

Abstract
Modified muscle use can result in muscle inflammation that is triggered by unidentified events. In the present investigation, we tested whether the activation of the complement system is a component of muscle inflammation that results from changes in muscle loading. Modified rat hindlimb muscle loading was achieved by removing weight-bearing from the hindlimbs for 10 days followed by reloading through normal ambulation. Experimental animals were injected with the recombinant, soluble complement receptor sCR1 to inhibit complement activation. Assays for complement C4 or factor B in sera showed that sCR1 produced large reductions in the capacity for activation of the complement system through both the classical and alternative pathways. Analysis of complement C4 concentration in serum in untreated animals showed that the classical pathway was activated during the first 2 hours of reloading. Analysis of factor B concentration in untreated animals showed activation of the alternative pathway at 6 hours of reloading. Administration of sCR1 significantly attenuated the invasion of neutrophils (−49%) and ED1+ macrophages (−52%) that occurred in nontreated animals after 6 hours of reloading. The presence of sCR1 also reduced significantly the degree of edema by 22% as compared to untreated animals. Together, these data show that increased muscle loading activated the complement system which then briefly contributes to the early recruitment of inflammatory cells during modified muscle loading.
Previous studies have provided evidence that modifications in mechanical loading induce structural damage, necrosis, inflammation, and functional impairment in muscle. 1-3 However, the mechanisms that underlie the invasion of inflammatory cells and subsequent muscle necrosis that occur after modified muscle use are not understood. Muscle inflammation and necrosis that occur in other muscle pathologies such as inflammatory and autoimmune diseases, 4-7 chemically induced skeletal muscle necrosis, 8 and ischemic skeletal and cardiac muscle injuries 9,10 have been shown to result, at least in part, from the actions of the complement system. Because muscle injuries and pathology that are known to involve the complement system histologically resemble muscle inflammation and injury caused by modified muscle use, 2,8 we have tested the contribution of the complement system in muscle pathogenesis that occurs after modified muscle loading.
The activation of the complement system by the classical or the alternative pathway produces complement fragments that can play important roles in the inflammatory response. For example, leukocyte iC3b receptor CR3 (Mac-1; CD11b/CD18) has been demonstrated to be involved in leukocyte adherence to endothelium via covalent fixation of complement fragment iC3b to endothelium 11 whereas C3a and C5a can enhance expression of neutrophil-endothelial cell adhesion receptor, induce vascular leakage, attract leukocytes by chemotaxis, 12 and stimulate the release of histamine 13 and lytic enzymes in the necrotic area. 14 In addition to exerting its biological activity on immunocompetent cells, the complement system can generate the membrane attack complex which involves complement activation and binding of C5b-9 that can lead to a loss in membrane integrity and necrosis of the targeted cells. 15 Thus, many of the characteristics of muscle experiencing modified use, such as leukocyte diapedesis, 2 muscle membrane defects, 16 and necrosis, 1 are all consistent with changes expected of complement-mediated events in muscle.
If muscle damage that occurs during reloading results in part from complement activation, then blocking the complement cascade could provide a means to reduce muscle injury during modified muscle use. Numerous strategies have been used in previous investigations to inhibit complement activation that occurs in pathological processes. For example, the depletion of serum complement 17 and blockage of complement cascade via injections of recombinant endogenous regulatory proteins 18 or through specific antibodies directed against the anaphylatoxin substances C3a and C5a 19 have been shown to reduce the accumulation of neutrophils and deposition of complement in ischemic tissue. The truncated, soluble form of complement receptor-1 (sCR1) is the most effective agent for the suppression of ischemia/reperfusion injury in myocardium, 18 skeletal muscle, 20 and gut 21 in which the activation of complement system is particularly well documented after changes in blood flow. The sCR1 retains the ability of the native receptor to bind to activated C3 (C3b) and C4 (C4b) thereby blocking complement activation. Thus, administration of sCR1 to animals before modification of muscle loading could prevent that portion of muscle inflammation and necrosis that is attributable to complement activation.
In this study, we have tested whether the complement system plays a role in inflammatory cell invasion and muscle fiber injury that occurs during muscle loading after a period of unloading by assaying whether sCR1 administration can attenuate muscle injury or inflammation. In addition, we have assessed the pathway through which complement activation occurs during modified muscle use by assaying for changes in the serum concentrations of factor B and C4 which provide indices of the activation of alternative and classical pathways. Inflammation, fiber necrosis, edema, and complement activation were measured after muscle unloading and in animals experiencing muscle reloading for 2, 6, or 24 hours because that time course encompasses that in which extensive inflammation and fiber damage have been shown to occur. 2 The results of this study support the hypothesis that the complement system is activated through both the classical and alternative pathways and participates in the recruitment of leukocytes during early stages of increased muscle use.
Materials and Methods
Experimental Protocol
Adult, 9- to 10-week-old, female Wistar rats were housed individually, maintained on a 12 hour light/dark cycle, and allowed to acclimate to their new environment for a few days. The hindlimbs of the rats were suspended for 10 days by using the technique of Morey-Holton and Wronski. 22 Thirty minutes before reloading, experimental and control rats were injected intraperitoneally with sCR1 (20 mg/kg) or an equal volume of phosphate-buffered saline (PBS), respectively. Complement inhibition by sCR1 was maintained in the 24-hour-reloaded group by injecting half doses of sCR1 every 8 hours. Recombinant, endotoxin-free, human sCR1 at a concentration of 3.9 or 4.75 mg/ml in PBS was kindly provided by AVANT Immunotherapeutics, Inc. (Needham, MA). After 10 days of suspension, the animals were removed from the suspension apparatus and allowed to ambulate voluntarily for 0, 2, 6, or 24 hours, designated as the reloading period. All animal treatments followed protocols approved by the University of California, Los Angeles, Animal Research Committee.
Blood Collection and Tissue Preparation
Following the experimental protocol, rats were anesthetized with sodium pentobarbital (50 mg/kg) and blood was obtained by cardiac puncture and collected in a polypropylene tube. After 10 minutes at room temperature, blood samples were centrifuged at 800 × g for 10 minutes and the serum was collected and stored at −70°C. After blood collection, soleus muscles were excised with the tendons intact and maintained near resting length by stapling the tendons to balsa wood coated with a layer of OCT compound. The tissues were then frozen in melting isopentane cooled in liquid nitrogen. Samples were stored at −70°C. Soleus muscles were sectioned at 10 μm, adhered to slides coated with chromium potassium sulfate and gelatin, and stored at −20°C.
Immunohistochemistry
Sections were processed for immunohistochemistry with the following antibodies: 1) anti-ED1+ and anti-ED2+ (diluted 1:100; Bioproducts for Science, Indianapolis, IN) which recognize antigens specific for different subpopulations of monocytes and macrophages; 2) anti-W3/13 (diluted 1:100; Bioproducts for Science) which binds to leukosialin present on neutrophils, monocytes, and T-lymphocytes; and 3) anti-complement C3 (diluted 1:1,000; Cappel Research Products, Durham, NC). Anti-W3/13 is used as a marker for neutrophils in the present investigation because previous studies have shown that neutrophils are the only leukosialin expressing cells to invade muscle in significant numbers in the present model of muscle inflammation. 16 The sections were then washed in PBS and incubated with biotinylated anti-mouse or anti-goat IgG (diluted 1:200; Vector Laboratories, Burlingame, CA) for 1 hour at room temperature. After incubation for 2 hours at room temperature in biotinylated second antibody, tissue samples were washed with PBS and incubated with streptavidin-conjugated horseradish peroxidase (1:1,000; Vector Laboratories). After three washes, the antigen-antibody-chromogen complex was developed using a peroxidase substrate kit (AEC; Vector Laboratories). The concentration of inflammatory cells labeled with each antibody was measured in two sections separated by 1 mm in each soleus muscle and examined by light microscopy using Nomarski optics. The number of labeled cells in each section was counted and the total area of the section determined and multiplied by its thickness to express the number of each cell type/mm3.
Quantitation of Fiber Necrosis and Estimation of Muscle Edema
The percentage of total muscle fibers in cross-sections that were invaded by ED1+ cells was determined by light microscopy as an index of muscle necrosis. Tissue edema was measured by observing muscle cross-sections with a microscope equipped with an eyepiece micrometer containing a 10 × 10 sampling grid. Five fields were sampled from each section by counting the percentage of grid sampling points that overlay extracellular tissue, excluding vascular lumina, and that value used as an index of tissue edema. Sample sites were selected randomly, but excluded sites containing sectioning artifacts.
Factor B and C4 Analysis
Factor B is exclusive to the alternative complement pathway and it is commonly used as an indicator of the degree of its activation. 23 Factor B concentration was monitored using an assay for hemolysis of sheep red blood cells. Briefly, factor B concentration was determined by mixing the following reagents: 10 μl of factor B deficient serum (Sigma, St. Louis, MO), 5 μl of sheep red blood cells (10 9 cells/ml; Rockland, Gilbertsville, PA), 1 μl of rat serum, and a volume of gelatin veronal buffer (GVB+) (3.5 mmol/L sodium barbital, 140 mmol/L NaCl, 0.1% gelatin, pH 7.4) containing 20 mmol/L MgCl2 and 80 mmol/L EGTA calculated to give a final reaction volume of 55 μl. After 15 minutes at 37°C with agitation, the reaction was stopped by the addition of 0.5 ml of ice-cold GVB+ and centrifuged at 800 × g to pellet the unlysed erythrocytes. The amount of hemoglobin was estimated by measuring O.D.412. Controls included tubes in which serum was not added to estimate spontaneous lysis, or with water added instead of GVB+ to obtain 100% hemolysis.
C4 analysis was used to test whether the classical pathway was activated during muscle reloading. C4 analysis was based on measurement of lysis of opsonized sheep red blood cells. Opsonization was performed by incubating cells suspended in GVB2+ (3.5 mmol/L sodium barbital, 140 mmol/L NaCl, 0.1% gelatin, pH 7.4, containing 0.15 mmol/L CaCl2, and 0.5 mmol/L MgCl2) with anti-sheep red blood cells for 30 minutes at 37°C. Opsonized red cells (100 μl) were mixed with 5 μl of complement C4-deficient serum from guinea pig (diluted 1:20; Sigma), 20 μl, 40 μl, and 60 μl of experimental rat serum samples (diluted 1:200), and a volume of GVB2+ to give a final reaction volume of 250 μl. The mixture was incubated at 37°C for 30 minutes and the reaction was stopped by adding 0.5 ml of ice-cold GVB2+. Samples were then centrifuged and the hemoglobin released was estimated as described above. Similar to the factor B assay, control tubes were included to estimate spontaneous lysis and 100% hemolysis. For quantification of factor B and C4 concentration, the percentage of hemolysis was determined by expressing the amount of hemolysis obtained from each experimental rat serum relative to 100% hemolysis.
Deposition of Complement C3 on Degenerating Fibers
Bupivacaine was injected in soleus muscle to activate the complement system and initiate deposition of complement C3 on necrotic fibers. 8 The muscles were then used as positive controls for C3 immunohistochemistry. Soleus muscles were surgically exposed under generalized anesthesia and 0.2% bupivacaine in PBS was injected in the middle of the muscle belly with a 271/2-gauge needle. The incision was then sutured and covered with wet cotton gauze for 1 hour. After the chemical injury, soleus muscles were dissected and processed for immunohistochemistry analysis as described above.
Statistical Analysis
The values obtained in sCR1-treated animals were compared to the values obtained from PBS-treated animals for all parameters quantified. The significance of differences between groups was tested by one-way analysis of variance (P < 0.05). If significant differences within the experimental treatments were present, groups within the experimental treatment were compared by the Mann-Whitney nonparametric test to locate groups that differed significantly at P < 0.05. Each value for factor B and complement was the average of two independent measurements. Where the error bars are not visible in the figures, this is because of very small error bars.
Results
Effects of Complement Inhibition on Muscle Inflammation
The sequence of inflammatory and necrotic changes in muscle observed in the present investigation is similar to previous findings, 24 although significant changes occurred earlier during reloading in the present study. For example, ED1+ cell concentrations were significantly elevated by 6 hours of reloading in the present study, but were not significantly increased until 24 hours in previous findings. 24 ED2+ cell concentrations were elevated at 24 hours reloading in the present investigation, but not until 48 hours in previous work. The advanced course of inflammation in the present investigation may be attributable to the additional animal handling during sCR1 or PBS injections, because animals in the previous study were not subjected to intraperitoneal injections.
The concentration of neutrophils increased significantly in all groups experiencing reloading when compared to suspended animals (Figures 1 and 2) ▶ ▶ . We confirmed that W3/13+ cells in muscle were neutrophils by isolating mononucleated cells from reloaded soleus muscle and then performing indirect immunohistochemistry for W3/13 using the isolated cells. The only W3/13+ cells isolated from the reloaded solei contained multilobed nuclei characteristic of neutrophils.
Figure 1.
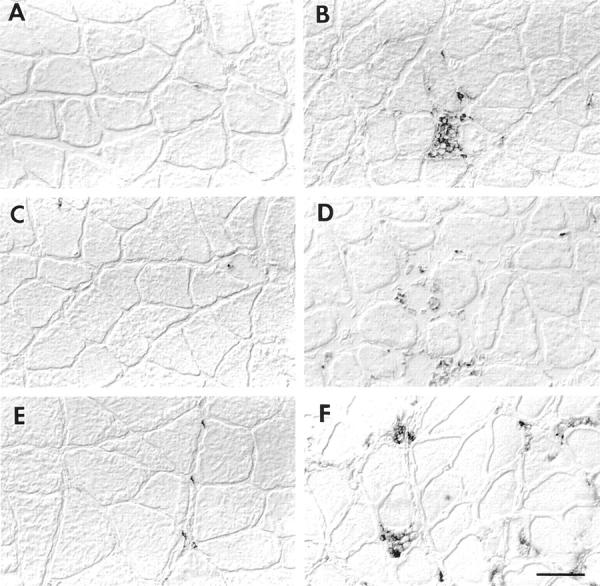
Immunolabeling of neutrophils and ED1+ and ED2+ macrophages. Left column shows cross-sections of soleus muscles from ambulatory control labeled with anti-W3/13 (A), anti-ED1+ (C), or ED2+ (E). Right column shows soleus muscle after 6 hours of reloading, labeled with anti-W3/13 (B), anti-ED1+ (D), and anti-ED2+ (F). Scale bar, 50 μm.
Figure 2.

Neutrophil concentration in soleus muscles during modified loading. Black bar, ambulatory control animals; white bar, suspended animals; stippled bars, PBS-treated animals; and striped bars, sCR1-treated animals for each of the reloading time points indicated. n = 6 for all groups. *, significantly different from suspended animals. #, significantly different from time-matched PBS-treated animals. Error bars, SE.
The administration of sCR1 significantly reduced muscle inflammation and necrosis by 27% at the 6-hour reloading time point when compared to time-matched, PBS-injected controls (Figure 2) ▶ . Similarly, sCR1 reduced ED1+ cell concentration by 46% at 6 hours of reloading, but did not significantly affect the much larger increase that was observed at 24 hours of reloading (Figure 3) ▶ . Inhibition of complement by sCR1 was also apparent in the sCR1-mediated decrease in ED1+ cell invasion of muscle fibers at 6 hours, but not at 24 hours of reloading (Figure 4) ▶ . There was no significant increase in ED2+ cell concentration in either PBS or sCR1-treated animals at 2 or 6 hours of reloading, compared to animals experiencing unloading only (Figure 5) ▶ . However, at 24 hours of reloading, both PBS-treated and sCR1-treated animals showed elevated concentrations of ED2+ cells in soleus muscles. Administration of sCR1 did not affect the increase in ED2+ cell concentration that occurred at 24 hours of reloading. Administration of sCR1 also reduced the degree of edema (−22%) at 6 hours of reloading, as compared to PBS-treated controls (Figure 6) ▶ .
Figure 3.

ED1+ cell concentration in soleus muscles during modified loading. Symbols and number of animals per group are the same as in Figure 2 ▶ .
Figure 4.

Muscle fiber necrosis as percentage of total fibers in soleus cross-section. Fibers invaded by ED1+ macrophages were counted as necrotic. Symbols and number of animals per group are the same as in Figure 2 ▶ .
Figure 5.
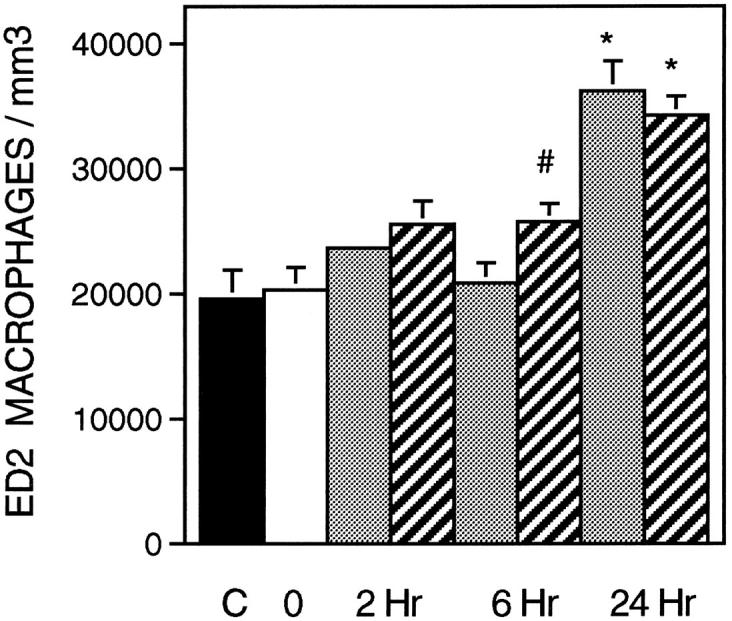
ED2+ cell concentration in soleus muscles during modified loading. Symbols and number of animals per group are the same as in Figure 2 ▶ .
Figure 6.

Index of edema in soleus muscles as estimated by measuring the percentage of muscle cross-section that was occupied by extracellular matrix. Black bar, ambulatory control (n = 6); stippled bar, PBS-treated animals reloaded for 6 hours (n = 9); striped bar, = sCR1-treated animals reloaded for 6 hours (n = 6).
Complement Activation during Muscle Reloading
Complement activation was determined by measuring the concentration of complement C4 and factor B, which are molecules whose functions are restricted to the classical and alternative pathways, respectively. If the complement system is activated, complement C4 and factor B are proteolytically cleaved and their fragments used as substrate for the subsequent reaction. Thus, a decrease in concentration of either factor in the bioassay used here reflects their cleavage in vivo as the classical or alternative pathway is activated. Analysis of complement C4 concentration in serum showed that there was a significant decrease after 2 hours of reloading when compared to suspended animals, indicating that the classical pathway was activated during the first hours of reloading. The concentration of C4 returned to the ambulatory control level after 24 hours of reloading. The presence of sCR1 in serum (mean concentration, 80.3 μg/ml) abrogated the activation of the classical pathway and reduced significantly the lysis of sensitized sheep red blood cells (Figure 7) ▶ .
Figure 7.
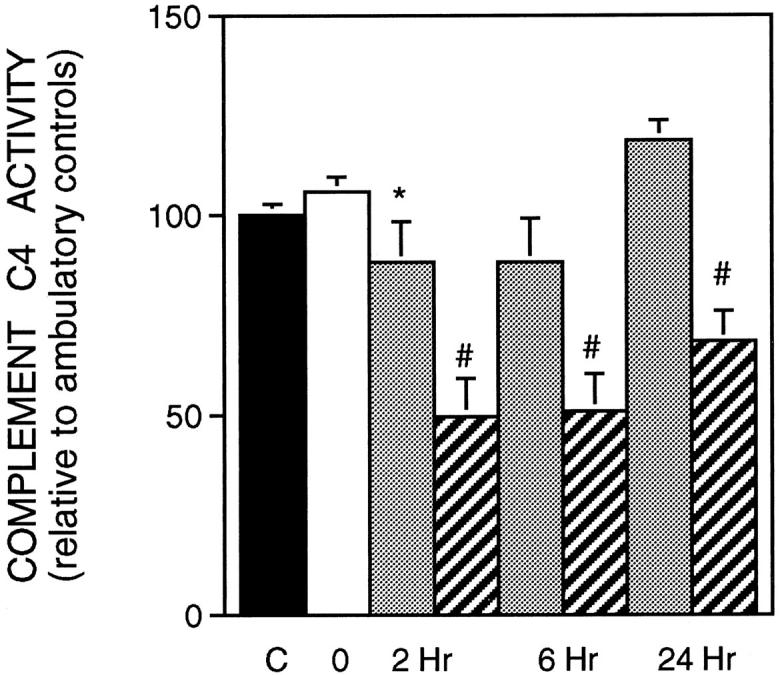
Complement C4 concentration in serum. Symbols are the same as in Figure 2 ▶ . Ambulatory control (n = 6); suspended animals (n = 6); PBS-treated-animals reloaded for 2 hours (n = 6); sCR1-treated animals reloaded for 2 hours (n = 4); PBS-treated animals reloaded for 6 hours (n = 6); sCR1-treated animals reloaded for 6 hours (n = 3); PBS-treated animals reloaded for 24 hours treated (n = 6); sCR1-treated animals reloaded for 24 hours (n = 4).
Factor B concentration was significantly reduced in the PBS-treated group after 6 hours of reloading, when compared to suspended animals. The alternative pathway was also inhibited by the presence of sCR1 in serum where a reduction by >50% of hemolysis was observed in serum from rats reloaded for 2, 6, or 24 hours (Figure 8) ▶ . Staining of muscle sections show that deposition of complement C3 was observed in the positive control sections in which the soleus muscles were injected with bupivacaine for 1 hour. Complement C3 was also clearly detectable on the surface of muscle fibers at 2 hours of the reloading, but only faintly detectable at 24 hours of reloading (Figure 9) ▶ .
Figure 8.

Concentration of factor B in serum. Symbols are the same as in Figure 2 ▶ . Ambulatory control (n = 15); suspended animals (n = 14); PBS-treated animals reloaded for 2 hours (n = 21); sCR1-treated animals reloaded for 2 hours (n = 4); PBS-treated animals reloaded for 6 hours (n = 6); sCR1-treated animals reloaded for 6 hours (n = 3); PBS-treated animals reloaded for 24 hours treated (n = 6); sCR1-treated animals reloaded for 24 hours (n = 4).
Figure 9.
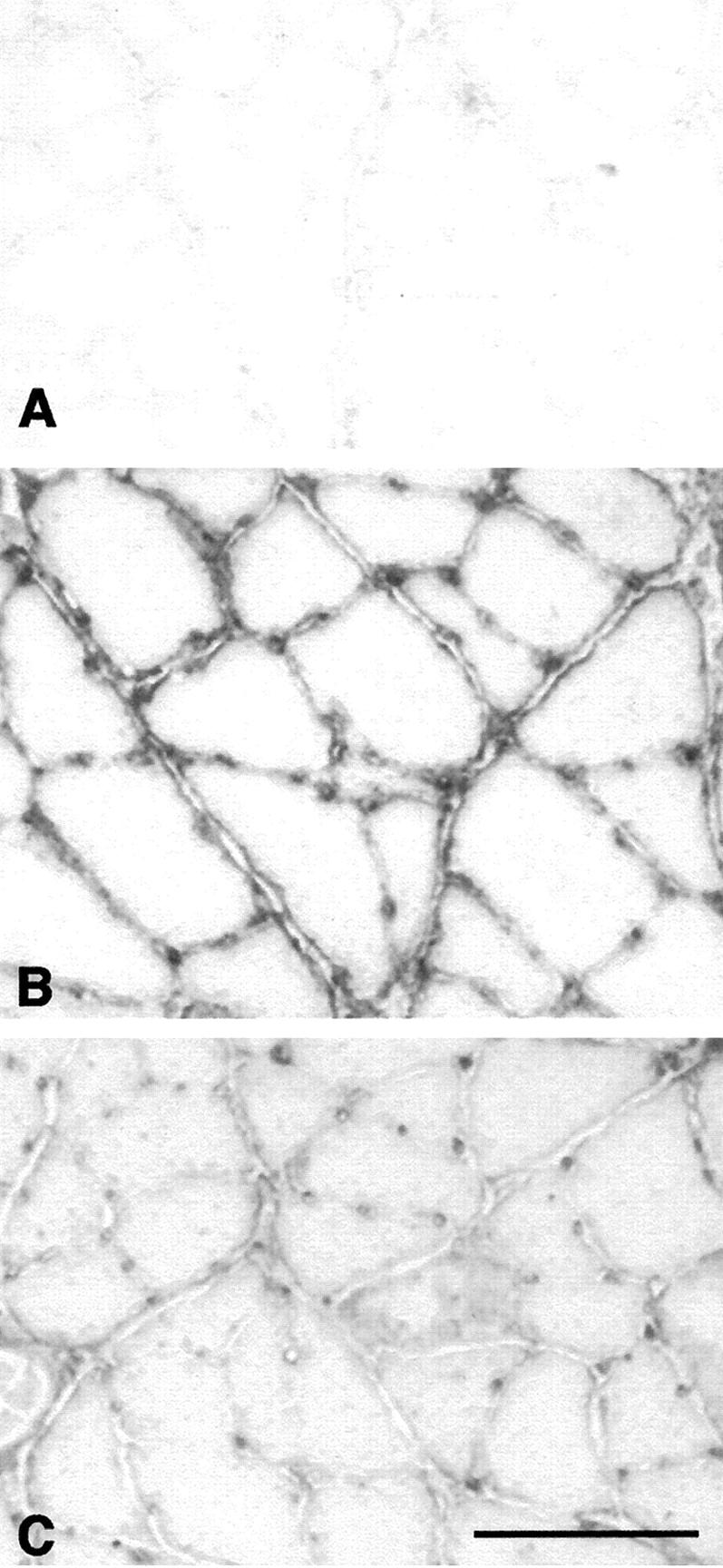
Immunolabeling of complement C3 on cross-sections of soleus muscles. A: sample from rat muscle experiencing unloading only. B: sample from rat experiencing 2 hours of reloading that shows C3 at the surface of muscle fibers. C: sample from rat experiencing 24 hours reloading that shows a reduction in C3 at the surface of muscle fibers, compared to 2 hour reloaded animals. Scale bar, 80 μm.
Discussion
Complement-mediated cytolysis has been implicated in playing a significant role in myopathies. For example, complement activation occurs in dermatomyositis, 25 myasthenia gravis, 6 X-linked vacuolated myopathy, 26 and after periods of ischemia and reperfusion. 9,10 Increases in complement cleavage products C3a and C4a in sera have also been observed after prolonged exercise 27 and C3 has been reported to increase in rat muscle after a 14-day space flight, 16 which suggests that complement-mediated inflammation and necrosis may occur during modified muscle use. Although the role for complement in muscle inflammation and injury has been confirmed in ischemia/reperfusion injuries by demonstrating that complement inhibitors can reduce injury and inflammation, 18,20,21 a role for complement in muscle injury and inflammation during modified use has not been experimentally tested previously.
The present findings show that muscle use after periods of muscle unloading increases activation of the complement system through both the classical and alternative pathways, and that activation increases muscle inflammation. Two mechanisms are known to be capable of initiating activation of the classical pathway: 1) an antibody-dependent mechanism in which specific immunoglobulins bind antigen and that complex leads to complement activation; 28 or 2) an antibody-independent mechanism in which cytosolic particles leaking from necrotic cells react with C1 without requiring antibody-antigen complex formation, which then activates the complement system. 29,30 The antibody-dependent mechanism is the best characterized and has been shown to be involved in other models of muscle injury. For example, recombination-activation gene 2-deficient mice, which are totally deficient in antibody, show less muscle injury after ischemia/reperfusion injuries in which complement is normally activated. 9 In injuries that result in rapid activation of complement via antibody-dependent mechanisms, it is expected that novel antigenic determinants are presented to pre-existing immunoglobulins 9 because activation occurs too rapidly to result from the production of new immunoglobulins to a unknown antigen. 31
The antibody-independent mechanism of activation is not well understood but has been demonstrated to occur in other models of muscle injury, such as cardiac ischemia/reperfusion. 32 In this case, mitochondrial proteins leaking from the injured cardiac cells can directly or indirectly cause the activation of complement without requiring antibody binding. 32 If antibody-independent activation of the classical pathway occurs in the present model system, it implies that some activation molecule is released during the reloading process, although we cannot determine the identity or source of the activating molecule with the present data. However, a potential source would be the muscle fibers that experience increased loading. Mechanical loads have been shown previously to lead to the creation of membrane lesions in muscle that permit the escape of cytosolic molecules into the extracellular space. 33 Previous investigations have shown that mitochondrial membrane proteins 32 and intermediate filament proteins 30 are capable of antibody-independent activation of complement, and both of these types of protein would be present in relatively high concentrations in the reloaded soleus muscles.
The results of this study also show that complement activation contributes to the recruitment of leukocytes to muscle at 6 hours of reloading. Two mechanisms by which complement activation may directly affect leukocyte invasion are by producing substances such as C5a that attract inflammatory cells to the site of injury, or by stimulating the expression of adhesion proteins required for leukocyte diapedesis. 34-36 If complement activation occurs through antibody-independent activation of the classical pathway, it would be likely that complement fragments serve a chemotactic role in the inflammation examined here. Because complement will be distributed throughout the extracellular space at relatively high concentrations whereas the putative activating molecule would be present in a concentration gradient that was highest in the reloaded muscle, this would produce a gradient of activated complement and complement fragments that could attract inflammatory cells to the injured muscle. However, this possibility can be confirmed only if the identity of the activating molecule were known.
The possible involvement of complement activation in stimulating diapedesis is also consistent with the reduction in the concentration of inflammatory cells in the muscles of reloaded animals treated with sCR1. For example, C5a or C5b-9 can induce expression of P- and E-selectins, intercellular adhesion molecule-1, and CD11b/CD18 integrin 34-38 which can in turn increase diapedesis, but this mechanism of stimulating leukocyte diapedesis would be inhibited by sCR1 treatments. However, other adhesion molecules involved in diapedesis, such as l-selectin and intercellular adhesion molecule-2 are not known to be influenced by complement, which may underlie the ability of some inflammatory cells to invade reloaded muscle even in the presence of sCR1.
The finding that administration of sCR1 reduces muscle edema indicates that complement also plays a role in muscle edema in this model of modified muscle use. Several previous reports support the likelihood that sCR1 inhibition of edema is attributable to blocking C5a production. For example, C5a can induce degranulation of mast cells to increase vascular permeability. 39 However, another investigation 16 has shown that modified muscle use can result in edema that is not associated with either an increase in mast cell numbers or increased mast cell degranulation. Although it is not known whether mast cell-independent edema is also complement-mediated, it may be partly attributable to direct damage of the vascular endothelium by complement.
In summary, we conclude that: 1) the complement system is activated through both the classical and alternative pathways during modified muscle use; 2) complement inhibition by sCR1 reduces the number of neutrophils and ED1+ macrophages, which suggests that complement fragments are involved in promoting the adhesion and/or migration of leukocytes toward injured muscle; and 3) complement activation contributes to muscle edema and muscle fiber necrosis during modified muscle use.
Footnotes
Address reprint requests to James G. Tidball, Department of Physiological Science, 5833 Life Science Building, University of California, Los Angeles, CA 90095-1527. E-mail: jtidball@physci.ucla.edu.
Supported by grant NAG5–4837 from NASA.
References
- 1.Krippendorf BB, Riley DA: Temporal changes in sarcomere lesions of rat adductor longus muscles during hindlimb reloading. Anat Rec 1994, 238:304-310 [DOI] [PubMed] [Google Scholar]
- 2.St. Pierre B, Tidball JG: Differential response of macrophage subpopulation to soleus muscle reloading after rat hindlimb suspension. J Appl Physiol 1994, 77:290-297 [DOI] [PubMed] [Google Scholar]
- 3.Leterme D, Falempin M: Contractile properties of rat soleus motor units following 14 days of hindlimb unloading. Pflugers Arch Eur J Physiol 1996, 432:313-319 [DOI] [PubMed] [Google Scholar]
- 4.Spuler S, Engel AG: Unexpected sarcolemmal complement membrane attack complex deposits on nonnecrotic muscle fibers in muscular dystrophies. Neurology 1998, 50:41-46 [DOI] [PubMed] [Google Scholar]
- 5.Sewry CA, Dubowitz V, Abraha A, Luzio JP, Campbell AK: Immunocytochemical localisation of components C8 and C9 in human diseased muscle. The role of complement in muscle fibre damage. J Neurol Sci 1987, 81:141-153 [DOI] [PubMed] [Google Scholar]
- 6.Tsujihata M, Yoshimura T, Satoh A, Kinoshita I, Matsuo H, Mori M, Nagataki S: Diagnostic significance of IgG, C3 and C9 at the limb muscle motor end-plate in minimal myasthenia gravis. Neurology 1989, 39:1359-1363 [DOI] [PubMed] [Google Scholar]
- 7.Mascaro JM, Hausmann G, Herrero C, Grau JM, Cid M, Palou J: Membrane attack complex deposits in cutaneous lesions of dermatomyosites. Arch Dermatol 1995, 131:1386-1392 [DOI] [PubMed] [Google Scholar]
- 8.Orimo S, Hiyamuta E, Arahata K, Sugita H: Analysis of inflammatory cells and complement C3 in bupivacaine-induced myonecrosis. Muscle Nerve 1991, 14:515-520 [DOI] [PubMed] [Google Scholar]
- 9.Weiser MR, Williams JP, Moore FD, Kobzik L, Ma M, Hechtman HB, Carroll MC: Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med 1996, 183:2343-2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vakeva A, Morgan BP, Tikkanen I, Helin K, Laurila P, Meri S: Time course of complement activation and inhibitor expression after ischemic injury of rat myocardium. Am J Pathol 1994, 144:1357-1368 [PMC free article] [PubMed] [Google Scholar]
- 11.Marks RM, Todd RF, Ward PA: Rapid induction of neutrophil-endothelial adhesion by endothelial complement fixation. Nature 1989, 339:314-317 [DOI] [PubMed] [Google Scholar]
- 12.Kajita T, Hugli TE: C5a-induced neutrophilia—a primary humoral mechanism for recruitment of neutrophils. Am J Pathol 1990, 137:467-477 [PMC free article] [PubMed] [Google Scholar]
- 13.Meuer S, Ecker U, Hadding U, Bitter-Suermann D: Platelet-serotonin release by C3a and C5a: two independent pathways of activation. J Immunol 1981, 126:1506-1509 [PubMed] [Google Scholar]
- 14.Chenoweth DE, Hugli TE: Demonstration of specific C5a receptor on intact human polymorphonuclear leukocytes. Proc Natl Acad Sci USA 1978, 75:3943-3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schafer H, Mathey D, Hugo F, Bhakdi S: Deposition of the terminal C5b-9 complement complex in infarcted areas of human myocardium. J Immunol 1986, 137:1945-1949 [PubMed] [Google Scholar]
- 16.Riley DA, Ellis S, Giometti CS, Hoh JF, Ilyina-Kakueva EI, Oganov VS, Slocum GR, Bain JL, Sedlak FR: Muscle sarcomere lesions and thrombosis after spaceflight and suspension unloading. J Appl Physiol 1992, 73:33S-43S [DOI] [PubMed] [Google Scholar]
- 17.Maroko PR, Carpenter CB, Chiariello M, Fishbein MC, Randvany P, Kostman JD, Hale SL: Reduction by cobra venom factor of myocardial necrosis after coronary artery occlusion. J Clin Invest 1978, 61:661-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisman HF, Bartow T, Leppo MK, Marsh HC, Carson GR, Concido MF, Boyle MDP, Roux KH, Weisfeldt ML, Fearon DT: Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischaemic myocardial inflammation and necrosis. Science 1990, 249:146-151 [DOI] [PubMed] [Google Scholar]
- 19.Mulligan MS, Schmid E, Beck-Schimmer B, Till GO, Friedl HP, Brauer RB, Hugli TE, Miyasaka M, Wagner RL, Johnson KJ, Ward PA: Requirement and role of C5a in acute inflammatory injury in rats. J Clin Invest 1996, 98:503-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindsay TF, Hill J, Ortiz F, Rudolph A, Valeri CR, Hechtman HB, Moore FD: Blockage of complement activation prevents local and pulmonary albumin leak after lower torso ischaemia-reperfusion. Ann Surg 1992, 216:677-683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill J, Lindsay TF, Ortiz F, Yeh GC, Hechtman HB, Moore FD: Soluble complement receptor 1 ameliorates the local and remote organ injury after intestinal ischemia-reperfusion injury in the rat. J Immunol 1992, 149:1723-1728 [PubMed] [Google Scholar]
- 22.Morey-Holton E, Wronski J: Animal models for stimulating weightlessness. Physiologist 1981, 24(Suppl.):S45-S48 [Google Scholar]
- 23.Rubin BB, Smith A, Liauw S, Isenman D, Romaschin AD, Walker PM: Complement activation and white cell sequestration in postischemic skeletal muscle. Am J Physiol 1990, 259:H525-H531 [DOI] [PubMed] [Google Scholar]
- 24.Tidball JG, Berchenko E, Frenette J: Macrophage invasion does not contribute to muscle membrane injury during inflammation. J Leukoc Biol 1999, 65:492-498 [PubMed] [Google Scholar]
- 25.Kissel JT, Halterman RK, Rammohan KW, Mendell JR: The relationship of complement-mediated microvascularity to the histologic features and clinical duration of disease in dermatomyositis. Arch Neurol 1991, 48:26-30 [DOI] [PubMed] [Google Scholar]
- 26.Villanova M, Louboutin JP, Chateau D, Eymard B, Sagniez M, Tomé FMS, Fardeau M: X-linked vacuolated myopathy: complement membrane attack complex on surface membrane of injured muscle fibers. Ann Neurol 1995, 37:637-645 [DOI] [PubMed] [Google Scholar]
- 27.Dufaux B, Order U: Complement activation after prolonged exercise. Clin Chim Acta 1989, 179:45-50 [DOI] [PubMed] [Google Scholar]
- 28.Porter RR, Reid KB: Activation of the complement system by antibody-antigen complexes: the classical pathway. Adv Protein Chem 1979, 33:1-71 [DOI] [PubMed] [Google Scholar]
- 29.Pinckard RN, Olson MS, Giclas PC, Terry R, Boyer JT, O’Rourke RA: Consumption of classical complement components by heart subcellular membranes in vitro and in patients after acute myocardial infarction. J Clin Invest 1975, 56:740-750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linder E, Lehto VP, Stenman S: Activation of complement by cytoskeletal intermediate filaments. Nature 1979, 278:176-178 [DOI] [PubMed] [Google Scholar]
- 31.Kelley RE, Olson MS, Pinckard RN: Characterization of an anti-heart mitochondria autoantibodies produced in dogs following myocardial infarction. Circ Res 1974, 35:862-870 [DOI] [PubMed] [Google Scholar]
- 32.Ciglas PC, Pinckard RN, Olson MS: In vitro activation of complement by isolated human heart subcellular membranes. J Immunol 1979, 122:146-151 [PubMed] [Google Scholar]
- 33.Clarke MS, Feeback DL: Mechanical load induces sarcoplasmic wounding and FGF release in differentiated human skeletal muscle cultures. FASEB J 1996, 10:502-509 [DOI] [PubMed] [Google Scholar]
- 34.Foreman KE, Vaporciyan AA, Bonish BK, Jones ML, Johnson KJ, Glovsky MM, Eddy SM, Ward PA: C5a-induced expression of P-selectin in endothelial cells. J Clin Invest 1994, 94:1147-1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mulligan MS, Schmid E, Till GO, Hugli TE, Friedl HP, Roth RA, Ward PA: C5a-dependent upregulation in vivo of lung vascular P-selectin. J Immunol 1997, 158:1857-1861 [PubMed] [Google Scholar]
- 36.Fletcher MPG, Stahl G, Longhurst J: C5a-induced myocardial ischemia: role for CD18-dependent PMN localization and PMN-platelet interactions. Am J Physiol 1993, 265:H1750-H1761 [DOI] [PubMed] [Google Scholar]
- 37.Kilgore KS, Shen J, Miller BF, Ward PA, Warren JS: Enhancement by the complement membrane attack complex of TNF-induced endothelial cell expression of E-selectin and ICAM-1. J Immunol 1995, 155:1434-1441 [PubMed] [Google Scholar]
- 38.Rinder CS, Rinder HM, Smith BR, Fitch JCK, Smith MJ, Tracey JB, Matis LA, Squinto SP, Rollins SA: Blockage of C5a and C5b-9 generation inhibits leukocyte and platelet activation during extracorporeal circulation. J Clin Invest 1995, 96:1564-1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson AR, Hugli TE, Muller-Eberhard HJ: Release of histamine from rat mast cells by complement peptides C3a and C5a. Immunology 1975, 6:1067-1080 [PMC free article] [PubMed] [Google Scholar]