast to several cell lines, 28,49 hepatocytes maintain their GSH level during the process of active cell death.
Indirect Triggering of TNF-R1 by High-Dose Hepatotoxins: Apoptosis Is Independent of Caspases and Prevented by GSH Depletion
It was shown that the toxicity of α-amanitin or ActD involves a local effect of TNF on TNF-R1 of hepatocytes. By this mechanism, hepatocytes underwent apoptosis as assessed by morphological evidence and DNA laddering. 34 Because the contribution of caspases in these models had not been examined, we investigated whether caspase-3-like proteases become active after administration of α-amanitin or actinomycin D. We detected no increase in DEVD-afc-cleaving activity at the time points examined (without figure), and even 20 hours after treatment of mice, no respective enzyme activity was observed (Table 1) ▶ . To exclude a possible contribution of caspases other than DEVD-afc-cleaving enzymes, we chose a pharmacological approach, ie, repetitive treatment of mice with the irreversible, nonspecific caspase inhibitor z-VAD-fmk (10 mg/kg at t = −1 hour, 5 mg/kg at t = +1 and +3 hours). Although this inhibitor completely abrogated liver injury elicited by administration of αCD95 in a parallel control experiment (7,860 ± 270 U/L ALT 8 hours after 2 μg per animal αCD95, 35 ± 10 U/L ALT 8 hours after 2 μg per animal αCD95 + z-VAD-fmk-treatment, n = 3 per group), z-VAD-fmk had no impact on liver toxicity elicited by ActD (1,180 ± 260 U/L ALT 8 hours after 2 mg/kg ActD, 1,040 ± 110 U/L ALT 8 hours after 2 mg/kg ActD + z-VAD-fmk-treatment, n = 3 per group). These data suggest that apoptotic signaling in these models is not transmitted through caspase activation, but by alternative signaling pathways. 6
Again, the extent of liver injury after administration of α-amanitin or ActD, was dramatically decreased by >90% in mice that had been pretreated with the GSH depletor phorone (Table 1) ▶ . This experiment demonstrates that also in these caspase-independent, TNF-R1-mediated liver injury models, the toxicity of classical hepatotoxins is attenuated under conditions of low hepatic GSH.
Con A-Mediated Liver Injury: Depletion of GSH Modulates Cytokine Release and Inhibits Endothelial and Hepatocyte Cell Death
Intravenous injection of Con A in naive mice induces the polyclonal activation of T cells, which release proinflammatory cytokines (eg, TNF, IFN-γ, GM-CSF), followed by acute inflammatory liver injury that resembles immune-mediated hepatitis in humans. 35,50 Mice pretreated with the glutathione depletor phorone were protected against liver injury induced by Con A, as demonstrated by an suppression of ALT release by 94% 8 hours after Con A challenge (Table 1) ▶ . As described, 37 no activation of hepatic caspase-3-like enzymes was detectable. To demonstrate a protection independent of the compound phorone, we additionally depleted glutathione by other means in this model: 1) the aromatic compound 1-cis-chloro-2,4-dinitrobenzene (100 mg/kg, intraperitoneally) 19 depleted hepatic glutathione to 20 ± 9% of control (n = 3 per group) within 30 minutes and reduced Con A-mediated liver injury (25 mg/kg, i.v.) from 4,200 ± 490 U/L to 100 ± 20 U/L ALT 8 hours after challenge; 2) the selective, irreversible inhibitor of GSH synthesis, d,l-buthionin-S,R-sulfoximin (buthionine-sulfoximine, 890 mg/kg, intraperitoneally), 12 reduced the glutathione content of the liver to 43 ± 10% 9 hours after buthionine-sulfoximine treatment compared to untreated controls, and also conferred protection in this experiment (100 ± 60 U/L ALT 8 hours after Con A) when given 9 hours before Con A. Thus, the observed protection is unlikely to be restricted to phorone, but is because of a decrease of hepatic glutathione levels as such.
Phorone preferentially depletes hepatic glutathione, but, to a lesser extent, can also affect the glutathione content of other organs and cells. 19 We observed a drop in glutathione to 25 ± 6% in spleen cells isolated 30 minutes after injection of phorone, and it has been shown that glutathione alterations affect various lymphocyte functions in vitro and ex vivo. 9,39,40 Therefore, we tested whether phorone treatment affects the systemic release of cytokines in the Con A model. Examining the time course of IFN-γ-release, we found that the release of this pivotal cytokine 51 was strongly suppressed in animals treated with phorone and Con A compared to Con A-treated control mice (Figure 2A) ▶ , ie, an inhibition to 19% of control levels at 3 hours. In phorone-treated mice, we also noticed a remarkable delay in the release of TNF (Figure 2A) ▶ , which is produced by macrophages and T cells in this model. 36,50 As a functional consequence, this event was paralleled by a delayed activation of the transcription factor NF-κB in the liver (Figure 2B) ▶ . NF-κB activation was detectable 1 hour after Con A treatment in response to TNF as described, 52 but not before 3 hours in mice that additionally received phorone and thus were completely protected from liver damage. We further investigated the release of IL-2, a marker for T cell proliferation, and of IL-4, whose release is also crucial in the Con A model. 53,54 Similar to TNF, the release of both IL-2 and IL-4 was delayed, but after 3 hours, there was no difference in the circulating levels of these cytokines between Con A-treated mice and animals that received Con A and phorone (Figure 2C) ▶ . To elucidate whether this immunomodulation by phorone treatment might be responsible for the protection, we varied the time of phorone injection relative to the Con A challenge. Even when phorone was given 3 hours after Con A, the prevention of liver damage was almost complete, ie, a reduction by 97% compared to Con A controls, and significant reduction of ALT release was still seen by phorone application at +5 hours (Figure 2D) ▶ .
Figure 2.

Influence of glutathione depletion on Con A-induced cytokine release, NF-κB activation, and curative protection by phorone in murine Con A-mediated liver injury. Mice were injected with Con A (25 mg/kg) with and without pretreatment with phorone (t = −1 hour). A: The systemic release of IFN-γ and TNF was followed throughout a period of 5 hours (n = 3 per group). B: The activation of NF-κB was assessed at the time points indicated in Con A-treated, Con A and phorone-treated, and in a control mouse as indicated; as a control, the specificity of the DNA-protein complex was competed with an excess of unlabeled NF-κB sequence. C: The systemic release of IL-2 and IL-4 was followed over a period of 3 hours (n = 3 per group). D: Mice were treated with Con A (25 mg/kg), and 8 hours later, the release of ALT in the plasma was determined. Eight groups of mice additionally received phorone (250 mg/kg) at the time points indicated relative to Con A challenge (n = 3 per group) and Con A control (n = 9). *, P value < 0.05 versus Con A-treated controls based on ANOVA, followed by the Dunnett multiple comparison test.
We further investigated this phenomenon in detail histologically: 8 hours after Con A injection, large areas, especially in the periportal zone, displayed pronounced sinusoid destruction (Figure 1C) ▶ . The histology of the damaged areas resembled that of ischemic liver injury, including pyknotic nuclei, hepatocyte necrosis, and virtual disappearance of sinusoidal endothelial cells (SEC), and severe erythrocyte agglutination had taken place. In contrast, liver specimens from phorone-treated mice were histologically indiscernible from control livers. Notably, specimen taken from mice that received phorone 3 hours after Con A injection displayed erythrocyte agglutination, but no necrotic foci, and SEC were still present in these samples (Figure 1D) ▶ . The examination of the sinusoidal structure by electron microscopy supported these findings. In contrast to control liver specimens (Figure 3A) ▶ , SEC in livers from Con A-treated mice (8 hours) detached from neighboring hepatocytes, some sinusoids appeared entirely denuded, and agglutinating erythrocytes filled the sinusoids and were found also in the space of Disse (Figure 3B) ▶ . However, we did not detect any hepatocytes that displayed typical morphological features of apoptosis. Pretreatment of mice with phorone entirely abrogated the destruction of sinusoids (Figure 3C) ▶ . Delayed administration of phorone 3 hours after Con A selectively prevented SEC damage (Figure 3D) ▶ , and, as a consequence, hepatocyte death as judged by the reduction of ALT release (Figure 2D) ▶ , but failed to inhibit erythrocyte agglutination (Figure 3D) ▶ .
Figure 3.
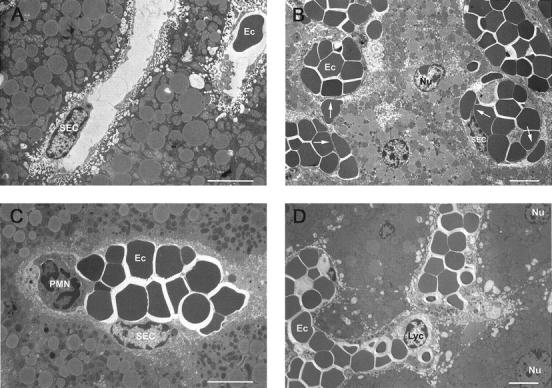
Transmission electron micrographs of hepatic sinusoids in murine Con A-mediated liver injury. A: Untreated control; B: Con A (25 mg/kg), 8 hours; C: Con A, pretreatment with phorone (−1 hour, 250 mg/kg); D: Con A, injection of phorone 3 hours after Con A. Scale bar, 6 μm. SEC, sinusoidal endothelial cell; detached SEC are marked with arrows; Ec, erythrocyte; Nu, hepatocyte nucleus; PMN, polymorphonuclear granulocyte; Lyc, lymphocyte.
These data demonstrate that in the Con A model, an experimentally induced deficiency of glutathione 1) severely affects T cell response in vivo, 2) delays, but not inhibits, NF-κB activation in the liver, which is protected from damage under this circumstance notwithstanding; and 3) directly prevents SEC and hepatocyte cell death.
Prevention of Liver Damage but Not Lethality in an Endotoxic Shock Model
We finally investigated the influence of hepatic glutathione alterations in a commonly used murine model of hyperinflammatory shock, ie, administration of high-dose lipopolysaccharide to naive mice. Here, liver injury occurs by necrosis of hepatocytes, despite its dependence on TNF. 33,38,55,56 Mice were injected with 10 mg/kg of LPS and the histological examination of a liver specimen taken 20 hours later demonstrated a typical, dispersed single-cell necrosis of hepatocytes, which did not display any zonation (Figure 1E) ▶ . The nuclei of necrotic cells appeared karyolytic, pronounced hepatocyte membrane lysis occurred, and a mild infiltration of granulocytes was observed. In mice that had received LPS and phorone, no necrotic hepatocytes were found, and the overall histology of the liver seemed normal (Figure 1F) ▶ . Likewise, the ALT release in mice treated with phorone and LPS was entirely abrogated in contrast to LPS-treated animals, and this model obviously does not involve the activation of hepatic caspase-3-like caspases (Table 1) ▶ . 33 The observed protection was not because of inhibition of cytokine release, because plasma peak level concentrations of TNF and IL-1 were not significantly altered in phorone-treated mice (Figure 4) ▶ .
Figure 4.

Circulatory release of IL-1β (A) and TNF (B) after LPS treatment of mice. Mice received 10 mg/kg LPS, and one group additionally was treated with phorone 1 hour after LPS. The release of IL-1β and TNF 2 hours after challenge was determined by enzyme-linked immunosorbent assay (n = 3 per group, P < 0.05 versus untreated controls based on ANOVA, followed by the Dunnett multiple comparison test). LPS versus LPS and phorone was considered not significant.
We compared the survival outcome in four animal models (Figure 5) ▶ , and found that hepatoprotection by phorone treatment entirely prevented lethality in two apoptotic, caspase-dependent models (GalN/LPS, GalN/TNF), as described for CD95-mediated liver injury. 25 In contrast, mortality in the Con A model and in LPS shock was not influenced by the hepatoprotective effect of phorone (Figure 5B) ▶ , indicating that liver damage in these two experimental settings is, in contrast to models of apoptotic, caspase-dependent liver injury models, not directly linked to lethality.
Figure 5.
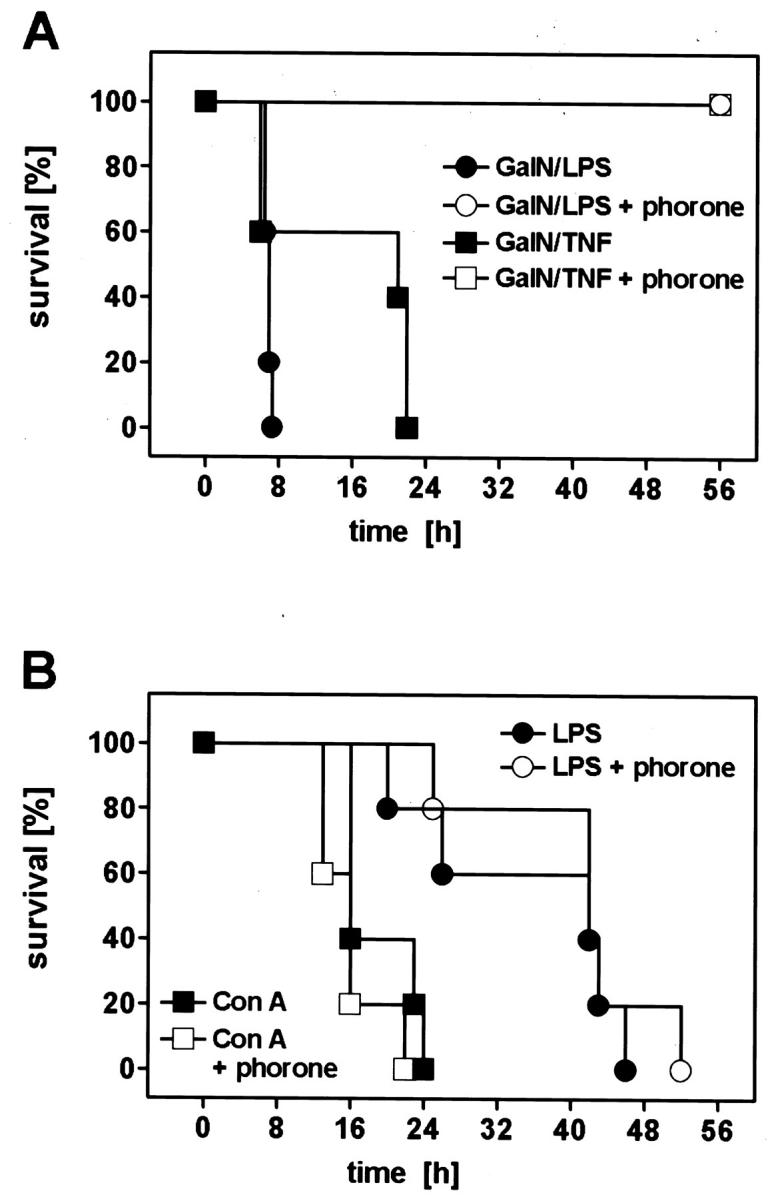
Prevention of lethality by phorone in murine GalN/LPS- and GalN/TNF-mediated liver damage (A), but not in the LPS-shock- and Con A-model (B). A: Animals were injected with GalN/LPS (700 mg/kg; 10 μg/kg) or GalN/TNF (700 mg/kg; 5 μg/kg), respectively, and two additional groups received phorone (250 mg/kg, t = −1 hour for GalN/TNF, t = +1 hour for GalN/LPS). The survival time was monitored for the time indicated. B: Mice were treated with either Con A (50 mg/kg), or high-dose LPS (10 mg/kg), and two additional groups received phorone (250 mg/kg, t = −1 hour for Con A, t = +1 hour for LPS). The survival time was monitored for the time indicated.
Discussion
Possible Mechanistic Interactions of Cell Death and GSH Deficiency
Direct Inhibition of Receptor-Dependent Hepatocyte Death
Examining various murine models of cytokine-mediated liver injury, this study demonstrates that the destruction of liver tissue does not take place when glutathione is not available in sufficient amounts. This holds true for models involving either apoptosis or necrosis as the primary hepatocyte insult (Table 1 ▶ , Figure 1 ▶ ). In each of these models, the activation of death receptors, ie, TNF-R1 or CD95, is necessary for the induction of liver damage. 2,6,33,34 To allow a concise discussion of the mechanistic implications of our findings, we present the key points of our hypotheses in the form of two graphs in Figure 6 ▶ .
Figure 6.

Hypothetical interaction sites of GSH deficiency (A) with death receptor-mediated signaling in the hepatocyte (B) with liver injury mechanisms initiated by intravenous Con A. DISC, death-inducing signaling complex; HC, hepatocyte. For further explanation, see text.
Within the signaling cascade of death receptors, we tentatively position the interaction of GSH with the execution of apoptosis at the death-inducing signaling complex or upstream signaling events, and before the downstream signals including impairment of mitochondrial function (Figure 6A) ▶ . The following arguments support this theory: 1) in a previous study, we localized the site of death signal inhibition in CD95-mediated liver injury between CD95 engagement and activation of downstream caspase-3-like caspases; 25 2) mitochondrial dysfunction serves as an amplifier and thus marks the point of no return in several cell death paradigms, including TNF-mediated apoptosis; 57,58 3) consistently, TNF-mediated hepatocyte demise was aggravated by lowering mitochondrial GSH in vitro, ie, by downstream potentiation of the death signal; 59 4) GSH depletion prevented hepatocyte death in a sustained fashion, and allowed long-term survival in three models of lethal, apoptotic liver injury (Figure 5) ▶ . 25 Further investigations in vitro will discriminate whether the death-inducing signaling complex formation or upstream death-promoting signals, ie, caspase-8, kinases, or alternative pathways, display a relevant dependence on an intact thiol status of the respective cell.
Multiple Mechanistic Interference Sites in Con A-Mediated Liver Injury
The Con A model differs from the other experimental models of TNF-dependent liver injury with regard to TNF receptor subtype redundancy, 60 postreceptor triggering of cell death programs, 6,37,52,61 the role of endothelial cells, 62 and the involvement of further cytokines in addition to TNF, 36,63 ie, IL-4 53 and IFN-γ, 51 which all amplify the overactivation of immune response and causally contribute to liver injury (Figure 6B) ▶ .
Because T cell function, 9,39 cytokine release, 64 and NF-κB activation 65 are modulated by GSH, it was probable that an insufficient availability of GSH would prevent liver damage after Con A injection. A diminished T cell responsiveness, ie, partial suppression or delayed release of circulating TNF, IFN-γ, or IL-4, and a delayed induction of NF-κB (Figure 2, A–C) ▶ , alone might already explain protection. To examine whether IFN-γ suppression could be the reason for prevention of Con A hepatotoxicity, 51 we supplemented phorone/Con A-treated mice with IFN-γ (50 μg/kg, i.v., t = −15 minutes before Con A). Although this treatment restored the level of systemic IFN-γ comparable to that in control Con A-treated mice (7,330 ± 900 pg/ml at 30 minutes after IFN-γ treatment), it failed to restore sensitivity to Con A (not shown). This implies that T cell suppression alone is unlikely to be a sufficient explanation for the protective action of GSH depletors against Con A hepatotoxicity (Figure 6B) ▶ .
Although the observed suppression of early pro-inflammatory cytokine release after depletion of GSH (Figure 2, A and C) ▶ is likely to contribute to desensitization toward Con A, it also seems feasible that post-TNF-R-signaling events leading to hepatocyte death are disrupted under this condition. In particular, two signal transduction events known to be crucial in the Con A model, ie, transactivation of NF-κB and activation of caspase-1, 37,52,66 are known to be controlled by thiols. 24,25,65 Therefore, a further and independent effect of a lack of GSH could be located in the insufficient activation of these two factors at the hepatocyte level as depicted in Figure 6B ▶ .
Endothelial damage has been poorly investigated in Con A hepatotoxicity. Fluorescein isothiocyanate-labeled Con A given in vivo is immediately and exclusively found in hepatic sinusoids, 36 and a selective damage of SEC was reported to be an early event in liver destruction. 62 In contrast, endothelial damage in other organs has not been reported. By intravital microscopy, it was demonstrated that after Con A injection, T cells primarily adhere to the endothelium in the periportal area, 67 where liver damage was most pronounced after 8 hours (Figure 1C) ▶ . In extension of these observations, we here present electron microscopic evidence for a prominent SEC cell destruction after Con A injection, whereas no typical signs of hepatocyte apoptosis were visible (Figure 3B) ▶ . Without previous knowledge of the experimental model, we would have characterized the lesions as being similar to those seen after an ischemic insult (Figure 1C) ▶ .
Mode of Hepatocyte Demise in Con A-Mediated Liver Injury
Initially, the actual mode of hepatocyte cell demise in the Con A model has been characterized as apoptotic, 36 and this notion was repeatedly confirmed by the TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling) assay. 66,68 Predominantly necrotic hepatocyte death was also reported 69 (this study). To appreciate the significance of these previous findings, three relevant methodological aspects must be discussed nowadays: 1) the detection of oligosomal DNA fragmentation and DNA laddering in crude liver homogenate fails to discriminate between apoptosis of hepatocytes and other hepatic cell populations; 32 2) the TUNEL assay does not unequivocally discriminate between apoptotic and necrotic cells; 70 and 3) none of the previous studies investigated the morphology by electron microscopy. Based on these three independent methodological reservations, we conclude from the absence of hyperchromatic hepatocyte nuclei in many liver sections from Con A experiments (Figure 3B) ▶ that apoptotic hepatocyte death is of minor importance in this model. This conclusion is supported by the recent findings that Con A-induced liver injury is neither associated with hepatic caspase activation, nor sensitive toward inhibition by the potent broad-spectrum caspase inhibitor z-VAD-fmk. 37 Considering the observation of early SEC damage, we propose that hepatocyte demise in the Con A model occurs in a necrotic way and as a result of endothelial disruption and ischemic conditions, as outlined in Figure 6B ▶ . The observation that in endothelial cells GSH is decreased by diethyl maleate 71 demonstrates that these cells contain active GSH transferases, ie, it is very likely that phorone will also deplete endothelial GSH. The mode of SEC death on Con A injection and its modulation by GSH requires attention and detailed further experiments.
Although phorone treatment prevented liver damage in the Con A model, it failed to rescue mice from a lethal Con A challenge (Figure 5B) ▶ , indicating that Con A causes animal death independent of hepatic damage. We interpret this situation as a generalized, fatal condition comparable to LPS- or superantigen-induced lethal shock. 72 Our experimental findings allow to identify two different events as possible causal lethal conditions after Con A injection (Figure 6B) ▶ , which were both insensitive toward phorone treatment: 1) severe hemorrhage, as evidenced by histology (Figure 1, C and D ▶ ; Figure 3, B–D ▶ ) and by a dramatic increase in spleen weight 8 hours after Con A treatment (untreated control: 90 ± 10 mg; Con A (25 mg/kg): 230 ± 20 mg; 250 mg/kg phorone + Con A (25 mg/kg): 210 ± 20 mg; n = 3 per group), and 2) exceedingly high levels of proinflammatory circulating cytokines from 3 hours onwards (Figure 2, A and C) ▶ .
Impaired Apoptosis in GSH Deficiency: Significance in Liver Disease
Receptor-mediated hepatocyte death in an autocrine or paracrine fashion has been recognized to be an important event in toxic liver injury, and damaged or toxin-stressed hepatocytes rapidly up-regulate their death receptors TNF-R1 or CD95. 1,34,73,74 When the total initial tissue lesion is below the threshold that causes organ failure, apoptotic hepatocytes are usually removed by professional phagocytes or by neighboring parenchymal cells. 2,38 Hence, any dysregulation of this active cell death program may have negative consequences for the organ.
The hepatic metabolism of the analgesic drug acetaminophen depletes liver GSH via phase II conjugation. 13 When overdosed, hepatocytes are damaged by formation of reactive metabolites. 55,75 In this situation of toxic stress and lack of GSH, the possibility raises that because of the blocked apoptotic program, necrotic hepatocyte death will predominate. Consistently, it was shown that CD95-mediated liver apoptosis was blocked in acetaminophen-treated mice. 76 Two further examples for liver disease associated with pathological decreased hepatic GSH levels are Wilson’s disease and chronic alcohol consumption. 15-17,77 In both cases, the involvement of death receptor activation has been described, 74,78 and little apoptosis, but rather necrotic liver damage and cirrhosis have been seen. 79,80
In summary, our data demonstrate that a hepatic GSH deficiency can disturb the physiological balance between apoptosis and necrosis, inflammation, and proliferation. This consideration implies that a dysregulation of receptor-mediated cell death should be considered as a determinant for the clinical progression of liver disease, eg, for the development of inflammation and fibrosis.
Acknowledgments
We thank Mrs. U. Gebert, Mrs. M. Häberlin, and Ms. T. Menard for their excellent technical assistance; Dr. A. K. Kiemer for performing the electrophoretic mobility shift assay; and Dr. G. R. Adolf for providing recombinant murine TNF and IFN-γ.
Footnotes
Address reprint requests to Professor A. Wendel, Faculty of Biology, University of Konstanz, POB M667, D-78457 Konstanz, Germany. E-mail: albrecht.wendel@uni-konstanz.de.
Supported by the Deutsche Forschungsgemeinschaft (research group: “Mechanisms of Endogenous Tissue Destruction,” grant We 686/18).
References
- 1.Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W, Krammer PH, Runke LL: Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med 1995, 182:1223-1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leist M, Gantner F, Künstle G, Wendel A: Cytokine-mediated hepatic apoptosis. Rev Physiol Biochem Pharmacol 1998, 133:109-155 [DOI] [PubMed] [Google Scholar]
- 3.Patel T, Roberts LR, Jones BA, Gores GJ: Dysregulation of apoptosis as a mechanism of liver disease: an overview. Semin Liver Dis 1998, 18:105-114 [DOI] [PubMed] [Google Scholar]
- 4.Valente M, Calabrese F: Liver and apoptosis. Ital J Gastroenterol Hepatol 1999, 31:73-77 [PubMed] [Google Scholar]
- 5.Galle PR: Apoptosis in liver disease. J Hepatol 1997, 27:405-412 [DOI] [PubMed] [Google Scholar]
- 6.Bradham CA, Plumpe J, Manns MP, Brenner DA, Trautwein C: Mechanisms of hepatic toxicity I. TNF-induced liver injury. Am J Physiol 1998, 275:G387-G392 [DOI] [PubMed] [Google Scholar]
- 7.Jones TW, Thor H, Orrenius S: Cellular defense mechanisms against toxic substances. Arch Toxicol Suppl 1986, 9:259-271 [DOI] [PubMed] [Google Scholar]
- 8.Meister A: On the antioxidant effects of ascorbic acid and glutathione. Biochem Pharmacol 1992, 44:1905-1915 [DOI] [PubMed] [Google Scholar]
- 9.Dröge W, Schulze-Osthoff K, Mihm S, Galter D, Schenk H, Eck HP, Roth S, Gmünder H: Functions of glutathione and glutathione disulfide in immunology and immunopathology. FASEB J 1994, 8:1131-1138 [PubMed] [Google Scholar]
- 10.Uhlig S, Wendel A: The physiological consequences of glutathione variations. Life Sci 1992, 51:1083-1094 [DOI] [PubMed] [Google Scholar]
- 11.Hall AG: The role of glutathione in the regulation of apoptosis. Eur J Clin Invest 1999, 29:238-245 [DOI] [PubMed] [Google Scholar]
- 12.Anderson ME: Glutathione and glutathione delivery compounds. Adv Pharmacol 1997, 18:65-78 [DOI] [PubMed] [Google Scholar]
- 13.Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB: Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973, 187:211-217 [PubMed] [Google Scholar]
- 14.Keller GA, Barke R, Harty JT, Humphrey E, Simmons RL: Decreased hepatic glutathione levels in septic shock. Predisposition of hepatocytes to oxidative stress: an experimental approach. Arch Surg 1985, 120:941-945 [DOI] [PubMed] [Google Scholar]
- 15.Summer KH, Eisenburg J: Low content of hepatic reduced glutathione in patients with Wilson’s disease. Biochem Med 1985, 34:107-111 [DOI] [PubMed] [Google Scholar]
- 16.Lieber CS: Hepatic and metabolic effects of ethanol: pathogenesis and prevention. Ann Med 1994, 26:325-330 [DOI] [PubMed] [Google Scholar]
- 17.Videla LA, Valenzuela A: Alcohol ingestion, liver glutathione and lipoperoxidation: metabolic interrelations and pathological implications. Life Sci 1982, 31:2395-2407 [DOI] [PubMed] [Google Scholar]
- 18.Jaeschke H, Wendel A: Diurnal fluctuation and pharmacological alteration of mouse organ glutathione content. Biochem Pharmacol 1985, 34:1029-1033 [DOI] [PubMed] [Google Scholar]
- 19.Plummer JL, Smith BR, Sies H, Bend JR: Chemical depletion of glutathione in vivo. Methods Enzymol 1981, 77:50-59 [DOI] [PubMed] [Google Scholar]
- 20.Mehmetcik G, Alptekin N, Toker G, Uysal M: Mitochondrial lipid peroxides and antioxidant enzymes in the liver following phorone-induced glutathione depletion. Res Commun Mol Pathol Pharmacol 1997, 96:353-356 [PubMed] [Google Scholar]
- 21.Prescott LF: Glutathione: a protective mechanism against hepatotoxicity. Biochem Soc Trans 1982, 10:84-85 [DOI] [PubMed] [Google Scholar]
- 22.Wendel A, Tiegs G: Manipulation of liver glutathione status—a double-edged sword. Vina J eds. Glutathione: Metabolism and Physiological Functions. 1989, :pp 21-28 CRC Press, Boca Raton [Google Scholar]
- 23.Boggs SE, McCormick TS, Lapetina EG: Glutathione levels determine apoptosis in macrophages. Biochem Biophys Res Commun 1998, 247:229-233 [DOI] [PubMed] [Google Scholar]
- 24.Hampton MB, Orrenius S: Redox regulation of apoptotic cell death. Biofactors 1998, 8:1-5 [DOI] [PubMed] [Google Scholar]
- 25.Hentze H, Künstle G, Volbracht C, Ertel W, Wendel A: CD95-mediated murine hepatic apoptosis requires an intact glutathione status. Hepatology 1999, 30:177-185 [DOI] [PubMed] [Google Scholar]
- 26.Jones JJ, Fan J, Nathens AB, Kapus A, Shekhman M, Marshall JC, Parodo J, Rotstein OD: Redox manipulation using the thiol-oxidizing agent diethyl maleate prevents hepatocellular necrosis and apoptosis in a rodent endotoxemia model. Hepatology 1999, 30:714-724 [DOI] [PubMed] [Google Scholar]
- 27.Thornberry NA, Lazebnik Y: Caspases: enemies within. Science 1998, 281:1312-1316 [DOI] [PubMed] [Google Scholar]
- 28.van den Dobbelsteen DJ, Nobel CSI, Schlegel J, Cotgreave IA, Orrenius S, Slater FG: Rapid and specific efflux of reduced glutathione during apoptosis induced by anti-Fas/APO-1 antibody. J Biol Chem 1996, 271:15420-15427 [DOI] [PubMed] [Google Scholar]
- 29.Manna SK, Kuo MT, Aggarwal BB: Overexpression of gamma-glutamylcysteine synthetase suppresses tumor necrosis factor-induced apoptosis and activation of nuclear transcription factor-kappa B and activator protein-1. Oncogene 1999, 18:4371-4382 [DOI] [PubMed] [Google Scholar]
- 30.Leist M, Gantner F, Künstle G, Bohlinger I, Tiegs G, Bluethmann H, Wendel A: The 55-kD tumor necrosis factor receptor and CD95 independently signal murine hepatocyte apoptosis and subsequent liver failure. Mol Med 1996, 2:109-124 [PMC free article] [PubMed] [Google Scholar]
- 31.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME: Apoptosis signaling by death receptors. Eur J Biochem 1998, 254:439-459 [DOI] [PubMed] [Google Scholar]
- 32.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A: Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol 1995, 146:1220-1234 [PMC free article] [PubMed] [Google Scholar]
- 33.Mignon A, Rouquet N, Fabre M, Martin S, Pages JC, Dhainaut JF, Kahn A, Briand P, Joulin V: LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med 1999, 159:1308-1315 [DOI] [PubMed] [Google Scholar]
- 34.Leist M, Gantner F, Naumann H, Bluethmann H, Vogt K, Brigelius-Flohe R, Nicotera P, Volk HD, Wendel A: Tumor necrosis factor-induced apoptosis during the poisoning of mice with hepatotoxins. Gastroenterology 1997, 112:923-934 [DOI] [PubMed] [Google Scholar]
- 35.Tiegs G, Hentschel J, Wendel A: A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest 1992, 90:196-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G: Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology 1995, 21:190-198 [DOI] [PubMed] [Google Scholar]
- 37.Künstle G, Hentze H, Germann PG, Tiegs G, Meergans T, Wendel A: Concanavalin A hepatotoxicity in mice: TNF-mediated organ failure independent of caspase-3-like protease activation. Hepatology 1999, 30:1241-1251 [DOI] [PubMed] [Google Scholar]
- 38.Bohlinger I, Leist M, Gantner F, Angermüller S, Tiegs G, Wendel A: DNA fragmentation in mouse organs during endotoxic shock. Am J Pathol 1996, 149:1381-1393 [PMC free article] [PubMed] [Google Scholar]
- 39.Gmünder H, Dröge W: Differential effects of glutathione depletion on T cell subsets. Cell Immunol 1991, 138:229-237 [DOI] [PubMed] [Google Scholar]
- 40.Robinson MK, Rodrick ML, Jacobs DO, Rounds JD, Collins KH, Saporoschetz IB, Mannick JA, Wilmore DW: Glutathione depletion in rats impairs T-cell and macrophage immune function. Arch Surg 1993, 128:29-34 [DOI] [PubMed] [Google Scholar]
- 41.Bergmeyer HU: Methods of Enzymatic Analysis, vol 3. Weinheim, Verlag Chemie, 1983, pp 1–605
- 42.Tietze F: Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 1969, 27:502-522 [DOI] [PubMed] [Google Scholar]
- 43.Thornberry NA: Interleukin-1beta converting enzyme. Methods Enzymol 1994, 244:615-631 [DOI] [PubMed] [Google Scholar]
- 44.Schreiber E, Matthias P, Müller MM, Schaffner W: Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 1989, 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones RA, Johnson VL, Buck NR, Dobrota M, Hinton RH, Chow SC, Kass GE: Fas-mediated apoptosis in mouse hepatocytes involves the processing and activation of caspases. Hepatology 1998, 27:1632-1642 [DOI] [PubMed] [Google Scholar]
- 46.Enari M, Talanian RV, Wong WW, Nagata S: Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature 1996, 380:723-726 [DOI] [PubMed] [Google Scholar]
- 47.Jaeschke H, Fisher MA, Lawson JA, Simmons CA, Farhood A, Jones DA: Activation of caspase 3 (CPP32)-like proteases is essential for TNF-alpha-induced hepatic parenchymal cell apoptosis and neutrophil-mediated necrosis in a murine endotoxin shock model. J Immunol 1998, 160:3480-3486 [PubMed] [Google Scholar]
- 48.Peristeris P, Clark BD, Gatti S, Faggioni R, Mantovani A, Mengozzi M, Orencole SF, Sironi M, Ghezzi P: N-acetylcysteine and glutathione as inhibitors of tumor necrosis factor production. Cell Immunol 1992, 140:390-399 [DOI] [PubMed] [Google Scholar]
- 49.Ghibelli L, Fanelli C, Rotilio G, Lafavia E, Coppola S, Colussi C, Civitareale P, Ciriolo MR: Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J 1998, 12:479-486 [DOI] [PubMed] [Google Scholar]
- 50.Gantner F, Leist M, Küsters S, Vogt K, Volk HD, Tiegs G: T cell stimulus-induced crosstalk between lymphocytes and liver macrophages results in augmented cytokine release. Exp Cell Res 1996, 229:137-146 [DOI] [PubMed] [Google Scholar]
- 51.Küsters S, Gantner F, Künstle G, Tiegs G: Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology 1996, 111:462-471 [DOI] [PubMed] [Google Scholar]
- 52.Trautwein C, Rakemann T, Brenner DA, Streetz K, Licato L, Manns MP, Tiegs G: Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology 1998, 114:1035-1045 [DOI] [PubMed] [Google Scholar]
- 53.Toyabe S, Seki S, Iiai T, Takeda K, Shirai K, Watanabe H, Hiraide H, Uchiyama M, Abo T: Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J Immunol 1997, 159:1537-1542 [PubMed] [Google Scholar]
- 54.Nishikage T, Seki S, Toyabe S, Abo T, Kagata Y, Iwai T, Hiraide H: Inhibition of concanavalin A-induced hepatic injury of mice by bacterial lipopolysaccharide via the induction of IL-6 and the subsequent reduction of IL-4: the cytokine milieu of concanavalin A hepatitis. J Hepatol 1999, 31:18-26 [DOI] [PubMed] [Google Scholar]
- 55.Losser MR, Payen D: Mechanisms of liver damage. Semin Liver Dis 1996, 16:357-367 [DOI] [PubMed] [Google Scholar]
- 56.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Krönke M, Mak TW: Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 1993, 73:457–467 [DOI] [PubMed]
- 57.Bradham CA, Qian T, Streetz K, Trautwein C, Brenner DA, Lemasters JJ: The mitochondrial permeability transition is required for tumor necrosis factor alpha-mediated apoptosis and cytochrome c release. Mol Cell Biol 1998, 18:6353-6364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1311 [DOI] [PubMed] [Google Scholar]
- 59.Colell A, Garcia-Ruiz C, Miranda M, Ardite E, Mari M, Morales A, Corrales F, Kaplowitz N, Fernandez-Checa JC: Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998, 115:1541-1551 [DOI] [PubMed] [Google Scholar]
- 60.Küsters S, Tiegs G, Alexopoulou L, Pasparakis M, Douni E, Künstle G, Bluethmann H, Wendel A, Pfizenmaier K, Kollias G, Grell M: In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur J Immunol 1997, 27:2870-2875 [DOI] [PubMed] [Google Scholar]
- 61.Trautwein C, Rakemann T, Malek NP, Plumpe J, Tiegs G, Manns MP: Concanavalin A-induced liver injury triggers hepatocyte proliferation. J Clin Invest 1998, 101:1960-1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Knolle PA, Gerken G, Löser E, Dienes HP, Gantner F, Tiegs G, Meyer zum Büschenfelde KH, Lohse AW: Role of sinusoidal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 1996, 24:824–829 [DOI] [PubMed]
- 63.Mizuhara H, O’Neill E, Seki N, Ogawa T, Kusunoki C, Otsuka K, Satoh S, Niwa M, Senoh H, Fujiwara H: T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med 1994, 179:1529-1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gosset P, Wallaert B, Tonnel AB, Fourneau C: Thiol regulation of the production of TNF-alpha, IL-6 and IL-8 by human alveolar macrophages. Eur Respir J 1999, 14:98-105 [DOI] [PubMed] [Google Scholar]
- 65.Mihm S, Galter D, Dröge W: Modulation of transcription factor NF kappa B activity by intracellular glutathione levels and by variations of the extracellular cysteine supply. FASEB J 1995, 9:246-252 [DOI] [PubMed] [Google Scholar]
- 66.Ksontini R, Colagiovanni DB, Josephs MD, Edwards CKR, Tannahill CL, Solorzano CC, Norman J, Denham W, Clare-Salzler M, MacKay SL, Moldawer LL: Disparate roles for TNF-alpha and Fas ligand in concanavalin A-induced hepatitis. J Immunol 1998, 160:4082-4089 [PubMed] [Google Scholar]
- 67.Hokari R, Miura S, Fujimori H, Koseki S, Tsuzuki Y, Kimura H, Higuchi H, Serizawa H, Granger DN, Ishii H: Altered migration of gut-derived T lymphocytes after activation with concanavalin A. Am J Physiol 1999, 277:G763-G772 [DOI] [PubMed] [Google Scholar]
- 68.Tagawa Y, Kakuta S, Iwakura Y: Involvement of Fas/Fas ligand system-mediated apoptosis in the development of concanavalin A-induced hepatitis. Eur J Immunol 1998, 28:4105-4113 [DOI] [PubMed] [Google Scholar]
- 69.Kimura K, Ando K, Ohnishi H, Ishikawa T, Kakumu S, Takemura M, Muto Y, Moriwaki H: Immunopathogenesis of hepatic fibrosis in chronic liver injury induced by repeatedly administered concanavalin A. Int Immunol 1999, 11:1491-1500 [DOI] [PubMed] [Google Scholar]
- 70.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R: In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 1995, 21:1465-1468 [DOI] [PubMed] [Google Scholar]
- 71.Cotgreave IA, Constantin-Teodosiu, Moldeus P: Nonxenobiotic manipulation and sulfur precursor specificity of human endothelial cell glutathione. J Appl Physiol 1991, 70:1220–1227 [DOI] [PubMed]
- 72.Glauser MP: The inflammatory cytokines. New developments in the pathophysiology and treatment of septic shock. Drugs 1996, 52:9-17 [DOI] [PubMed] [Google Scholar]
- 73.Müller M, Strand S, Hug H, Heinemann EM, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR: Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest 1997, 99:403-413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strand S, Hofmann WJ, Grambihler A, Hug H, Volkmann M, Otto G, Wesch H, Mariani SM, Hack V, Stremmel W, Krammer PH, Galle PR: Hepatic failure and liver cell damage in acute Wilson’s disease involve CD95 (APO-1/Fas) mediated apoptosis. Nat Med 1998, 4:588-593 [DOI] [PubMed] [Google Scholar]
- 75.Wendel A: Measurement of in vivo lipid peroxidation and toxicological significance. Free Radic Biol Med 1987, 3:355-358 [DOI] [PubMed] [Google Scholar]
- 76.Lawson JA, Fisher MA, Simmons CA, Farhood A, Jaeschke H: Inhibition of Fas receptor (CD95)-induced hepatic caspase activation and apoptosis by acetaminophen in mice. Toxicol Appl Pharmacol 1999, 156:179-186 [DOI] [PubMed] [Google Scholar]
- 77.Ferenci P: Wilson’s disease. Ital J Gastroenterol Hepatol 1999, 31:416-425 [PubMed] [Google Scholar]
- 78.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG: Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999, 117:942-952 [DOI] [PubMed] [Google Scholar]
- 79.Stromeyer FW, Ishak KG: Histology of the liver in Wilson’s disease: a study of 34 cases. Am J Clin Pathol 1980, 73:12-24 [DOI] [PubMed] [Google Scholar]
- 80.Hall PD: Pathological spectrum of alcoholic liver disease. Alcohol Alcohol Suppl 1994, 2:303-313 [PubMed] [Google Scholar]