

Abstract
The immune response to influenza A virus is characterized by an influx of both macrophages and T lymphocytes into the lungs of the infected host, accompanied by induced expression of a number of CC chemokines. CC chemokine receptors CCR5 and CCR2 are both expressed on activated macrophages and T cells. We examined how the absence of these chemokine receptors would affect pulmonary chemokine expression and induced leukocyte recruitment by infecting CCR5-deficient mice and CCR2-deficient mice with a mouse-adapted strain of influenza A virus. CCR5−/− mice displayed increased mortality rates associated with acute, severe pneumonitis, whereas CCR2−/− mice were protected from the early pathological manifestations of influenza because of defective macrophage recruitment. This delay in macrophage accumulation in CCR2−/− mice caused a subsequent delay in T cell migration, which correlated with high pulmonary viral titers at early time points. Infected CCR5−/− mice and CCR2−/− mice both exhibited increased expression of the gene for MCP-1, the major ligand for CCR2−/− and a key regulator of induced macrophage migration. These studies illustrate the very different roles that CCR5 and CCR2 play in the macrophage response to influenza infection and demonstrate how defects in macrophage recruitment affect the normal development of the cell-mediated immune response.
Chemoattractant cytokines, or chemokines, are small, inducible, pro-inflammatory molecules involved in the recruitment of leukocytes to sites of injury or infection. More recently, research has highlighted the role of chemokines in the regulation of certain aspects of hematopoiesis 1 and the normal trafficking of residential leukocytes to both lymphoid and nonlymphoid organs. 2,3 The biological effects of chemokines are mediated through their interaction with members of a large group of seven transmembrane-spanning (serpentine), G-protein-coupled receptors. 4 Although virtually every tissue in the body can produce chemokines, chemokine receptor expression is generally limited to activated leukocyte populations. CXC chemokine receptors were initially identified on neutrophils, and have thus been traditionally associated with acute inflammation. In contrast, CC chemokine receptors are expressed on a wide variety of cells, including eosinophils, basophils, monocytes, macrophages, and T lymphocytes. 5,6 Therefore, it is not surprising that CC chemokines have been implicated in a number of more chronic diseases including atherosclerosis, 7-9 arthritis, 10 glomerulonephritis, 11 and allograft rejection. 12
The influx of mononuclear inflammatory cells into infected host tissues has long been recognized as the hallmark of the host defense mechanism against viral infection. 13,14 This cell-mediated immunity is the predominant mechanism for containment and recovery from primary viral infections. Consequently, patients who are immunocompromised, including those who are elderly or HIV-infected, are more susceptible to the extreme manifestations of viral infection. 15 In general, the pathogenesis of lytic viral infections, such as influenza infections, can be divided into two phases, the cellular events that precede T cell invasion and those that follow it. The initial attack of influenza A on respiratory epithelial cells causes the extravasation of small numbers of blood-derived neutrophils, followed by larger numbers of blood monocytes/macrophages into the infected lung. During this period, viral replication continues in the epithelial cells and infection spreads to the collecting macrophages. 16-18 These processes alone are not able to clear the virus from the lung. By day 7, CD8+ cytotoxic T cells (CTLs) from nearby mediastinal lymph nodes (MLNs) begin to accumulate in the infected lungs and the efficient process of T-cell mediated viral clearance begins. 15,16,19 Although this cell-mediated mechanism is required for host protection and recovery, the overwhelming inflammatory cell accumulation can be harmful to the infected host by aggravating clinical symptoms and contributing to potentially lethal lung pathology. 20,21 Although it is still unclear as to which inflammatory proteins mediate influenza A-induced leukocyte recruitment, virologists have recently begun to focus on the potential role of chemokines in this process. 14,22
A number of in vitro studies have associated influenza A infection with increased monocyte and epithelial cell expression of several CC chemokines, including RANTES, 17,23,24 MIP-1α, and MCP-1, 17,24,25 and the CXC chemokine, IP-10. 25 Additionally, MIP-1α-deficient mice infected with the influenza A virus displayed a marked reduction in virus-induced pneumonitis, but a significant increase in viral titers at days 6 and 7 postinfection. 22 This work implicated MIP-1α as a major T cell chemoattractant functioning in the lungs of influenza-infected mice. Although it is known that macrophages are responsible for the production of inflammatory mediators (including MIP-1α) that regulate the T cell response to influenza A virus infection, 26 it is not known which, if any, chemokines promote the early activation and migration of these macrophages to infected lungs.
In recent years insight into the pathogenesis of influenza A has been gained through the use of murine transgenic and gene knockout animal models. 21,22,27-31 In this report, we have explored the roles of two chemokine receptors, CCR5 and CCR2, in influenza A-induced pulmonary macrophage recruitment. CCR2-deficient mice, in particular, as well as MCP-1-deficient mice have been previously reported to have significant defects in macrophage recruitment. 32-35 In these studies, we infected mice deficient in either CCR5, a receptor for MIP-1α, MIP-1β, and RANTES, or CCR2, the primary receptor for MCP-1, with a mouse-adapted influenza A virus (PR/8/34). We examined the pathological manifestations of the virus as well as events occurring simultaneously in the regional lymph nodes. We found that CCR5 deficiency leads to a hyperacute inflammatory response that is often fatal, whereas CCR2 deficiency leads to a milder inflammatory response with reduced lung pathology and increased survival rates.
Materials and Methods
Animals
CCR2−/−, MIP-1α−/−, and +/+ control mice were generated and maintained as previously described 22,32 and were on mixed 129/Ola X C57BL/6J genetic background. Wild-type control mice carry the CCR gene cluster derived from the 129/Ola strain. CCR2−/− mice were bred with MIP-1α−/− mice to obtain mice that were heterozygous for both mutations (CCR2+/−/MIP-1α+/−). The double heterozygotes were then intercrossed to generate mice which were doubly deficient (CCR2−/−/MIP-1α−/−). CCR2 and MIP-1α genotypes were determined by methods that have been described previously. 21,32 CCR5−/− mice were generated by targeted replacement of the entire CCR5 coding region with a neomycin-resistance gene in embryonic stem cells. Correctly targeted embryonic stem cells were selected and male chimeras were generated and mated to C57BL/6J females. F1 heterozygotes were mated to obtain homozygous CCR5−/− mice (WA Kuziel, TC Dawson, RL Reddick, and N Maeda, manuscript in preparation). All experimental mice were bred and maintained in pathogen-free conditions until viral inoculation. All animal experiments were done in accordance with National Institutes of Health guidelines and protocols approved by the Animal Care and Use Committee at the University of North Carolina at Chapel Hill.
Virus Infection and Quantitation
Influenza A/Puerto Rico/8/34 (A/PR/8/34) virus grown in the allantoic cavity of 10-day-old embryonated hen eggs was a gift from Dr. John F. Sheridan, Ohio State University. At 6 to 10 weeks of age, mice were anesthetized with a ketamine-xylazine solution, and intranasally infected with 5 hemeagglutinating units (HAU) of the influenza A/PR/8/34 stock. Virus was isolated from the lungs of infected mice 5 days postinfection by first weighing, then grinding the tissue into minimal essential medium, followed by a series of freeze/thaw cycles and high-speed centrifugation of the ground tissue. The supernatant was collected and 10-fold serial dilutions of 0.1-ml aliquots were added in replicates of six to 96-well plates of confluent Madin Darby canine kidney cells. Plates were incubated for 24 hours at 37°C. Viral titers (expressed as TCID50) were determined as previously described. 36 Individual group titers were compared to the control titers with unpaired Student’s t-tests.
Histological Analysis of the Lungs
At days 2, 3, 5, 16, and 25 postinfection, animals were sacrificed and one lung was resected, inflated, and embedded in freezing embedding medium (Tissue-Tek, Torrance, CA), then snap-frozen in isopentane cooled with liquid nitrogen. Frozen serial sections from all animals were stained with hematoxylin and eosin (H&E) and examined microscopically. Each specimen was assigned a score of 0 to 4+ based on the severity of lung pathology. 22 Additional cryosections selected for immunohistochemical study were acetone-fixed and incubated with an antibody against one of several mouse leukocyte markers. Sections were incubated with a series of peroxidase-labeled secondary antibodies, developed with 9-amino-3-ethylene-carbazole, and counterstained with Mayer’s hematoxylin (Sigma Diagnostics, St. Louis, MO). Primary antibodies that were used included: a mouse macrophage/monocyte marker, MOMA-2 (Serotec, Oxford, UK); a mouse neutrophil marker, NIMPR40 (kindly received from Dr. Peter Heeringa, University of North Carolina, Chapel Hill); and lymphocyte markers CD8a, CD4, and CD3e (Pharmingen, San Diego, CA).
RNase Protection Assay
Total RNA from lungs and MLNs of uninfected mice (day 0) and infected mice at days 2 and 3 postinfection was prepared using TRIzol Reagent (Life Technologies, Inc., Grand Island, NY). Chemokine mRNA levels were determined using the RiboQuant Multipurpose Ribonuclease Protection Assay (RPA) System with the mCK-5 probe set (Pharmingen, San Diego, CA). The dried gel was exposed to X-ray film and developed for 24 hours at −70°C. Bands were detected and densitometrically quantitated using RiboQuant software. All chemokine values were normalized to the housekeeping gene Gapdh. Each data point represents a sample of pooled RNA from two individual animals. Group values were compared independently to control values using unpaired Student’s t-tests.
Flow Cytometry and Cytospin Preparations
Cell suspensions from the MLNs and bronchoalveolar lavage (BAL) fluid of infected mice were obtained 5 days postinfection and pooled by genotype. Cells were stained with the following anti-mouse mAbs: PE anti-CD3, FITC anti-CD4, or FITC anti-CD8 (Pharmingen, San Diego, CA). After staining, cells were sorted and counted by FACS analysis on a FACScan machine using LYSYS II, Version 1.1 software (Becton Dickinson, San Jose, CA). Pooled BAL cells which were not used for FACS analysis were spun for 7 minutes at 1,000 rpm onto serum-coated slides using a cytocentrifuge (StatSpin; Shandon Scientific, Runcorn, UK). After Giemsa-Wright staining, leukocyte differentials were determined by averaging three counts of at least 250 cells each.
Results
Survival Rates and Influenza A Viral Clearance in CCR5−/− and CCR2−/− Mice
To asses the impact of CCR5 and CCR2 on susceptibility to influenza A virus infection, age-matched control (+/+), CCR5−/−, CCR2−/−, and CCR2−/−/MIP-1α−/− (doubly-deficient) mice were inoculated intranasally with 5 HAU of mouse-adapted influenza A/PR/8/34. As shown in Figure 1A ▶ , at this dose, 30% of control mice succumbed to the virus by day 10 postinfection. However, CCR5 deficiency resulted in a sharp increase in mortality early in the course of infection; 42% of all CCR5−/− mice died by day 6. CCR5−/− mice that survived this early death recovered and lived well beyond day 16 postinfection. In striking contrast to these results, CCR2-deficient mice were substantially less susceptible to death from influenza A infection. As a group, these animals exhibited only 10% mortality through day 16 postinfection. A strain of mice deficient in both CCR2 and MIP-1α was the most protected, exhibiting no mortality during the observation period for this experiment. Because all of the animals used are hybrids between 129/Ola and C57BL/6 strains, we cannot rule out completely a possibility that nonselected alleles have been sorted differently in each group. However, considering that the loci unlinked to CCR5-CCR2 are equally randomized and that the wild-type controls carry CCR5-CCR2 loci from 129/Ola strain, it is unlikely that the random sorting of alleles can account for the dramatic differences in pneumonitis observed in these groups of animals.
Figure 1.

The effect of CCR5 and CCR2 deletion on influenza associated mortality and viral titers. A: Mortality of +/+ (▪), CCR5−/− (•), CCR2−/− (▴), or CCR2−/−/MIP-1α−/− (♦ mice (n = 19 per group) infected intranasally with 5 HAU of influenza A (strain A/PR/8/34) and monitored for 16 days. Data represent two independent experiments combined. B: Influenza A titers in +/+, CCR5−/−, CCR2−/−, and CCR2−/−/MIP-1α−/− mice measured at day 5 after infection with 10 HAU of virus. Data are expressed as log[TCID50] per gram of tissue. *P < 0.01 versus +/+ group by Student’s t-test.
To determine whether these differences in influenza virus susceptibility correlated with T-cell-mediated viral clearance, we measured lung viral titers at day 5 postinfection (Figure 1B) ▶ . Although some of the CCR5−/− mice died prematurely, there was no significant difference in the viral load harbored by the surviving CCR5−/− mice when compared to the control mice. In contrast, CCR2-deficient animals, which exhibited an increased survival rate, had 10- to 100-fold higher viral titers than the +/+ controls. Previous results obtained by Cook et al, 22 showed that deletion of the MIP-1α gene leads to a similar increase in viral titers after influenza virus infection. Our data using the CCR2−/−/MIP-1α−/− double-knockout mice confirm this phenotype and seem to indicate that the additional deletion of CCR2 exacerbates this effect by causing even higher viral titers at this time point. These results suggest that the absence of both CCR2 and MIP-1α may independently cause delays in the migration of key leukocyte populations after influenza infection, which may subsequently cause a delay in viral clearance. In addition, our results along with those from Cook et al, 22 show that influenza-induced mortality does not directly correlate with increases in virus replication.
Altered Severity of Pulmonary Pathology in the Absence of CCR2 and CCR5
The above results confirm that the sometimes fatal, downstream pathological events that occur after influenza infection are clearly not because of an overwhelming viral burden in the lungs. This implies that the harsh pulmonary pathology may be caused by the responding inflammatory cells rather than by the cytopathic effects of the virus itself. Indeed, the early increase in mortality seen in the surviving CCR5−/− mice was associated with unusually massive leukocyte accumulations and severe pulmonary tissue damage as early as day 2 postinfection (Figure 2C) ▶ , compared to relatively mild inflammation in the control animals (Figure 2A) ▶ . Likewise, the protective effect of the CCR2 deficiency as seen in the survival study was confirmed by histological examination of the intact lungs from infected mice. At days 3 and 5 postinfection, CCR2−/− mice had a marked reduction in pulmonary inflammatory cell infiltration (Figure 3, E and F) ▶ , compared to control mice (Figure 3, A and B) ▶ . Additionally, the thickening of the septal walls, disarray/disorganization of the bronchial epithelial cells, and the areas of consolidation that were present in control lungs at day 5 postinfection (Figure 3B) ▶ were noticeably absent in CCR2−/− mice at day 5 postinfection (Figure 3F) ▶ . However, an extended time course revealed that CCR2−/− animals eventually developed some of these histopathological characteristics. When histological sections from all groups for five time points were scored 0 to 4+ based on the amount of inflammation and the severity of tissue damage, the histological scores of some of the CCR2−/− mice at day 16 were as high as those of CCR5−/− mice at day 2 (Figure 4) ▶ . However, no deaths occurred in the CCR2−/− animals, suggesting that cell populations involved may be different in these two groups of mice although the histological scores look similar. All surviving animals were able to recover from the infection by day 25 postinfection. These results indicate that CCR5 deficiency accelerates the onset of the pathological manifestations of influenza A virus, whereas CCR2 deficiency causes a delay in the onset of the normal pathological manifestations of influenza A virus.
Figure 2.
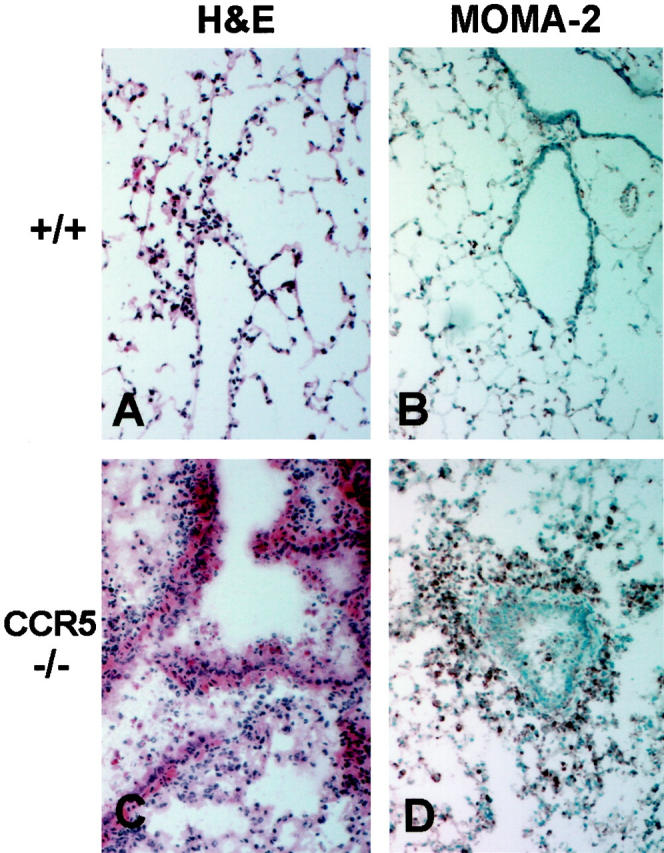
Inflammatory response to influenza A in lungs of control and CCR5-deficient mice. All histology sections are from lung tissues taken at day 2 postinfection with 5 HAU of influenza A; original magnification, ×100. A and B: Sections from +/+ lungs show H&E staining of only a few inflammatory cells characteristic for this early time point (A) and MOMA-2 staining of a few, sparse resident alveolar macrophages (B). C and D: Sections from CCR5−/− lungs show H&E staining of massive inflammatory cell infiltration and interstitial pneumonitis (C) and intense MOMA-2 staining of the infiltrating macrophages (D).
Figure 3.
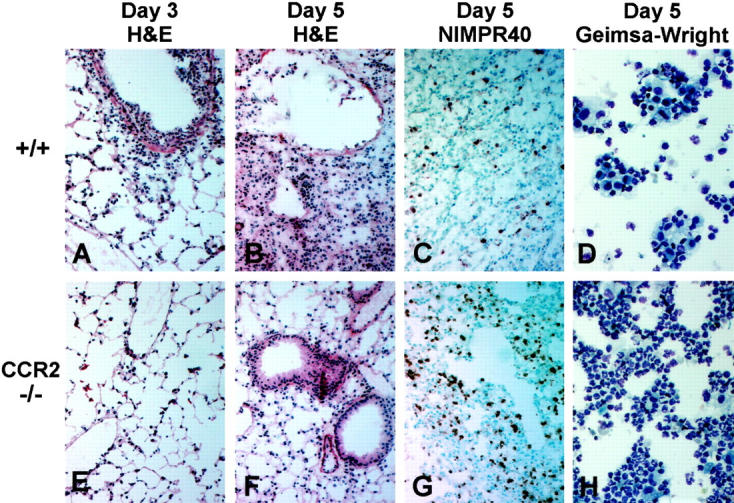
Inflammatory response to influenza A in lungs of control and CCR2-deficient mice. All histology sections are from lung tissue taken at days 3 and 5 postinfection with 5 HAU of influenza A; original magnification, ×100. Cytological preparations are BAL samples taken at day 5 postinfection; original magnification, ×400. A–C: Sections from +/+ lungs show H&E staining of the acute inflammatory cell infiltrate typically seen 3 days postinfection (A), the areas of chronic inflammation and tissue consolidation, thickening of the septal walls, and disorganization of the bronchial epithelial cells typically seen 5 days postinfection (B), and NIMPR40 staining of the relatively few neutrophils seen at this time point in +/+ mice (C). D: Giemsa-Wright staining of the BAL cells obtained from the +/+ mice 5 days postinfection. E–G: Sections from CCR2−/− lungs show H&E staining of less cellular infiltration 3 days postinfection (E), absence of the epithelial cell damage and consolidating pneumonitis that is seen in control mice at day 5 postinfection (F), and relatively intense NIMPR40 staining of the accumulating neutrophils, which is the dominant leukocyte population present in CCR2−/− lungs 5 days postinfection (G) . H: Giemsa-Wright staining of CCR2−/− BAL cells obtained at day 5 postinfection.
Figure 4.

Time course of pulmonary pathology scores for surviving +/+, CCR5−/−, and CCR2−/− mice after influenza virus infection. All lung histology sections were scored (blinded) 0+ to 4+ based on the severity of inflammation and tissue damage. For this experiment, CCR5−/− mice were not analyzed at day 5 postinfection. Each data point represents the average of 3 to 5 mice per group for each time point. *, P = 0.002 versus +/+ group, and †, P = 0.05 versus +/+ group both by Student’s t-test.
Defective Macrophage Function in Infected CCR5−/− and CCR2−/− Mice
To determine which leukocyte subpopulation was prematurely responding to the influenza A virus infection in CCR5−/− mice, immunohistochemical analysis was performed. As expected at day 2 postinfection, both control mice and CCR2−/− mice had NIP/R40-positive neutrophils present in the lungs (data not shown). However, macrophages were the predominant cell type infiltrating at day 2 postinfection in CCR5−/− lungs, as illustrated by an intense positive MOMA-2 staining (Figure 2D) ▶ compared to light staining in the control lungs (Figure 2B) ▶ . Neither CD4+ T cells, nor CD8+ T cells were present at day 2 postinfection in either the +/+, CCR2−/−, or CCR5−/− mice (data not shown).
We and others have previously reported that mice deficient in either MCP-1 or CCR2 have pronounced defects in macrophage function when immunologically challenged. 32-35 This also proved to be true of CCR2−/− mice when challenged with the influenza A virus. Although the acute neutrophilic response was unaltered in the CCR2 knockout mice, these mice failed to mount the expected mononuclear attack that begins with infiltration of macrophages by day 3 postinfection in control mice. In fact, even at day 5 postinfection, the few identifiable leukocytes that were present in CCR2−/− lungs were still neutrophils (Figure 3G) ▶ . This was confirmed by a cytological preparation of bronchoalveolar lavage (BAL) cells 5 days postinfection, which clearly shows that polymorphonuclear leukocytes (neutrophils) continue to be the dominating cell type at this time point in CCR2-deficient mice (Figure 3H) ▶ . When the BAL differentials were counted, only 10 ± 0.7% of the BAL cells in CCR2−/− mice were monocytes/macrophages, compared with 60 ± 1.5% in control mice at the same time point (Figure 5) ▶ . These experiments illustrate that the delay in the pathogenesis of influenza in the CCR2−/− mice is caused by their inability to properly recruit macrophages during an inflammatory attack.
Figure 5.

Differential leukocyte counts of BAL cells obtained from influenza-infected +/+ and CCR2−/− mice. Data represents the average of 3 counts (of approximately 300 cells per count) from one pooled sample per group (each group = 5 mice pooled).
MCP-1 Modulation of the Macrophage Response to Influenza A Virus
Although the defective macrophage recruitment in CCR2−/− mice was not surprising, the hyperacute macrophage accumulation in the CCR5−/− animals was unexpected. To investigate the mechanism of this aberrant response, we examined pulmonary chemokine expression levels of several relevant CC chemokine genes after influenza virus infection. Image densitometry of an RNase protection assay revealed a threefold increase in RANTES RNA expression at day 2 postinfection in CCR5−/− mice compared to control animals or CCR2-deficient mice (Figure 6A) ▶ . Excess RANTES might attract additional macrophages through binding to its alternate receptor, CCR1, on these cells. IP-10 expression was increased in all animals with slightly more increase in the CCR5−/− mice, which could also be contributing to the macrophage response by their binding to other, intact receptors. More likely, however, the excess macrophage accumulation in CCR5−/− mice results from increased expression of MCP-1, a key regulator of induced macrophage migration. Both the CCR5−/− and CCR2−/− animals exhibited a twofold increase in day 2 postinfection pulmonary MCP-1 RNA levels relative to the levels in control mice (Figure 6A) ▶ . As would be expected, the overexpression of MCP-1 correlates with an increased migration of macrophages in CCR5−/− mice, but not in mice lacking CCR2, the major receptor for MCP-1. There were no significant differences in eotaxin, MIP-1α, MIP-1β, MIP-2, or TCA-3 levels in the CCR2−/− or CCR5−/− mice when compared to controls.
Figure 6.

Time course of chemokine mRNA expression levels in the lung (A) and the MLN (B). RNA was obtained from uninfected (day 0) tissues and from postinfection (day 2, day 3) tissues of +/+, CCR5−/−, and CCR2−/− mice. RNA was prepared and chemokine mRNA was detected by a multiprobe RNase protection assay and quantified by image densitometry, as described in Materials and Methods. Each data point represents a sample of pooled RNA from two individual animals and chemokine mRNA amounts were expressed relative to that of GAPDH.
Delayed T Cell Response in CCR2−/− Mice
The cellularity of MLNs of mice infected with influenza A virus increases rapidly, with maximal total cell counts reached by day 5 postinfection. All major lymphocyte subsets are involved in this expansion, including CD4+ and CD8+ T cells. 37 At day 5 postinfection, ∼23% of total MLN cells in control mice were CD8+ (consistent with previous reports). These numbers were modestly reduced in the MLNs of CCR2−/− mice to ∼19% CD8+ T cells (Table 1) ▶ . Additionally, the percentage of total (CD3+) T cells was substantially reduced in the CCR2−/− MLNs (37% versus 53% in +/+). As shown in Figure 6B ▶ , this decrease in T cell numbers in MLNs of CCR2−/− mice correlates with a decrease in MLN expression of RANTES. These data imply that, relative to control MLNs, MLNs of CCR2−/− mice are less activated, and that fewer CTLs are homing to or dividing in the MLNs of these animals. Subsequently, there will be fewer primed CD8+ T cells to localize to the site of virus growth. In fact, FACS analysis did reveal a marked reduction in the percentage of CD8+ T cells in the BAL fluid of CCR2−/− mice compared to controls (data not shown). This complements the results of immunohistochemical analyses that indicated an absence of CD3+ or CD8+ T cells in the intact lungs of CCR2−/− mice at day 5 postinfection (data not shown). Altogether, these experiments demonstrate that proper signaling through CCR2 is necessary for lymphocyte accumulation in secondary lymphoid organs and for the subsequent trafficking to influenza infected tissues.
Table 1.
T Cell Distribution in MLN of Influenza-Infected Mice
| MLN cell population | Percent cells staining for | ||
|---|---|---|---|
| CD8 | CD4 | CD3 | |
| +/+* | 23 | 30 | 53 |
| CCR2−/−* | 19 | 17 | 37 |
*Group values represent the average of two samples with four pooled MLN per sample.
Discussion
Infection of CCR5- and CCR2-deficient mice with influenza A virus has yielded several unexpected and striking results regarding influenza-induced immunity and the associated pathology. First, CCR5−/− mice show an accelerated macrophage accumulation in the lungs and increased mortality. Second, the acute inflammation seen in CCR5−/− mice is opposite from the protective effects that is seen in mice deficient in the CCR5 ligand, MIP-1α. Third, the virus-infected CCR2−/− mice have increased survival rates compared to controls, in the presence of an early block in pulmonary macrophage accumulation and an increased viral load. All of these results highlight the importance of macrophages in generating an appropriate immune response to influenza infection and in the development of associated lung pathology. Macrophages may both directly and indirectly contribute to influenza associated lung pathology. A direct contribution of macrophages to influenza-induced lung pathology is illustrated by their ability to express inducible nitric-oxide synthase (NOS2) after becoming infected. 38 This causes macrophage secretion of potentially injurious inducible nitric-oxide synthase-derived nitric oxide metabolites. Deletion of the inducible nitric-oxide synthase gene confers protection against influenza-induced lethal lung pathology, without altering CTL activity. 28 Macrophages can also indirectly control the progression of influenza pathogenesis by regulating T cell response. Just before the induction of apoptotic cell death, 17,39 infected macrophages secrete cytokines such as TNFα, IL-1, IL-6, and M-CSF, and chemokines such as MIP-1α, RANTES, and IP-10 all of which could potentially contribute to lung pathology by inducing the activation and migration of CTLs to the site of an influenza infection. 17,21,26,40-42 Thus, the macrophage response to the influenza virus leads to the rapid onset of a proinflammatory signaling cascade and enhanced activation and immigration of lymphocytes into the lung. Once present in the lung, these lymphocytes, specifically CTLs, can recognize and directly lyse virally infected cells, thereby eliminating the virus and initiating recovery of the tissue. 43
Inflammation in response to primary influenza virus infection not only involves macrophages and CTLs, but also involves chemokine-dependent trafficking of dendritic cells and natural killer cells in-between the site of infection, the circulation, and the draining lymph nodes. CC chemokine receptors CCR5 and CCR2 are expressed on all of these cell types, 41,44 and several recent reports describe the production of various CCR5 and CCR2 ligands by mononuclear and epithelial cells after being infected by influenza A virus. 17,23,24,45 Thus, CCR5 and/or CCR2 could potentially modulate both the afferent and efferent phases of the immune response to influenza virus infection.
In our studies, we show that CCR5 is not absolutely required for macrophage recruitment into influenza-infected lungs or for control of viral replication. CCR5 deficiency does, however, lead to a more negative clinical outcome relative to controls, with approximately half of all influenza-infected CCR5-deficient mice dying prematurely as a result of early, overwhelming macrophage accumulation in the lungs. Although not proven in our experiments, this accelerated macrophage accumulation is likely to be linked to enhanced expression of MCP-1 and RANTES (Figure 6A) ▶ , and possibly of IP-10, and their binding to other intact chemokine receptors on the CCR5-deficient macrophages. In other experiments, we have also observed enhanced inflammatory cell recruitment in the lungs of CCR5−/− mice after primary infection with Cryptococcus neoformans. 46 The relationship between the absence of CCR5 signaling and increased chemokine expression is not clear at this time, but alterations in leukocyte development or trafficking patterns directly related to CCR5 deficiency may be involved.
The enhanced lung inflammation and pathology of CCR5−/− mice stands in sharp contrast to the reduced inflammation and pathology observed in mice deficient in MIP-1α, a CCR5 ligand. 22 These results together suggest that during influenza infections, MIP-1α primarily functions through another of its known receptors (CCR1, CCR4), and that its interactions with CCR5 have limited, if any, importance for the pulmonary mononuclear response to influenza virus infection. These observations underscore the complexity of the chemokine system and the difficulty of relating the interplay of chemokines and their receptors in vitro, with leukocyte function and biological consequences in vivo.
It has been well established that the loss of functional CCR2 does not alter blood monocyte levels, but severely impairs the ability of blood monocytes/macrophages to respond to certain types of injurious stimuli or specific pathogenic agents. 7,32-34 Our studies implicate influenza A virus as yet another such pathogen whereby mononuclear leukocyte activation and migration is mediated by a CCR2/MCP-1-dependent mechanism. Despite increased levels of MCP-1 RNA in the lungs of infected CCR2−/− mice, there is substantially reduced pulmonary macrophage accumulation in these animals. This correlates with reduced mortality even though the viral titers were 10- to 100-fold higher than in control or CCR5−/− mice at an early time point. These results suggest that the pathological manifestations of influenza infections are caused by the massive accumulation of inflammatory cells and not by the intrinsic cytopathic effects of the virus itself. Consistent with our previous experiments showing that the CCR2-deficient mice have enhanced early accumulation and delayed clearance of neutrophils in response to intraperitoneal thioglycollate injection, 32 neutrophils persisted in the influenza virus infected lung of the CCR2−/− mice. The delay in macrophage response resulting in reduced phagocytosis is at least partly responsible for the prolonged presence of neutrophils in lungs of the CCR2−/− mice. It is also likely that the neutrophilic persistence seen at later time points in our flu experiments are because of the consistent migration of new neutrophils into the lung. No evidence of defective apoptosis of neutrophils has been demonstrated in CCR2−/− mice.
Our data with the CCR2-deficient mice further show that the delay in the macrophage response leads to a subsequent delay in CTL development and/or migration to the infected lungs. This delay in T cell response is likely responsible for the increased viral load 5 days postinfection. It has been shown that CCR2 expression is inducible on T lymphocytes, 47-48 and T lymphocytes can respond to MCP-1 at high levels in vitro. 49 Thus, the apparent lack of T cell involvement in the lungs of CCR2−/− mice could, at least in part, be directly related to the absence of CCR2 from circulating T cells. However, this is unlikely to account for the majority of the delays in leukocyte accumulation in the CCR2−/− mice because the delays are seen at very early time points, when monocytes, not T cells, are widely regarded as the primary responders to induced MCP-1 expression in vivo. It is also important to note that whereas the CCR2 deficiency confers pathological protection at early time points, CCR2-deficient mice eventually mount a pulmonary mononuclear response that is sufficient to allow for tissue recovery with no significant rise in mortality. Obviously, there is some compensatory mechanism that might involve other chemokines and chemokine receptors, such as MIP-1α or IP-10. We observed increased expression of IP-10 (Figure 6A) ▶ , and IP-10 expression is markedly increased after influenza A virus infection of cultured human monocytes. 25 Like MCP-1, IP-10 promotes chemotaxis of monocytes and T lymphocytes, 50 but it functions through CXCR3. 4 Another intriguing possibility is the binding of MCP-1 to receptors other than CCR2. High levels of local MCP-1, resulting from the increased MCP-1 mRNA produced after infection, may help facilitate the binding of MCP-1 to alternative chemokine receptors.
Mice deficient in both CCR2 and MIP-1α show the best survival and the highest viral titers among all groups used in this study. This is consistent with the idea that MCP-1 and MIP-1α make distinct and additive contributions to the pulmonary inflammation induced by influenza virus infections. Absence of CCR2 clearly blocks the majority of the early pulmonary macrophage accumulation, whereas others have shown that MIP-1α is the major regulator of T-cell trafficking into lymph nodes and specific tissues in response to a variety of antigenic stimuli, including influenza A virus. 22,51-52
Altogether, our data demonstrate that the cell-mediated mechanism normally used in influenza-infected host tissues includes initial macrophage activation and recruitment via MCP-1 expression, followed by a quick and precise CTL attack against the influenza A virus. We show that elimination of either CCR5 or CCR2 significantly affects the course immune mechanisms as well as the clinical outcome, although in profoundly different ways. Altering the homing or phenotypic characteristics of the alveolar macrophages can cause an overwhelming, sometimes fatal early leukocyte response, as in the CCR5−/− mice. In contrast, reducing the migratory capability of the peripheral blood monocytes/macrophages confers protection from severe influenza-induced tissue damage at early time points, as seen in the CCR2−/− mice. Although there is clear overlap in CC chemokine receptor expression patterns in this case (and many others), the availability of chemokines and chemokine receptors in vivo has been repeatedly shown to be a highly specific, finely controlled, and tightly regulated process, making therapeutic disruption a distinct possibility. Our data indicate that an efficacious therapeutic agent might be one that targets the beginning stages of the early leukocyte inflammatory cascade, or more particularly, one that can specifically alter normal macrophage function. However, decreasing macrophage function could cause an increased susceptibility to secondary bacterial infections. 53 This possibility must be carefully considered during future investigations into the pathogenesis of the influenza A virus.
Acknowledgments
We thank Eileen McMahon, Peter Heeringa, Tene Osahar, John Cowhig, Qing Shi, Felicia Walton, and Patti Sheridan for their expert technical assistance; and Oliver Smithies, Jeff Hodgin, Suzanne Kirby, and Peter Heeringa for helpful advice and for critical reading of our manuscript.
Footnotes
Address reprint requests to Dr. Nobuyo Maeda, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, CB #7525, 703 Brinkhous-Bullitt Building, Chapel Hill, NC 27599-7525. E-mail: nobuyo@med.unc.edu.
Supported by National Institutes of Health grants HL42630 and HL54123.
References
- 1.Cook DN: The role of MIP-1α in inflammation and hematopoiesis. J Leukoc Biol 1996, 59:61-66 [DOI] [PubMed] [Google Scholar]
- 2.Baggiolini M: Chemokines and leukocyte traffic. Nature 1998, 392:565-568 [DOI] [PubMed] [Google Scholar]
- 3.Ward SG, Bacon K, Westwick J: Chemokines and T lymphocytes: more than an attraction. Immunity 1998, 9:1-11 [DOI] [PubMed] [Google Scholar]
- 4.Zlotnik A, Morales J, Hedrick JA: Recent advances in chemokines and chemokine receptors. Crit Rev Immunol 1999, 19:1-47 [PubMed] [Google Scholar]
- 5.Devalaraja MN, Richmond A: Multiple chemotactic factors: fine control or redundancy? Trends Pharm Sci 1999, 20:151-156 [DOI] [PubMed] [Google Scholar]
- 6.Wells TNC, Power CA, Proudfoot AEI: Definition, function and pathophysiological significance of chemokine receptors. Trends Pharm Sci 1998, 19:376-380 [DOI] [PubMed] [Google Scholar]
- 7.Dawson TC, Kuziel WA, Osahar TA, Maeda N: Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 1999, 143:205-211 [DOI] [PubMed] [Google Scholar]
- 8.Boring L, Gosling J, Cleary M, Charo IF: Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394:894-897 [DOI] [PubMed] [Google Scholar]
- 9.Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ: Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell 1998, 2:275-281 [DOI] [PubMed] [Google Scholar]
- 10.Barnes DA, Tse J, Kaufhold M, Owen M, Hesselgesser J, Strieter R, Horuk R, Perez HD: Polyclonal antibody directed against human RANTES ameliorates disease in the Lewis rat adjuvant-induced arthritis model. J Clin Invest 1998, 101:2910-2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lloyd C, Gutierrez-Ramos J-C: The role of chemokines in tissue inflammation and autoimmunity in renal diseases. Curr Opin Nephrol Hypertens 1998, 7:281-287 [DOI] [PubMed] [Google Scholar]
- 12.Russell ME, Adams DH, Wyner LR, Yamashita Y, Halnon NJ, Karnovsky MJ: Early and persistent induction of monocyte chemoattractant protein 1 in rat cardiac allografts. Proc Natl Acad Sci USA 1993, 90:6086-6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virelizier JL, Allison AC, Schild GC: Immune responses to influenza virus in the mouse and their role in control of the infection. Br Med Bull 1979, 35:65-68 [DOI] [PubMed] [Google Scholar]
- 14.Friedland JS: Chemokines in viral disease. Res Virol 1996, 147:131-138 [DOI] [PubMed] [Google Scholar]
- 15.Bender BS, Small PA, Jr: Influenza: pathogenesis and host defense. Semin Respir Infect 1992, 7:38-45 [PubMed] [Google Scholar]
- 16.Doherty PC: Immune responses to viruses. Rich RR Fleisher TA Schwarz BD Shearer WT Strober W eds. Clinical Immunology, Principles and Practice, 1996, vol 1.:pp 535-549 Mosby-Year Book, Inc., St. Louis [Google Scholar]
- 17.Hofmann P, Sprenger H, Kaufmann A, Bender C, Hasse M, Gemsa D: Susceptibility of mononuclear phagocytes to influenza A virus infection and possible role in the antiviral response. J Leukoc Biol 1997, 61:408-414 [DOI] [PubMed] [Google Scholar]
- 18.Fujisawa H, Sumiaki T, Taniguchi M, Zinnaka Y, Nomoto K: Protective mechanisms against pulmonary infection with influenza virus. I. Relative contribution of polymorphonuclear leukocytes and of alveolar macrophages to protection during the early phase of intranasal infection. J Gen Virol 1987, 68:425-432 [DOI] [PubMed] [Google Scholar]
- 19.Shaw MW, Arden NH, Maassab HF: New aspects of influenza viruses. Clin Microbiol Rev 1992, 5:74-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannon MJ, Openshaw PJM, Askonas BA: Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med 1988, 168:1163-1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moskophidis D, Kioussis D: Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J Exp Med 1998, 188:223-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, Smithies O: Requirement of MIP-1α for an inflammatory response to viral infection. Science 1995, 269:1583-1585 [DOI] [PubMed] [Google Scholar]
- 23.Matsukura S, Kokubu F, Kubo H, Tomita T, Tokunaga H, Kadodura M, Yamamoto T, Kuroiwa Y, Ohno T, Suzaki H, Adachi M: Expression of RANTES by normal airway epithelial cells after influenza virus A infection. Am J Respir Cell Mol Biol 1998, 18:255-264 [DOI] [PubMed] [Google Scholar]
- 24.Sprenger H, Meyer RG, Kaufmann A, Bubfeld D, Rischkowsky E, Gemsa D: Selective induction of monocyte and not neutrophil-attracting chemokines after influenza A virus infection. J Exp Med 1996, 184:1191-1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bubfeld D, Kaufmann A, Meyer RG, Gemsa D, Sprenger H: Differential mononuclear leukocyte attracting chemokine production after stimulation with active and inactivated influenza A virus. Cell Immunol 1998, 186:1-7 [DOI] [PubMed] [Google Scholar]
- 26.Wijburg OLC, Dinatale S, Vadolas J, Van Rooijen N, Strugnell RA: Alveolar macrophages regulate the induction of primary cytotoxic T-lymphocyte responses during influenza virus infection. J Virol 1997, 71:9450-9457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karupiah G, Buller RML, Van Rooijen N, Duarte CJ, Chen J: Different roles for CD4+ and CD8+ T lymphocytes and macrophage subsets in the control of a generalized viral infection. J Virol 1996, 70:8301-8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD: Rapid interferon γ-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J Exp Med 1998, 188:1541-1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graham MB, Braciale TJ: Resistance to and recovery from lethal influenza virus infection in B lymphocyte-deficient mice. J Exp Med 1997, 186:2063-2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bender BS, Croghan T, Zhang L, Small PA, Jr: Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 1992, 175:1143-1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Graham MB, Dalton DK, Giltinan D, Braciale VL, Stewart TA, Braciale TJ: Response to influenza infection in mice with a targeted disruption in the interferon γ gene. J Exp Med 1993, 178:1725-1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuziel WA, Morgan SJ, Dawson TC, Griffin S, Smithies O, Ley K, Maeda N: Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc Natl Acad Sci USA 1997, 94:12053-12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurihara T, Warr G, Loy J, Bravo R: Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med 1997, 186:1757-1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RVJ, Broxmeyer HE, Charo IF: Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J Clin Invest 1997, 100:2552-2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard G, Rollins BJ: Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med 1998, 187:601-608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hermann G, Beck FM, Sheridan JF: Stress-induced glucocorticoid response modulates mononuclear cell trafficking during an experimental influenza viral infection. J Neuroimmunol 1995, 56:179-186 [DOI] [PubMed] [Google Scholar]
- 37.Allan W, Zsuzsanna T, Clearly A, Doherty PC: Cellular events in the lymph node and lung of mice with influenza: consequences of depleting CD4+ T cells. J Immunol 1990, 144:3980-3986 [PubMed] [Google Scholar]
- 38.Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM, Dietzschold B, Maeda H: Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci USA 1996, 93:2448-2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fesq H, Bacher M, Nain M, Gemsa D: Programmed cell death (apoptosis) in human monocytes infected by influenza A virus. Immunobiology 1994, 190:175-182 [DOI] [PubMed] [Google Scholar]
- 40.Hennet T, Ziltener HJ, Frei K, Peterhans E: A kinetic study of immune mediators in the lungs of mice infected with influenza A virus. J Immunol 1992, 149:932-939 [PubMed] [Google Scholar]
- 41.Houde M, Arora DJS: Stimulation of tumor necrosis factor secretion by purified influenza virus neuraminidase. Cell Immunol 1990, 129:104-111 [DOI] [PubMed] [Google Scholar]
- 42.Luster AD: Chemokines—chemotactic cytokines that mediate inflammation. N Engl J Med 1998, 338:436-445 [DOI] [PubMed] [Google Scholar]
- 43.Kagi D, Hengartner H: Different roles for cytotoxid T cells in the control of infection with cytopathic versus noncytopathic viruses. Curr Opin Immunol 1996, 8:472-477 [DOI] [PubMed] [Google Scholar]
- 44.Caux C, Lebecque S, Liu Y-J, Banchereau J: Developmental pathways of human myeloid dendritic cells. Lotze MT Thomson AW eds. Dendritic Cells: Biology and Clinical Applications. 1999, :pp 63-92 Academic Press, London [Google Scholar]
- 45.Fritz RS, Hayden FC, Calfee DP, Cass LM, Peng AW, Alvord WG, Strober W, Straus SE: Nasal cytokine and chemokine responses in experimental influenza A virus infection: results of a placebo-controlled trial of intravenous zanamivir treatment. J Infect Dis 1999, 180:586-593 [DOI] [PubMed] [Google Scholar]
- 46.Huffnagle GB, McNeil LK, McDonald RA, Murphy JW, Toews GB, Maeda N, Kuziel WA: The role of CCR5 in organ-specific and innate immunity to Cryptococcus neoformans. J Immunol 1999, 163:4642-4646 [PubMed] [Google Scholar]
- 47.Denholm EM, Stankus GP: Changes in the expression of MCP-1 receptors on monocytic THP-1 cells following differentiation to macrophages with phorbol myristate acetate. Cytokine 1995, 7:436-440 [DOI] [PubMed] [Google Scholar]
- 48.Loetscher P, Seitz M, Baggiolini M, Moser B: Interleukin-2 regulates CC chemokine receptor expression and chemotactic responsiveness in T lymphocytes. J Exp Med 1996, 184:569-577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oin S, LaRosa G, Campbell JJ, Smith-Heath H, Kassam N, Shi X, Zeng L, Buthcher EC, Mackay CR: Expression of monocyte chemoattractant protein-1 and interleukin-8 receptors on subsets of T cells: correlation with transendothelial chemotactic potential. Eur J Immunol 1996, 26:640-647 [DOI] [PubMed] [Google Scholar]
- 50.Taub DD, Lloyd AR, Conlon K, Wang JM, Ortaldo JR, Harada A, Matsushima K, Kelvin DJ, Oppenheim JJ: Recombinant human interferon-inducible protein 10 is a chemoattractant for human monocytes and T lymphocytes and promotes T cell adhesion to endothelial cells. J Exp Med 1993, 177:1809-1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim CH, Broxmeyer HE: Chemokine: signal lamps for trafficking of T and B cells for development and effector function. J Leukoc Biol 1999, 65:6-15 [DOI] [PubMed] [Google Scholar]
- 52.Tedla N, Wang HW, McNeil P, Girolamo ND, Hampartzoumian T, Wakefield D, Lloyd A: Regulation of T lymphocyte trafficking into lymph nodes during an immune response by the chemokines macrophage inflammatory protein (MIP)-1α and MIP-1β. J Immunol 1998, 161:5663-5672 [PubMed] [Google Scholar]
- 53.Kass EH, Green GM, Goldstein E: Mechanisms of antibacterial action in the respiratory system. Bacteriol Rev 1966, 30:488-496 [DOI] [PMC free article] [PubMed] [Google Scholar]