

Abstract
In the passive Heymann nephritis (PHN) model of membranous nephropathy, complement C5b-9 induces glomerular epithelial cell (GEC) injury and proteinuria, which is partially mediated by eicosanoids. This study addresses the role of cyclooxygenase (COX)-1 and -2 in C5b-9-mediated eicosanoid production in GEC. Unstimulated rat GEC in culture primarily express COX-1. When stimulated with sublytic C5b-9, COX-2 was significantly up-regulated, whereas COX-1 was not affected. Compared with control, complement-treated GEC produced 32% more prostaglandin (PG) E2 in the presence of exogenous substrate, and the increase was abolished with the COX-2-selective inhibitor, NS-398. Release of arachidonic acid from GEC phospholipids via C5b-9-induced activation of cytosolic phospholipase A2 was associated with a marked stimulation of PGE2 production, which was inhibited by 60% with NS-398. The results in cultured GEC were extended to GEC injury in vivo by examining COX-1 and -2 expression in PHN. Glomeruli from rats with PHN expressed significantly more COX-1 and COX-2, as compared with normal rats. PGE2 production in glomeruli of rats with PHN was about twofold greater than in control glomeruli, and the increase was partially inhibited with NS-398. Thus, in GEC in culture and in vivo, C5b-9-induced eicosanoid production is regulated by both isoforms of COX. The inducible COX-2 may be an important novel mediator of C5b-9-induced glomerular injury.
Membranous nephropathy is a common cause of nephrotic syndrome in adults, and approximately one-third of affected patients will develop end stage renal failure, necessitating renal replacement therapy. In membranous nephropathy, injury of the glomerular capillary wall leads to impaired glomerular permselectivity and proteinuria. Passive Heymann nephritis (PHN) 1 in the rat closely resembles human membranous nephropathy, and PHN has been used to study the pathogenesis of the human disorder. In PHN, the complement C5b-9 membrane attack complex induces nonlytic injury of glomerular visceral epithelial cells (GEC), in association with altered GEC morphology and proteinuria. 1,2 A number of studies have demonstrated that metabolites of arachidonic acid (eicosanoids) play an important role in the pathogenesis of proteinuria in membranous nephropathy. Specifically, prostaglandin (PG) and thromboxane (TX) A2 production is enhanced in glomeruli isolated from rats with PHN, and inhibition of cyclooxygenase (COX) or TX synthase, or shifting production of dienoic prostanoids to inactive metabolites using fish oil diet, reduces proteinuria in certain models of membranous nephropathy. 3-6 The effect of TXA2 on proteinuria may be through an increase in glomerular transcapillary pressure, since this parameter is elevated in rat membranous nephropathy and appears to be responsible for a portion of the enhanced urine protein excretion. 7,8
We have previously used rat GEC in culture to characterize biochemical changes induced by sublytic C5b-9, including arachidonic acid metabolism. We have shown that sublytic C5b-9 activates cytosolic phospholipase A2 (cPLA2) in a calcium- and protein kinase C-dependent manner. 9-11 Free arachidonic acid released by cPLA2 is further converted to bioactive eicosanoids, including prostaglandins (PGs) and TXA2. The metabolism of arachidonic acid to PGs is catalyzed by COX. 12 To date, two isoforms of COX, namely COX-1 and COX-2, have been characterized. Although both isoforms have similar structures, enzymatic properties, and intracellular distribution, their modes of regulation are distinct. In contrast to COX-1, which is constitutively expressed in most mammalian cells, COX-2 protein is not expressed in most tissues under normal physiological conditions, but is induced in certain cell types in response to growth factors, tumor promoters, hormones, bacterial endotoxin, and cytokines. 12 Thus, it has been proposed that COX-1 may be involved in PG synthesis for maintenance of normal physiology, whereas COX-2 may produce PGs for inflammatory processes or mitogenesis.
Although there is considerable evidence to support a major pathogenetic role for eicosanoids in membranous nephropathy, little is known about the regulation and relative importance of the two COX isoforms in this disease. In the current study, we used cultured rat GEC to characterize the expression and regulation of COXs by C5b-9, and we defined the role of the two isoforms in complement-mediated arachidonic acid metabolism. We also extended the results in cultured GEC to in vivo C5b-9-dependent GEC injury by further characterizing COX-mediated arachidonic acid metabolism in the PHN model of membranous nephropathy.
Materials and Methods
Materials
Tissue culture media, Trizol reagent, Random Primer DNA Labeling System, and RNase T1 were obtained from Gibco BRL (Burlington, ON). NuSerum was purchased from Collaborative Research (Bedford, MA). [3H]PGE2 (200 Ci/mmol), [α-32P]dCTP (3000 Ci/mmol), and [α-32P]CTP (3000 Ci/mmol) were purchased from New England Nuclear (Boston, MA). PGE2, anti-PGE2 antiserum, C8-deficient human serum (C8D), purified human C8, and RNase A were from Sigma Chemical Co. (St. Louis, MO). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting reagents were from BioRad Laboratories (Mississauga, ON). Goat anti-COX-1 antiserum was from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-COX-2 antiserum was from Cayman Chemical (Ann Arbor, MI). β-actin cDNA probe was purchased from Ambion, Inc. (Austin, TX). Plasmids containing coding regions of rat COX-1 and -2 were kindly provided by Dr. Brian Kennedy (Merck-Frosst, Point Claire-Dorval, QC), 13 and each coding region was subcloned into the mammalian expression vector pcDNA3 (Invitrogen, Carlsbad, CA).
Culture of GEC
Primary cultures of rat GEC were established from explants of rat glomeruli. 10,11 Characterization of GEC was published previously. 10,11 According to established criteria, the cells demonstrated polygonal shape and cobblestone appearance at confluency, cytotoxic susceptibility to low doses of aminonucleoside of puromycin, positive immunofluorescence staining for cytokeratin, and presence of junctional complexes and apical microvilli by electron microscopy. The standard medium used to maintain GEC cultures, K1, consisted of Dulbecco’s modified Eagle’s medium/Ham F-10 (1:1) containing 5.0% NuSerum and hormone supplements. A subclone of GEC that stably overexpresses cPLA2 (fivefold above the endogenous level), or neo-GEC (control) were used in this study. These clones were produced and characterized previously. 10,11 Studies were done with cells between passages 4 and 70. For total RNA preparation, GEC were cultured in serum-poor medium (Dulbecco’s modified Eagle’s medium/Ham F-10 (1:1) with 0.5% fetal calf serum) for 16 hours before experiments.
Incubation of GEC with Complement
Rabbit antiserum to GEC 10 was used to activate complement on GEC membranes. Briefly, GEC were incubated with antiserum (5% v/v) in serum-poor medium for 40 minutes at 22°C. GEC were then incubated with normal human serum (2.5–3.0% v/v in serum-poor medium) or with heat-inactivated (decomplemented) human serum (56°C, 30 minutes) in controls, for the indicated times at 37°C. In some experiments, antibody-sensitized GEC were incubated with C8D (2.5–5.0% v/v) reconstituted with or without purified human C8 (80 μg/ml undiluted serum). We have generally used heterologous complement to facilitate studies with complement-deficient sera and to minimize possible signaling via complement-regulatory proteins; however, in previous studies, results of several experiments involving arachidonic acid metabolism were confirmed with homologous (rat) complement. 10 Sublytic concentrations of complement (≤5% normal human serum) were established previously. 10 Previous studies have shown that in GEC, complement is not activated in the absence of antibody.
Northern Blot Hybridization
Northern blot hybridization was performed as described previously. 14 Total RNA was extracted from GEC using the Trizol reagent according to manufacturer’s protocol. RNA (15 μg) was separated by gel electrophoresis on 1% agarose gels containing 1.9% formaldehyde and transferred to a nylon membrane. Coding regions of rat COX-1 and -2 cDNAs were radiolabeled with [α-32P]dCTP using the Random Primer DNA Labeling System. Membranes were hybridized in hybridization buffer (1% bovine serum albumin, 7% SDS, 0.5 Mol/L phosphate buffer, pH 6.8, 1 mmol/L EDTA), containing 1–2 × 10 6 cpm/ml of radiolabeled probe for 16 hours at 42°C, followed by washing in buffer A (0.5% bovine serum albumin, 5% SDS, 40 mmol/L phosphate buffer, pH 6.8, 1 mmol/L EDTA) twice for 20 minutes at 65°C, and then in buffer B (1% SDS, 40 mmol/L phosphate buffer, pH 6.8, 1 mmol/L EDTA) 4 times for 20 minutes at 65°C. Membranes were exposed to X-ray film with an intensifying screen at −70°C for 48 to 72 hours. The amount of mRNA was quantitated using scanning densitometry (NIH Image software).
RNase Protection Assay
RNase protection assay was performed using the methods described by Feng et al, except that pcDNA3 was used to construct probes for COX-1 and -2. 15 A 341-bp fragment of rat COX-1 cDNA produced by BstXI (from bp 1297 to bp 1637 in the coding region) was blunted and subcloned into the EcoRV site in pcDNA3. A 241-bp fragment of rat COX-2 cDNA produced by BamHI and EcoRI (from bp 291 to bp 531 in the coding region) was subcloned in pcDNA3. XhoI was used to linearize both COX-1 and COX-2 templates for labeling. 32P-labeled antisense cRNA probes were synthesized by in vitro transcription, using linearized COX cDNA templates, or β-actin template, T7 RNA polymerase and [α-32P]CTP. Total RNA (5–8 μg) was hybridized with 1 × 10 5 cpm of each labeled antisense cRNA probe for 16 hours at 55°C. Unhybridized probes were digested with RNaseA (0.3 μg/ml) and RNase T1 (90 U/ml) for 1 hour at 30°C. Then, the RNases were digested with proteinase K (0.5 mg/ml) for 30 minutes at 37°C. After phenol/chloroform extraction and ethanol precipitation, the hybrids were denatured at 85°C for 3 minutes and electrophoresed on 6% polyacrylamide gels. After drying, gels were exposed to X-ray film at −70°C for 24 to 48 hours.
Immunoblotting
GEC or rat glomeruli were lysed in Laemmli buffer (62.5 mmol/L Tris, 2% SDS, 10% glycerol, 0.01% bromphenol blue, pH 6.8) and boiled for 5 minutes. After centrifugation at 14,000 × g, supernatants were collected, and protein content was quantified by a modified Lowry method (Protein DC-assay, BioRad). Equal amounts of protein were separated by 8% SDS-PAGE under reducing conditions. Proteins were then electrophoretically transferred to nitrocellulose membrane, blocked with 5% dry milk, and incubated with goat anti-COX-1 antiserum for 2 hours at 22°C, or with rabbit anti-COX-2 antiserum for 16 hours at 4°C. After 3 washes, membranes were incubated with respective secondary antibodies conjugated with alkaline phosphatase, and alkaline phosphatase activity was detected by the addition of bromochloroindolyl phosphate with nitro blue tetrazolium. Protein content was quantitated using scanning densitometry (NIH Image software).
Measurement of PGE2 and TXA2 Generation by GEC
For measurement of PGE2 production in the presence of exogenous arachidonic acid, GEC were incubated with antibody and complement, and then with arachidonic acid (15 μmol/L) in measurement buffer (145 mmol/L NaCl, 5 mmol/L KCl, 0.5 mmol/L MgSO4, 0.5 mmol/L CaCl2, 1 mmol/L Na2HPO4, 5 mmol/L glucose, 20 mmol/L Hepes, pH 7.4) for 20 minutes at 37°C. For measurement of PGE2 generation resulting from release of endogenous arachidonic acid, culture medium was supplemented with arachidonic acid (7.5–10 μmol/L) for 24 to 48 hours before experiments. GEC were incubated with antibody and complement in serum-poor medium. Supernatants were collected in 100 μmol/L indomethacin to prevent further metabolism, and were used for PGE2 measurement. In some experiments, lipids were extracted from cells and supernatants for PGE2 measurements, as described previously. 10 The amount of PGE2 released into supernatants was equivalent to that from cells plus supernatants, indicating that most of the PGE2 was released from cells into supernatants. PGE2 was measured by radioimmunoassay using anti-PGE2 antibody and [3H]PGE2, according to the manufacturer’s protocol. The range of the standard curve in the assay was 15–500 pg/0.1 ml of PGE2. Samples were incubated with [3H]PGE2 and anti-PGE2 antibody for 1 hour at 4°C, after which time unbound PGE2 was removed by the addition of activated charcoal. The radioactivity of the supernatant was counted in a β-scintillation counter, and PGE2 concentration was calculated from standard formulas. In some experiments, cells were lysed with 1% Triton X-100, and protein content was measured to normalize PGE2 concentration according to protein content. For measurement of TXA2, cells were stimulated as above, and the concentration of TXB2 (the stable metabolite of TXA2) in supernatants was determined by enzyme immunoassay kit (Cayman Chemical). The range of the standard curve in the assay was 7.8 to 1000 pg/ml of TXB2.
Transient Transfection of Cos-1 Cells
Culture of Cos-1 cells was described previously. 11 Transient transfection of Cos-1 cells was performed by the diethylaminoethyl-dextran method. 11 Briefly, cells in 35-mm culture dish were transfected with 0.5 μg of plasmid DNA encoding rat COX-1 or COX-2. Three days after the transfection, cells were incubated with measurement buffer containing arachidonic acid (15 μmol/L) and NS-398 for 30 minutes at 37°C. PGE2 in supernatants was measured by radioimmunoassay.
Induction of PHN
Anti-Fx1A was prepared as described previously. 16 Male Sprague-Dawley rats (150–175 g; Charles River, St. Constant, QC, Canada) were injected with 450 μl of sheep anti-Fx1A antiserum. This batch of antiserum caused minimal proteinuria in the heterologous phase (day 5) but induced significant proteinuria in the autologous phase (day 14). On day 14, rats were sacrificed and glomeruli were isolated by differential sieving, 16 yielding a glomerular preparation that was >95% pure. For Western blotting, glomeruli were lysed in Laemmli buffer as described above (Immunoblotting). For the RNase protection assay, glomerular RNA was prepared using the Trizol reagent. To measure PGE2 generation, glomeruli from each rat were resuspended in 2 ml of measurement buffer and divided into two aliquots. One aliquot was incubated with NS-398 for 15 minutes at 37°C and then centrifuged for 2 minutes at 1,000 × g at 22°C. The second aliquot was treated in an identical manner, except that dimethylsulfoxide was used instead of NS-398 (control). Supernatants were transferred to another set of tubes, which contained indomethacin (final concentration, 100 μmol/L) to terminate the reaction, and 100 μl of each supernatant was used for the measurement of PGE2 by radioimmunoassay.
Statistics
Data are presented as mean ± SEM. The t statistic was used to determine significant differences between two groups. One-way analysis of variance (ANOVA) was used to determine significant differences among groups. Two-way ANOVA was used to determine significant differences in multiple measurements among groups. Where significant differences were found, individual comparisons were made between groups using the t statistic, and adjusting the critical value according to the Bonferroni method.
Results
C5b-9 Induces COX-2 mRNA and Protein in GEC
The first series of experiments were designed to determine whether sublytic C5b-9 regulates expression of COX mRNA. Unstimulated neo-GEC expressed COX-1 mRNA at a level readily detectable by Northern hybridization (using 15 μg of total RNA), whereas COX-2 mRNA was not detectable under the same conditions (Figure 1A) ▶ . When neo-GEC were stimulated with a sublytic concentration of complement by serial exposure to anti-GEC antibody and normal serum, COX-2 mRNA was up-regulated significantly, peaking approximately 2 hours after the initiation of C5b-9 assembly and continuing for at least for 4 hours. When heat-inactivated (decomplemented) serum was used instead of normal serum (control incubations), COX-2 mRNA up-regulation was trivial, indicating that COX-2 up-regulation is most likely mediated by complement activation. The small up-regulation of COX-2 induced by heat-inactivated serum (at 40 minutes) may be attributed to some minor stimulatory component(s) in the serum, such as growth factors. At the same time points, there were no consistent complement-dependent changes in COX-1 mRNA levels. When the amounts of mRNA were quantified by densitometry at 100 minutes, cells treated with complement had COX-1 mRNA levels similar to control, whereas complement-treated cells had ∼4 times more COX-2 mRNA than control (Figure 1A) ▶ . In addition to the well-described COX-2 transcript of approximately 4.2 kb, a larger transcript (∼5.0 kb) was also observed in Northern blots. The origin of the 5.0-kb transcript has not been clearly established, but posttranscriptional regulation, such as alternate polyadenylation described in other cell types, 17 may account for this phenomenon. These results suggest that expression of the two COX isozymes in GEC is differentially regulated by complement (Figure 1A) ▶ .
Figure 1.
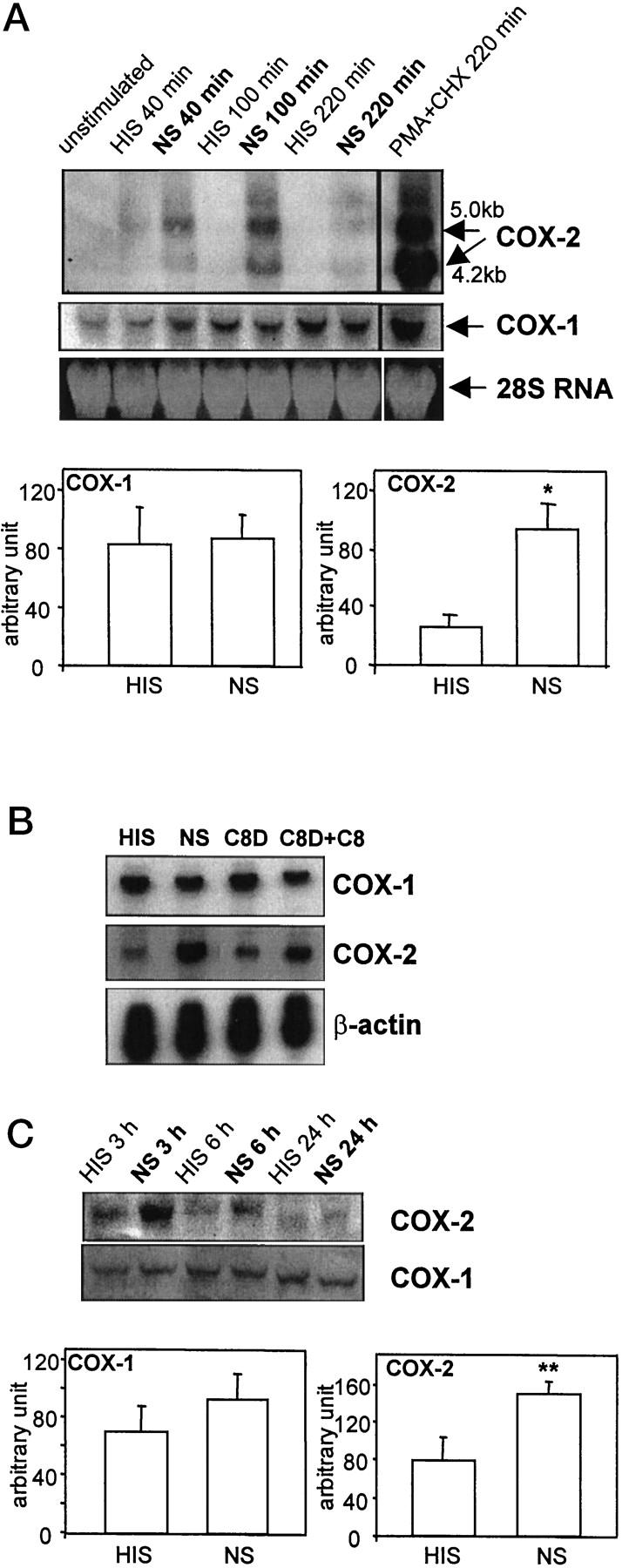
COX-2, but not COX-1, is up-regulated by C5b-9 in GEC. A: Neo-GEC were incubated with anti-GEC antiserum (5% v/v) for 40 minutes at 22°C and then with normal serum (NS, 2.5% v/v, to form C5b-9) or heat-inactivated serum (HIS, 2.5% v/v, control) for the indicated times at 37°C. As a positive control, GEC were incubated with phorbol myristate acetate (PMA, 100 nmol/L) + cycloheximide (CHX, 10 μg/ml). Total RNA (15 μg/lane) was analyzed by Northern blot hybridization. Ethidium bromide staining of the gel (28S RNA) demonstrates equal RNA loading. The bottom panels show quantification of mRNA by densitometry at 100 minutes. *P < 0.05 NS versus HIS (n = 3). B: Neo-GEC were incubated with anti-GEC antiserum, and then HIS, NS, C8-deficient serum (C8D), or C8D+C8 for 100 minutes. Total RNA (8 μg/lane) was analyzed by RNase protection assay. C: Neo-GEC were incubated with anti-GEC antiserum, and then with HIS or NS for the indicated times. Cell protein (100 μg/lane) was analyzed by Western blotting. The bottom panels show quantification of protein by densitometry at 3 hours. **P = 0.02, NS versus HIS (n = 4).
To demonstrate that C5b-9 assembly was actually required for COX-2 mRNA up-regulation, we incubated antibody-treated neo-GEC with C8D, with or without reconstitution with purified C8. C8D without C8 forms C5b-7, which was previously shown to be biologically inactive in GEC. 10 When evaluated by RNase protection assay, GEC incubated with C8D did not show significant COX-2 mRNA up-regulation, as compared with heat-inactivated serum. However, when C8D was reconstituted with purified C8, up-regulation of COX-2 mRNA was evident, indicating that formation of C5b-9 is required for COX-2 up-regulation (Figure 1B) ▶ . Quantification of COX-2 mRNA by densitometry showed that the relative amounts of COX-2 mRNA were heat-inactivated serum (HIS), 1.0; normal serum (NS), 2.2; C8D, 1.1; C8D+C8, 1.7 (average of 2 experiments). It was noted that the effects of C8D reconstituted with C8 were less potent, as compared with normal serum, probably because there appears to be some general loss of complement activity during the immunodepletion of C8 (unpublished observation).
We next examined if the complement-mediated COX mRNA regulation is reflected in protein expression. When neo-GEC were incubated with antibody and complement, COX-2 protein expression was up-regulated, as compared with GEC incubated with heat-inactivated serum (control). This up-regulation was transient, typically peaking at ∼3 hours of stimulation (Figure 1C) ▶ . COX-1 protein expression was not affected by complement for up to 24 hours of stimulation (Figure 1C) ▶ . When the amounts of protein were quantified by densitometry at 3 hours, cells treated with complement had similar amounts of COX-1 protein, as compared with control, whereas complement-treated cells had ∼1.6 times more COX-2 protein, as compared with control (Figure 1C) ▶ .
Role of COX Isozymes in Complement-Dependent Arachidonic Acid Metabolism in GEC
The results of mRNA analysis and immunoblotting suggested that COX-1 was the dominant COX isozyme in resting rat GEC in culture and in GEC incubated with heat-inactivated serum, but that COX-2 expression was up-regulated by sublytic C5b-9. To understand the role of the two COX isozymes in arachidonic acid metabolism in GEC, we next addressed the enzymatic activities of COX isozymes. In these experiments, exogenous arachidonic acid (15 μmol/L) was provided as substrate for COX. After stimulation of neo-GEC with sublytic concentrations of complement for 3 hours, PGE2 generation in the presence of exogenous arachidonic acid was increased by 32%, as compared with GEC that were incubated with heat-inactivated serum (control; Figure 2A ▶ ). A similar increase in PGE2 was observed after 24 hours of stimulation with complement, but the change was not statistically significant (not shown). These results indicate that GEC treated with complement have an increase in total COX enzyme activity. To determine which isozyme was responsible for the complement-mediated increase in COX activity, we examined the effect of the COX-2 selective inhibitor, NS-398. When exogenous arachidonic acid was added together with NS-398 (10−6 M), the complement-mediated increase in PGE2 generation was abolished, indicating that increased COX activity was due to the up-regulation of COX-2 (Figure 2A) ▶ , consistent with an increase in COX-2 protein (Figure 1C) ▶ . Nevertheless, in the presence of exogenous arachidonic acid, ∼75% of total PGE2 production in complement-treated cells was due to constitutive COX-1 activity, and COX-2 contributed ∼25% (Figure 2A ▶ , NS). A downward trend in PGE2 production was induced by NS-398 in GEC incubated with HIS (Figure 2A) ▶ . This result suggests that these GEC may contain a small amount of COX-2 activity and/or that NS-398 cross-reacted with COX-1 (see below).
Figure 2.

GEC incubated with complement have increased COX enzymatic activity. A: Subconfluent (80–90% confluent) monolayers of neo-GEC in 6-cm culture dishes were incubated with antibody and complement as in Figure 1 ▶ . At 3 hours, GEC were washed and incubated with buffer containing arachidonic acid (15 μmol/L), with or without NS-398 (1 μmol/L), for 20 minutes at 37°C. PGE2 in cells and supernatants was measured by radioimmunoassay. Values are mean ± SEM of 3 experiments performed in duplicate. Significant differences were present among groups (P < 0.002 ANOVA; *P < 0.02 NS versus HIS, **P < 0.005 NS versus NS+NS-398). PGE2 production in the control group (ie, HIS) was 1.4 ± 0.3 ng per plate. B: Cos-1 cells were transiently transfected with rat COX-1 or COX-2 cDNA. Three days after transfection, cells were incubated with exogenous arachidonic acid (15 μmol/L), with or without NS-398. PGE2 released into the supernatants was measured by radioimmunoassay. Values are mean ± SEM of 3 experiments performed in duplicate. Significant differences were present among groups (P < 0.0005 two-way ANOVA; *P < 0.0005 versus no NS-398).
NS-398 (10−6 M) was shown previously not to affect human or ovine COX-1 activity. 18-20 We verified the selectivity of NS-398 using Cos-1 cells transfected with rat COX-1 or COX-2 cDNA. When untransfected Cos-1 cells were incubated with exogenous arachidonic acid, the amount of PGE2 released to the supernatant was trivial (∼12 pg/0.1 ml), indicating that untransfected Cos-1 cells do not have significant endogenous COX activity. In contrast, Cos-1 cells transfected with COX-1 or COX-2 cDNA released significant amounts of PGE2 into supernatants when incubated with exogenous arachidonic acid (823 ± 166 pg/0.1 ml, n = 6, and 585 ± 85 pg/0.1 ml, n = 6, respectively). At 10−7 or 10−6 M NS-398, COX-1 activity tended to decrease by ∼10%, whereas COX-2 inhibition was ∼50 to 60% (Figure 2B) ▶ . Although it was possible to achieve almost complete inhibition of COX-2 with 10−5 M NS-398, this concentration of NS-398 also inhibited COX-1 significantly (∼40%).
In the above experiments, exogenous arachidonic acid was provided as substrate for COX. In the next series of experiments, we studied the effect of stimulus-dependent release of endogenous arachidonic acid on PGE2 generation. Previously, we reported that in GEC, sublytic C5b-9 activated cPLA2, leading to release of arachidonic acid and PGE2 generation, 10,11 whereas expression of other PLA2 isoforms, such as group IIA or group V secretory PLA2s, appears to be negligible, with or without stimulation by complement. 21 We have also reported that production of other eicosanoids, including PGF2α and TXA2, was also stimulated by complement. 22 To re-evaluate the role of cPLA2 and endogenous arachidonic acid release in complement-mediated eicosanoid production in view of the newly recognized regulation of COX-2 by complement (Figure 1) ▶ , we examined the profile of PGE2 generation in GEC that stably overexpress cPLA2. 10,11 When antibody-sensitized neo-GEC (vector-transfected cells) were stimulated with complement for up to 24 hours, only a small upward trend in PGE2 generation was noted, as compared with control (heat-inactivated serum; Figure 3A ▶ ). This result was consistent with previous studies and can be explained by the relatively small amount of arachidonic acid release due to the low expression level of endogenous cPLA2 in GEC. 10 In contrast, arachidonic acid release is amplified in complement-treated GEC that overexpress cPLA2, 10 and these cells released about five-to sixfold more PGE2 after 3 and 24 hours of complement stimulation, as compared with neo-GEC (Figure 3A) ▶ . Thus, release of arachidonic acid due to the activation of cPLA2 by complement is the critical rate-limiting step for eicosanoid generation.
Figure 3.
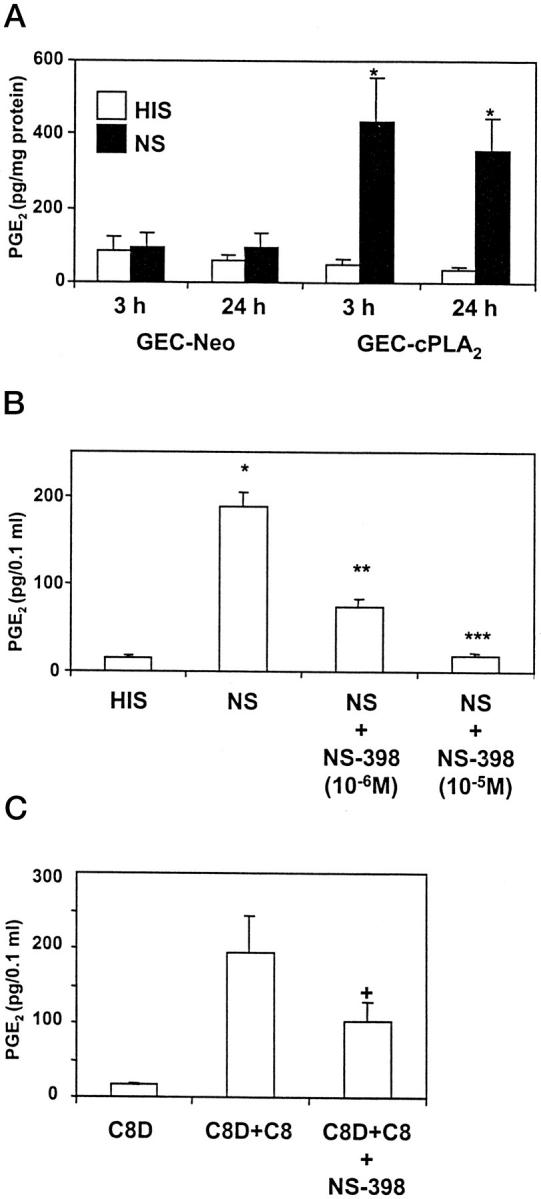
Effect of NS-398 on generation of PGE2 from endogenous arachidonic acid. A: GEC that stably overexpress cPLA2 (GEC-cPLA2) or neo GEC (vector-transfected) were incubated with antibody and complement (as in Figure 1 ▶ ) for 3 or 24 hours. PGE2 in supernatants was measured by radioimmunoassay. Values are mean ± SEM of 3 experiments performed in duplicate. Significant differences were present among groups (P < 0.0001 ANOVA; *P < 0.0001 NS versus HIS). B: GEC that overexpress cPLA2 were incubated with antibody and complement (3% NS) for 3 hours with or without NS-398, and PGE2 in supernatants was measured by radioimmunoassay. Values are mean ± SEM of 3 experiments performed in duplicate. Significant differences were present among groups; P < 0.0001 ANOVA; *P < 0.0001 NS versus HIS, **P < 0.0001 NS versus NS+NS-398 (10−6 M), ***P < 0.0001 NS versus NS+NS-398 (10−5 M). C: GEC that overexpress cPLA2 were incubated with antibody and 5% C8D, C8D+C8, or C8D+C8+NS-398 (1 μmol/L) for 3 hours. PGE2 in supernatants was measured by radioimmunoassay. Values are mean ± SEM of 3 experiments performed in duplicate. Significant differences were present among groups: P = 0.02 ANOVA; +P < 0.04 C8D+C8+NS-398 versus C8D+C8.
We then examined which isoform of COX is coupled to cPLA2-mediated arachidonic acid release. When GEC that overexpress cPLA2 were stimulated with complement for 3 hours in the presence of NS-398 (10−6 M), ∼60% of PGE2 generation was inhibited. NS-398, 10−5 M, inhibited complement-stimulated PGE2 production almost completely (Figure 3B) ▶ . In complement-stimulated GEC that overexpress cPLA2, TXA2 production as measured by its stable metabolite TXB2 (326 ± 52 pg/0.1 ml, n = 10) was increased, as compared with control cells (51 ± 10 pg/0.1 ml, n = 8). This complement-mediated TXA2 generation was also inhibited by NS-398 (10−6 M) by ∼30% (240 ± 55 pg/0.1 ml, n = 9), indicating that COX-2 also mediates complement-induced TXA2 generation.
To demonstrate that eicosanoid production was actually due to assembly of C5b-9, antibody-sensitized GEC were incubated with C8D with or without purified C8. C8D alone did not affect PGE2 generation; however, C8D+C8 stimulated PGE2 generation comparably to normal serum (Figure 3C) ▶ . PGE2 generation stimulated by C8D+C8 was also inhibited by NS-398 (Figure 3C) ▶ . Taken together, these results indicate that after stimulation with C5b-9, at least 50 to 60% of the arachidonic acid released from GEC phospholipids by cPLA2 is metabolized via the C5b-9-inducible COX-2. A smaller portion of arachidonic acid metabolism may be due to COX-1, but it was not possible to determine this precisely, because at doses of NS-398 that inhibited COX-2 completely, there was also cross-inhibition of COX-1.
Expression and Activity of COX Isozymes in PHN
It is important to determine whether results obtained in cultured GEC are relevant to in vivo GEC injury, ie, the PHN model of membranous nephropathy. In PHN, administration of heterologous anti-brush border antiserum (anti-Fx1A) leads to formation of glomerular subepithelial IgG deposits, activation of complement, assembly of C5b-9 in GEC plasma membranes, and GEC injury, which manifests as proteinuria. 1,2 Several studies have demonstrated an important role for eicosanoids, particularly the products of COX, in the glomerular permselectivity defect in PHN. 3-6
To determine which of the two COX isoforms may be involved in eicosanoid generation in PHN, we first examined the expression of COX-1 and -2 in glomeruli of normal rats and rats with PHN. Fourteen days after administration of anti-Fx1A antiserum, rats with PHN developed massive proteinuria (urine protein 352 ± 75 mg/day, n = 8, versus 9 ± 2 mg/day in age-matched normal rats, n = 8). On day 14, glomeruli isolated from control (normal) rats showed clear expression of COX-1 mRNA, whereas the level of COX-2 mRNA was relatively low but detectable (Figure 4) ▶ . Glomeruli from rats with PHN showed that COX-1 mRNA increased ∼1.5-fold, as compared with control, while COX-2 mRNA also increased ∼1.5-fold, although this change failed to reach statistical significance (Figure 4A) ▶ . When protein expression of COX-1 and -2 was examined by Western blotting, rats with PHN showed ∼1.4 times more COX-1 protein as compared with control and ∼1.8 times more COX-2 protein (Figure 4B) ▶ . Thus, by analogy to complement-treated GEC in culture, COX-2 is up-regulated in glomeruli of rats with PHN, and there is low basal expression of COX-2 in glomeruli and in cultured GEC. Similar to cultured GEC, COX-1 was expressed constitutively in glomeruli, but unlike GEC in culture, there appeared to be some COX-1 up-regulation in PHN.
Figure 4.
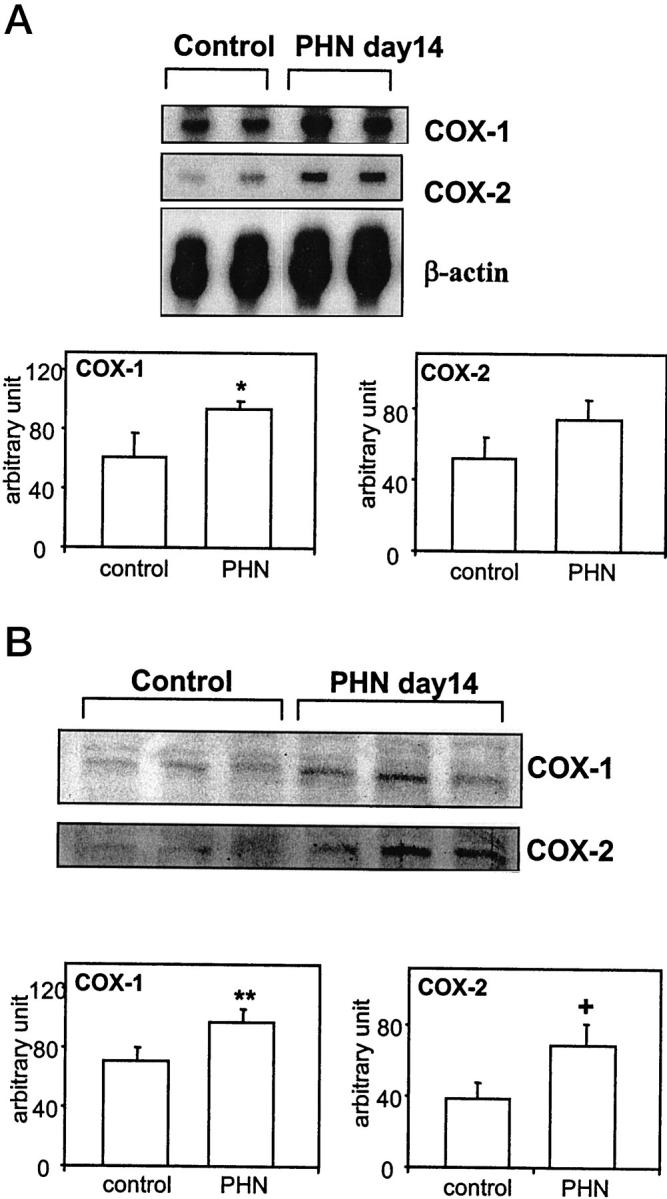
Expression of COX-1 and 2 in glomeruli from normal rats and rats with PHN (2 or 3 animals per group are presented). Rats were sacrificed on day 14, and glomeruli were prepared by differential sieving (see Materials and Methods). A: Total RNA (8 μg/lane) was analyzed by RNase protection assay. B: Glomerular protein (100 μg/lane) was analyzed by Western blotting. The bottom panels show quantification of mRNA (A) or protein (B) by densitometry. *P < 0.05 PHN versus control (n = 6). **P = 0.03, PHN versus control (n = 10–12). +P = 0.02, PHN versus control (n = 11–13).
It is well established that glomeruli from rats with PHN generate more PGE2 and TXA2, as compared with normal rats. 4-6 However, the contribution of each COX isoform to eicosanoid production has not been defined. To determine the contribution of COX-2 to eicosanoid generation, we incubated glomeruli in vitro with or without NS-398 and monitored the generation of PGE2. In keeping with previous reports, basal production of PGE2 was evident in control glomeruli (Figure 5) ▶ . Glomeruli from rats with PHN generated ∼2 times more PGE2, as compared with glomeruli from control rats (Figure 5) ▶ . NS-398 (10−7 M) inhibited PGE2 generation in PHN glomeruli significantly, but not completely, whereas at 10−5 M, there was almost complete inhibition (Figure 5) ▶ . At 10−7 M, NS-398 tended to decrease PGE2 production in control glomeruli, whereas inhibition was significant at 10−5 M. Therefore, a substantial portion of eicosanoid generation in PHN is mediated via COX-2, and the remainder may be mediated via COX-1. However, as in cultured GEC, it was not possible to quantitate precisely how much PGE2 was generated by each COX isoform because NS-398 inhibition of COX-2 is not complete at 10−7 M, and at the 10−5 M concentration, which inhibits COX-2 more effectively, NS-398 also inhibits COX-1 (Figure 2B) ▶ . PGE2 generation in control glomeruli may also be mediated, in part, via COX-2 (Figure 5) ▶ .
Figure 5.

Effect of NS-398 on PGE2 generation in glomeruli isolated from normal rats and rats with PHN (day 14). Glomeruli were prepared as in Figure 4 ▶ . A: Glomeruli were incubated in vitro with NS-398 (10−7 M) or vehicle (dimethylsulfoxide) for 15 minutes, and PGE2 released into the buffer was measured by radioimmunoassay. Values are mean ± SEM of 3 control rats and 5 rats with PHN. Significant differences were present among groups (P < 0.005 ANOVA; *P < 0.01 PHN versus control, **P = 0.02 PHN versus PHN+NS-398). B: Glomeruli were incubated as in A, except NS-398 was added at 10−5 M. Values are mean ± SEM of 4 control rats and 8 rats with PHN. Significant differences were present among groups: P < 0.0001 ANOVA; *P < 0.0001 PHN versus control, **P = 0.0001 PHN versus PHN+NS-398, ***P = 0.0006 control versus control+NS-398.
Complement-Conditioned Medium Does Not Up-Regulate COX-2
It has been demonstrated that COX-2 can be up-regulated by various growth factors and cytokines, and it was reported that complement stimulates the release of growth factors, such as basic fibroblast growth factor. 1 To evaluate the possibility that factors released from GEC on incubation with complement contribute to up-regulation of COX-2 in an autocrine/paracrine fashion, we examined the effect of complement-conditioned medium on COX-2 up-regulation. Media of GEC that had been incubated with antibody and normal serum, or heat-inactivated serum failed to induce a significant change in COX-2 protein level (Figure 6) ▶ . Analogous results were obtained when examining mRNA levels by Northern blot hybridization (Figure 6) ▶ . These results indicate that autocrine/paracrine factors are unlikely to be responsible for COX-2 up-regulation.
Figure 6.

Effect of complement-conditioned medium on COX-2 protein (A) and mRNA (B). GEC were incubated with antibody and normal serum (NS) or heat-inactivated serum (HIS) as in Figure 1 ▶ . At 3 hours (A) or 100 minutes (B), cells were harvested and conditioned media were collected. Conditioned media were added to untreated GEC, and the cells were further incubated for 3 or 6 hours (A) or 100 minutes (B). The two left lanes in each panel (Direct Effect) demonstrate that complement (NS) increased COX-2 protein (A) and mRNA (B) at 3 hours and 100 minutes, respectively. Media collected from the cells that had been incubated with NS or HIS (conditioned media) had no significant effect on levels of COX-2.
Discussion
The current study demonstrates that sublytic C5b-9 induces COX-2 mRNA and protein expression in GEC, the target of injury in experimental membranous nephropathy. The time course of COX-2 induction in cultured GEC was relatively rapid and transient (Figure 1) ▶ , and was similar to the pattern described in fibroblasts, although the amount of inducible COX-2 mRNA and protein expression in GEC was relatively small. 23 Induction of COX-2 protein was associated with an increase in COX-2 enzymatic activity. Thus, in complement-treated GEC, addition of exogenous arachidonic acid resulted in an increase in PGE2 production, which was blocked by the COX-2-selective inhibitor, NS-398 (Figure 2) ▶ . In addition, unstimulated GEC expressed COX-1 mRNA and protein constitutively, and the level of COX-1 expression was not affected by C5b-9 (Figure 1) ▶ . In keeping with our previous results showing that sublytic C5b-9 stimulates cPLA2 activity 9-11 the present study demonstrates that activation of cPLA2 is the critical rate-limiting step for complement-mediated PGE2 generation in GEC (Figure 3A) ▶ . At concentrations of complement that did not stimulate PGE2 production significantly in neo GEC, PGE2 synthesis was amplified in GEC that stably overexpress cPLA2 (Figure 3A) ▶ . Furthermore, this PGE2 generation was inhibited significantly in the presence of NS-398, indicating that a substantial portion of eicosanoid generation from endogenous arachidonic acid was mediated via COX-2 (Figure 3, B and C) ▶ .
There are several possible mechanisms that may account for the complement-mediated COX-2 up-regulation in GEC. Kilgore et al showed that C5b-9 up-regulates chemokine expression in human endothelial cells via nuclear factor-κB (NF-κB) activation. 24 The promoter region of the rat COX-2 gene contains NF-κB binding sites (−404 to −395, 5′-GGGGATTCCC-3′); 25 consequently, transcriptional regulation of COX-2 via complement-mediated activation of the NF-κB pathway is one possible mechanism. Bustos et al reported that endothelial cells exposed to xenoreactive antibodies and complement showed up-regulation of COX-2. 26 The authors did not specifically implicate C5b-9, and the time course of COX-2 mRNA induction was markedly delayed, compared with the current study, reaching a maximum at 24 hours. This delayed COX-2 induction required production of interleukin-1α, which acted as an autocrine factor. Considering the relatively rapid time course of COX-2 induction in GEC, it is unlikely that COX-2 induction in GEC was secondary to the production of other autocrine factor(s), and our results do not support the contribution of such factors (Figure 6) ▶ . We have previously shown that C5b-9 activates extracellular signal-related kinase-2 (ERK-2) in GEC. 9 Because various mitogen-activated protein kinases (MAPKs), including ERK-2, have been implicated in the transcriptional regulation of COX-2, 27,28 activation of MAPK pathways by C5b-9 is another possible mechanism for COX-2 up-regulation. Further studies will be required to address these possibilities.
Under certain conditions, a single cell can simultaneously express the two isoforms of COX, and several investigators have hypothesized that prostanoid synthesis through COX-1 and -2 may involve different arachidonic acid substrate pools, which may be coupled to distinct PLA2s and different extracellular stimuli. 12,29-32 For example, in both mitogen-stimulated fibroblasts and endotoxin-stimulated macrophages, PGE2 production was dependent on the induced expression and activity of the COX-2 gene and protein, despite the presence of constitutive, enzymatically active COX-1. 31 In contrast, the rapid PGD2 production in mast cells activated by the aggregated IgE receptor was dependent only on the presence and activity of constitutive COX-1. 33 Based on measurements of PGE2 production in the presence of exogenous arachidonic, in the current study, we have estimated that after GEC are stimulated with complement for 3 hours, COX-1 constitutes ∼75%,and COX-2 ∼25% of total COX activity (Figure 2) ▶ . However, after incubation with complement, PGE2 generation due to endogenously released arachidonic acid was inhibited with a COX-2-selective inhibitor by at least 60% (Figure 3B) ▶ , suggesting that endogenous arachidonic acid is preferentially metabolized by COX-2, ie, that there is preferential coupling between complement-induced cPLA2 activation, release of arachidonic acid from phospholipids, and metabolism via COX-2. Nevertheless, it is likely that arachidonic acid released by cPLA2 was also metabolized via COX-1. The specificity of currently-available COX-2- selective inhibitors for COX-2, as compared with COX-1, when evaluated by physiologically-relevant methods (eg, human whole blood assay), is limited, such that the concentrations of inhibitors that block COX-2 completely also partially inhibit COX-1. 34 Therefore, it is not possible to determine the exact percentage of contribution of each isoform in complement-mediated PGE2 generation.
In the kidney, it has been established that COX-1 is constitutively expressed in collecting duct cells and endothelial cells lining arteries and arterioles, and it contributes to the regulation of water reabsorption and vascular tone, respectively. 35 However, the expression and roles of COX isozymes in the glomerulus are not well understood. In normal adult human glomeruli, COX-2 was found in podocytes by immunohistochemistry, but COX-1 was not detected. 36 In normal adult rat glomeruli, COX-1 and COX-2 either were not detected or were detected weakly by immunohistochemistry, Western blotting or in situ hybridization. 37 Nevertheless, we and others have demonstrated that isolated normal rat glomeruli can clearly synthesize PGE2 and other prostanoids, indicating that there is basal arachidonic acid turnover and basal COX activity. In glomeruli, however, it is not possible to determine the cell type responsible for basal prostanoid production.
By analogy to GEC in culture, COX-2 was up-regulated in the PHN model of C5b-9-dependent GEC injury in vivo, but in addition, there was up-regulation of COX-1 (Figure 4) ▶ . Glomeruli from rats with PHN generated about twofold more PGE2, as compared with normal rats, and this PGE2 generation was partially inhibited by NS-398, indicating that most likely both COX isoforms contributed to C5b-9-mediated eicosanoid production in vivo (Figure 5) ▶ . We cannot provide a definite explanation for the discrepancy in COX-1 regulation between GEC in culture and glomeruli of rats with PHN. Nevertheless, several investigators have reported that COX-1 expression can be regulated by certain stimuli. In the human monocytic leukemia cell line THP-1 38 and the rat pheochromocytoma cell line PC12, 39 COX-1 was up-regulated with differentiation. In the mouse osteoblastic cells line MC3T3-E1 cells, COX-1 was up-regulated by basic fibroblast growth factor. 40 The precise mechanisms of COX-1 up-regulation are not understood. In these cases, COX-1 induction was delayed, as compared with COX-2 or immediate early oncogenes as c-fos, suggesting that up-regulation of COX-1 is controlled by mechanisms distinct from those of COX-2, such as synthesis of new proteins. 38-40 Possibly, the chronic exposure of GEC to C5b-9 in PHN (14 days, Figure 4 ▶ ) caused up-regulation of COX-1, which was not seen in the shorter experiments in culture (3–24 hours, Figure 1 ▶ ). Hirose et al also reported that the both isoforms of COX were up-regulated in rat anti-Thy-1 glomerulonephritis, a complement- and inflammatory cell-dependent rat model of glomerular mesangial injury. 41 Curiously, COX-2 appeared to be expressed exclusively in GEC, whereas COX-1 appeared to be expressed in glomerular epithelial and/or endothelial cells. It is also possible that COX-1 is up-regulated in cells other than GEC in glomeruli of rats with PHN.
A number of studies have examined the effects of PG or TXA2 synthesis inhibition on glomerular permselectivity in PHN. In two studies, the nonspecific COX inhibitor, indomethacin, or the TX synthase inhibitor, DP-1904, reduced autologous phase proteinuria significantly. 6 Indomethacin and the TX synthase inhibitor, OKY-046, reduced proteinuria in a model of PHN in the isolated perfused kidney. 3 In all of these studies, renal function was not affected significantly by the inhibitors. Substitution of eicosapentanoic and other ω-3 fatty acids for arachidonic acid can be achieved by enriching diets with fish oil. This maneuver shifts production of certain endogenous eicosanoids away from dienoic prostanoids to inactive metabolites. Administration of fish oil to rats with PHN suppressed production of TXA2, and reduced proteinuria significantly. 5 Together, the above studies support a role for arachidonic acid metabolites as mediators of proteinuria in PHN. Based on the present study, it may be useful to determine in the future if COX-2 selective inhibitors reduce proteinuria in membranous nephropathy, without affecting the normal physiological processes dependent on COX-1. However, such a study would require a COX-2 inhibitor that is considerably more specific than NS-398. While this manuscript was in preparation, Blume et al reported that the COX-2-selective inhibitor, flosulide, reduced proteinuria in rats with PHN. 42 However, the inhibition of proteinuria by flosulide was not associated with inhibition of glomerular eicosanoid generation. Actually, a high dose of flosulide inhibited protein expression of both COX-1 and -2, suggesting that the anti-proteinuric effect of flosulide may have been mediated by mechanisms other than inhibition of COX enzymatic activity. Furthermore, it should be noted that the actions of COX enzymes in kidney disease appear to be complex. For example, a recent report by Schneider et al showed that indomethacin and, to a lesser degree, COX-2-selective inhibitors significantly augmented monocyte/macrophage recruitment to glomeruli in two rat experimental models of glomerulonephritis, suggesting an important role for COX products as endogenous repressors of chemokine expression. 43 Also, mice deficient in COX-2 showed a postnatal maturation arrest of nephrons, developed glomerulosclerosis, and eventually died of uremia, 44,45 whereas administration of a COX-2-selective inhibitor to pregnant mice produced a similar phenotype. 46 Thus, a more precise understanding of the roles of COXs in different pathophysiologies and developmental stages will be necessary to use isoform-selective COX inhibitors safely and effectively.
Acknowledgments
We thank Lamine Aoudjit for technical assistance.
Footnotes
Address reprint requests to Tomoko Takano, M.D., Ph.D., Division of Nephrology, McGill University Health Centre, 3775 University Street, Room 236, Montreal, Quebec, Canada H3A 2B4. E-mail: ttomok@po-box.mcgill.ca.
Supported by research grants from the Medical Research Council of Canada and the Kidney Foundation of Canada. A. V. C. holds a scholarship from the Fonds de la Recherche en Santé du Québec. T. T. was the recipient of a fellowship from the Medical Research Council of Canada and currently holds a scholarship from the Medical Research Council of Canada.
References
- 1.Couser WG: Pathogenesis of glomerulonephritis. Kidney Int 1993, 44:S19-S26 [PubMed] [Google Scholar]
- 2.Salant DJ, Quigg RJ, Cybulsky AV: Heymann nephritis: mechanisms of renal injury. Kidney Int 1989, 35:976-984 [DOI] [PubMed] [Google Scholar]
- 3.Cybulsky AV, Lieberthal W, Quigg RJ, Rennke HJ, Salant DJ: A role for thromboxane in complement-mediated glomerular injury. Am J Pathol 1987, 128:45-51 [PMC free article] [PubMed] [Google Scholar]
- 4.Nagao T, Nagamatsu T, Suzuki Y: Effect of DP-1904, a thromboxane A2 synthase inhibitor, on passive Heymann nephritis in rats. Eur J Pharmacol 1996, 316:73-80 [DOI] [PubMed] [Google Scholar]
- 5.Weise WJ, Natori Y, Levine JS, O’Meara YM, Minto AW, Manning EC, Goldstein DJ, Abrahamson DR, Salant DJ: Fish oil has protective and therapeutic effects on proteinuria in passive Heymann nephritis. Kidney Int 1993, 43:359-368 [DOI] [PubMed] [Google Scholar]
- 6.Zoja C, Benigni A, Verroust P, Ronco P, Bertani T, Remuzzi G: Indomethacin reduces proteinuria in passive Heymann nephritis in rats. Kidney Int 1987, 31:1335-1343 [DOI] [PubMed] [Google Scholar]
- 7.Yoshioka T, Rennke HG, Salant DJ, Deen WM, Ichikawa I: Role of abnormally high transmural pressure in the permselectivity defect of glomerular capillary wall: a study in early passive Heymann nephritis. Circ Res 1987, 61:531-538 [DOI] [PubMed] [Google Scholar]
- 8.Gabbai FB, Gushwa LC, Wilson CB, Blantz RC: An evaluation of the development of experimental membranous nephropathy. Kidney Int 1987, 31:1267-1278 [DOI] [PubMed] [Google Scholar]
- 9.Cybulsky AV, Papillon J, McTavish AJ: Complement activates phospholipases and protein kinases in glomerular epithelial cells. Kidney Int 1998, 54:360-372 [DOI] [PubMed] [Google Scholar]
- 10.Cybulsky AV, Monge JC, Papillon J, McTavish AJ: Complement C5b-9 activates cytosolic phospholipase A2 in glomerular epithelial cells. Am J Physiol 1995, 269:F739-F749 [DOI] [PubMed] [Google Scholar]
- 11.Panesar M, Papillon J, McTavish AJ, Cybulsky AV: Activation of phospholipase A2 by complement C5b-9 in glomerular epithelial cells. J Immunol 1997, 159:3584-3594 [PubMed] [Google Scholar]
- 12.Smith WL, Garavito RM, DeWitt DL: Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and 2. J Biol Chem 1996, 271:33157-33160 [DOI] [PubMed] [Google Scholar]
- 13.Kennedy BP, Chan C-C, Culp SA, Cromlish WA: Cloning and expression of rat prostaglandin endoperoxide synthase (cyclooxygenase)-2 cDNA. Biochem Biophys Res Comm 1993, 197:494-500 [DOI] [PubMed] [Google Scholar]
- 14.Takano T, Fiore S, Maddox JF, Brady HR, Petasis NA, Serhan CN: Aspirin-triggered 15-epi-Lipoxin A4 (LXA4) and LXA4 stable analogs are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med 1997, 185:1693-1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng L, Sun W, Xia Y, Tang WW, Chanmugam P, Soyoola E, Wilson CB, Hwang D: Cloning two isoforms of rat cyclooxygenase: differential regulation of their expression. Arch Biochem Biophys 1993, 307:361-368 [DOI] [PubMed] [Google Scholar]
- 16.Salant DJ, Cybulsky AV: Experimental glomerulonephritis. Methods in Enzymology. Edited by G. DiSabato. New York, Academic Press, 1988, pp 421–461 [DOI] [PubMed]
- 17.Newton R, Seybold J, Liu SF, Barnes PJ: Alternate COX-2 transcripts are differentially regulated: implications for post-transcriptional control. Biochem Biophys Res Comm 1997, 234:85-89 [DOI] [PubMed] [Google Scholar]
- 18.Johnson JL, Wimsatt J, Buckel SD, Dyer RD, Maddipati KR: Purification and characterization of prostaglandin H synthase-2 from sheep placental cotyledons. Arch Biochem Biophys 1995, 324:26-34 [DOI] [PubMed] [Google Scholar]
- 19.Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, Chan H, Sigal E, Ramesha C: Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim Biophys Acta 1994, 1209:130-139 [DOI] [PubMed] [Google Scholar]
- 20.Futaki N, Takahashi T, Yokoyama M, Arai I, Higuchi S, Otomo S: NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins 1994, 47:55-59 [DOI] [PubMed] [Google Scholar]
- 21.Cybulsky AV, Takano T, Papillon J, McTavish AJ: Complement-induced phospholipase A2 activation in experimental membranous nephropathy. Kidney Int 2000, 57:1052-1062 [DOI] [PubMed] [Google Scholar]
- 22.Cybulsky AV, Salant DJ, Quigg RJ, Badalamenti J, Bonventre JV: Complement C5b-9 complex activates phospholipases in glomerular epithelial cells. Am J Physiol 1989, 257:F826-F836 [DOI] [PubMed] [Google Scholar]
- 23.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR: TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin systhase/cyclooxygenase homologue. J Biol Chem 1991, 266:12866-12872 [PubMed] [Google Scholar]
- 24.Kilgore KS, Schmid E, Shanley TP, Flory CM, Maheswari V, Tramontini NL, Cohen H, Ward PA, Friedl HP, Warren JS: Sublytic concentrations of the membrane attack complex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1 through nuclear factor-κB activation. Am J Pathol 1997, 150:2019-2031 [PMC free article] [PubMed] [Google Scholar]
- 25.Sirois J, Levy L, Simmons DL, Richards JS: Characterization and hormonal regulation of the promoter of the rat prostaglandin endoperoxidase synthase 2 gene in granulosa cells. J Biol Chem 1993, 268:12199-12206 [PubMed] [Google Scholar]
- 26.Bustos M, Coffman TM, Saadi S, Platt JL: Modulation of eicosanoid metabolism in endothelial cells in a xenograft model. Role of cyclooxygenase-2. J Clin Invest 1997, 100:1150–1158 [DOI] [PMC free article] [PubMed]
- 27.Guan Z, Buckman SY, Miller BW, Springer LD, Morrison AR: Interleukin-1β-induced cyclooxygenase-2 expression requires activation of both c-Jun NH2-terminal kinase and p38 MAPK signal pathways in rat renal mesangial cells. J Biol Chem 1998, 273:28670-28676 [DOI] [PubMed] [Google Scholar]
- 28.Xie W, Herschman HR: Transcriptional regulation of prostaglandin synthase 2 gene expression by platelet-derived growth factor and serum. J Biol Chem 1996, 271:31742-31748 [DOI] [PubMed] [Google Scholar]
- 29.Murakami M, Kambe T, Shimbara S, Kudo I: Functional coupling between various phospholipase A2s and cyclooxygenases in immediate and delayed prostanoid biosynthetic pathways. J Biol Chem 1999, 274:3103-3115 [DOI] [PubMed] [Google Scholar]
- 30.Tischfield JA: A reassessment of the low molecular weight phospholipase A2 gene family in mammals. J Biol Chem 1997, 272:17247-17250 [DOI] [PubMed] [Google Scholar]
- 31.Reddy ST, Herschman HR: Ligand-induced prostaglandin synthesis requires expression of the TIS10/PGS-2 prostaglandin synthase gene in murine fibroblasts and macrophages. J Biol Chem 1994, 269:15473-15480 [PubMed] [Google Scholar]
- 32.Balsinde J, Balboa MA, Dennis EA: Functional coupling between secretory phospholipase A2 and cyclooxygenase-2 and its regulation by cytosolic group IV phospholipase A2. Proc Natl Acad Sci USA 1998, 95:7951-7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami M, Matsumoto R, Austen KF, Arm JP: Prostaglandin endoperoxide synthase-1 and -2 couple to different transmembrane stimuli to generate prostaglandin D2 in mouse bone marrow-derived mast cells. J Biol Chem 1994, 269:22269-22275 [PubMed] [Google Scholar]
- 34.Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR: Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo- oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci USA 1999, 96:7563-7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith WL, Bell TG: Immunohistochemical localization of the prostaglandin-forming cyclooxygenase in renal cortex. Am J Physiol 1978, 235:F451-F457 [DOI] [PubMed] [Google Scholar]
- 36.Komhoff M, Grone H-J, Klein T, Seyberth HW, Nusing RM: Localization of cyclooxygenase-1 and 2 in adult and fetal human kidney: implication for renal function. Am J Physiol 1997, 272:F460-F468 [DOI] [PubMed] [Google Scholar]
- 37.Harris RC, McKanna JA, Akai Y, Jacobson HR, DuBois RN: Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest 1994, 94:2504-2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith CJ, Morrow JD, Roberts LJ, Marnett LJ: Differentiation of monocytoid THP-1 cells with phorbol ester induces expression of prostaglandin endoperoxide synthase-1 (COX-1). Biochem Biophys Res Commun 1993, 192:787-793 [DOI] [PubMed] [Google Scholar]
- 39.Kaplan MD, Olschowka JA, O’Banion MK: Cyclooxygenase-1 behaves as a delayed response gene in PC12 cells differentiated by nerve growth factor. J Biol Chem 1997, 272:18534-18537 [DOI] [PubMed] [Google Scholar]
- 40.Kawaguchi H, Pilbeam CC, Gronowicz G, Abreu C, Fletcher BS, Herschman HR, Raisz LG, Hurley MM: Transcriptional induction of prostaglandin G/H synthase-2 by basic fibroblast growth factor. J Clin Invest 1995, 96:923-930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirose S, Yamamoto T, Feng L, Yaoita E, Kawasaki K, Goto S, Fujinaka H, Wilson CB, Arakawa M, Kihara I: Expression and localization of cyclooxygenase isoforms and cytosolic phospholipase A2 in anti-Thy-1 glomerulonephritis. J Am Soc Nephrol 1998, 9:408-416 [DOI] [PubMed] [Google Scholar]
- 42.Blume C, Heise G, Muhlfeld A, Back D, Schror K, Gerhardz CD, Grabensee B, Heering P: Effect of flosulide, a selective cyclooxygenase 2 inhibitor, on passive Heymann nephritis in the rat. Kidney Int 1999, 56:1770-1778 [DOI] [PubMed] [Google Scholar]
- 43.Schneider A, Harendza S, Zahner G, Jocks T, Wenzel U, Wolf G, Thaiss F, Helmchen U, Stahl RA: Cyclooxygenase metabolites mediate glomerular monocyte chemoattractant protein-1 formation and monocyte recruitment in experimental glomerulonephritis. Kidney Int 1999, 55:430-441 [DOI] [PubMed] [Google Scholar]
- 44.Morhan SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Marhler JF, Kluckman KD, Ledford A, Lee CA, Smithies O: Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 1995, 83:473-482 [DOI] [PubMed] [Google Scholar]
- 45.Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, Gorry SA, Trzaskos JM: Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 1995, 378:406-409 [DOI] [PubMed] [Google Scholar]
- 46.Komhoff M, Breyer MD: Peri-natal inhibition of cyclooxygenase-2 impairs renal development in the mouse. J Am Soc Nephrol 1998, 9:364A (abstr.)