

Abstract
Recent evidence suggests that mutant huntingtin protein-induced energetic perturbations contribute to neuronal dysfunction in Huntington’s disease (HD). Given the ubiquitous expression of huntingtin, other cell types with high energetic burden may be at risk for HD-related dysfunction. Early-onset cardiovascular disease is the second leading cause of death in HD patients; a direct role for mutant huntingtin in this phenomenon remains unevaluated. Here we tested the hypothesis that expression of mutant huntingtin is sufficient to induce cardiac dysfunction, using a well-described transgenic model of HD (line R6/2). R6/2 mice developed cardiac dysfunction by 8 weeks of age, progressing to severe failure at 12 weeks, assessed by echocardiography. Limited evidence of cardiac remodeling (e.g. hypertrophy, fibrosis, apoptosis, β1 adrenergic receptor downregulation) was observed. Immunogold electron microscopy demonstrated significant elevations in nuclear and mitochondrial polyglutamine presence in the R6/2 myocyte. Significant alterations in mitochondrial ultrastructure were seen, consistent with metabolic stress. Increased cardiac lysine acetylation and protein nitration were observed, and were each significantly associated with impairments in cardiac performance. These data demonstrate that mutant huntingtin expression has potent cardiotoxic effects; cardiac failure may be a significant complication of this important experimental model of HD. Investigation of the potential cardiotropic effects of mutant huntingtin in humans may be warranted.
Keywords: huntingtin, Huntington’s disease, heart, mitochondria, polyglutamine, cardiovascular, transgenic, nitrotyrosine, acetylation
INTRODUCTION
Huntington’s disease (HD) is a devastating genetic disease, characterized primarily by a progressive loss of cognitive and motor function, leading to severe patient disability and death (Bates et al., 2002). HD is caused by the mutation of a single ubiquitously expressed gene product of unknown function, the huntingtin protein, which contains an expanded polyglutamine repeat domain in HD patients (Huntington’s Disease Collaborative Research Group, 1993). The mechanisms by which mutant huntingtin causes cellular dysfunction and death remain undefined; recent evidence has implicated metabolic and energetic dysfunction in HD neurons (including mitochondrial effects) (Brennan et al., 1985; Browne et al., 1997; Koroshetz et al., 1997; Brouillet et al., 1999; Sanchez-Pernaute et al., 1999; Schapira, 1999; Tabrizi et al., 1999; Ferrante et al., 2000; Jenkins et al., 2000; Tabrizi et al., 2000; Andreassen et al., 2001b; Panov et al., 2002; Panov et al., 2003; Choo et al., 2004; Brustovetsky et al., 2005; Milakovic and Johnson, 2005; Panov et al., 2005; Seong et al., 2005; Benchoua et al., 2006; Squitieri et al., 2006). Biochemical and imaging studies in both HD patients and mouse models demonstrate reduced mitochondrial complex activities and altered metabolic substrate distributions (N-acetylaspartate, lactate, phosphocreatine) in the basal ganglia (Brennan et al., 1985; Browne et al., 1997; Sanchez-Pernaute et al., 1999; Schapira, 1999; Tabrizi et al., 1999; Jenkins et al., 2000; Schapiro et al., 2004; Jenkins et al., 2005; Reynolds et al., 2005; Tsang et al., 2006). Despite increased food intake, HD patients can exhibit weight loss (Djousse et al., 2002; Hamilton et al., 2004; Trejo et al., 2004) and weight loss is a characteristic of transgenic mouse models of HD (Bates et al., 1998; Li et al., 2005; Stack et al., 2005). Agents that boost mitochondrial efficiency (e.g. creatine, coenzyme Q) are under investigation as potential therapeutic strategies in HD patients (Koroshetz et al., 1997; Matthews et al., 1998; Ferrante et al., 2000 Ferrante et al., 2001; Andreassen et al., 2001b; Schilling et al., 2001; Ferrante et al., 2002; Dedeoglu et al., 2003; Tabrizi et al., 2003; Verbessem et al., 2003; Ryu et al., 2005; Tabrizi et al., 2005; Hersch et al., 2006; Smith et al., 2006; Stack et al., 2006). While these studies strongly suggest that metabolic dysfunction may contribute to mutant huntingtin-mediated cell death, they fail to explain why the ubiquitous expression of mutant huntingtin has cytotoxic effects primarily in neurons (i.e. mutant huntingtin expression occurs in most cell types, not just neurons at risk). Indeed, levels of huntingtin expression do not directly correlate with neuronal vulnerability, with relatively resistant corticostriatal neurons and striatal cholinergic interneurons expressing high levels of huntingtin and vulnerable striatal projection neurons expressing variable levels of huntingtin (Fusco et al., 1999). Current hypotheses include loss of brain-derived neurotrophic factor synthesis by corticostriatal neurons, the high metabolic demand of neurons relative to other non-excitable cell types, or the slow turnover of neurons, as terminally differentiated cells, allowing for greater accumulation of huntingtin protein relative to other cell types (Bowling and Beal, 1995; Davies et al., 1999; Zuccato et al., 2001). Skeletal muscle cells (another differentiated excitable cell type with high metabolic demand) from HD patients demonstrate many of the same energetic disturbances evident in HD striatal neurons (Arenas et al., 1998; Sathasivam et al., 1999; Lodi et al., 2000; Luthi-Carter et al., 2002; Orth et al., 2003; Ribchester et al., 2004; Schapira and Lodi, 2004; Saft et al., 2005; Strand et al., 2005; Gizatullina et al., 2006). These findings raise the possibility that the ubiquitous expression of mutant huntingtin may place other cell types that are under high metabolic demand at risk.
While HD has been considered primarily a neurological disease state, multiple epidemiological studies have shown that cardiac failure is the second leading cause of death in HD patients (Chiu and Alexander, 1982; Lanska et al., 1988; Sorensen and Fenger, 1992). Cardiac failure is implicated as the cause of death in over 30% of HD patients, compared to less than 2% of age-matched non-HD patients in the general population (Chiu and Alexander, 1982; Lanska et al., 1988; CDC/NCHS and AHA, 1988–94). Energetic dysfunction and metabolic injury are causative factors in both acute and progressive cardiac disease states, and many of the same energetic changes that occur in HD neurons (mitochondrial dysfunction, altered substrate distributions) are typically seen in failing cardiac myocytes (Ingwall, 1993; Paolisso et al., 1994; Francis et al., 1995; Katz, 1998; Vogt and Kubler, 1999). However, the mechanisms of cardiac failure remain largely unstudied in both clinical settings and experimental models of HD. Here we tested the hypothesis that expression of the mutant huntingtin protein leads to cardiac dysfunction in a highly relevant animal model of HD, the R6/2 transgenic mouse. Further mechanistic studies assessed the HD heart for histopathological evidence of cardiac failure, determined the intra-cardiomyocyte distribution of mutant huntingtin, and investigated mechanistic roles for protein acetylation and protein oxidation in the development of HD-related cardiac failure.
METHODS
The R6/2 transgenic mouse model of Huntington’s disease
Male R6/2 mice (expressing exon 1 of the human huntingtin gene with an expanded CAG repeat, repeat length n ~150) and wildtype littermate controls were bred in our animal facility (Mangiarini et al., 1996). Breeder pairs were obtained from Jackson Laboratories (Bar Harbor, ME). Genotyping by polymerase chain reaction was conducted at 6 weeks of age to determine study groups. Mice (12–15 per group) were studied at 8, 10, and 12 weeks for the functional experiments, then tissues were collected at 12 weeks for histological experiments. All animal procedures are in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and have been approved by the Institutional Laboratory Animal Care and Use Committee of The Ohio State University.
Murine echocardiography
At 8, 10, and 12 weeks of age, in vivo cardiovascular performance was assessed in R6/2 mice and littermate controls (n = 12–15 for each group) using a Sonos 5500 echocardiography unit (Hewlett-Packard, Andover, MA), as previously described (Weinstein et al., 2000; Mihm et al., 2001a; Mihm et al., 2002). Mice were anesthetized by halothane inhalation (~1% halothane in 95/5% oxygen/CO2). Parameters were determined using the American Society for Echocardiography leading-edge technique in blinded fashion. These parameters allowed the determination of left ventricular (LV) fractional shortening (FS) by the equation: FS = [(LVIDd − LVIDs) / LVIDd] x 100%, where LVID refers to the LV internal dimension at diastole (d) and systole (s). Ascending aortic flow waveforms were recorded using a continuous wave Doppler flow probe oriented in a short axis, suprasternal manner. Velocity-time integrals (VTI) were calculated from these waveforms. After sacrifice, aortic root cross-sectional area was measured and cardiac output was calculated by the equation: CO = Heart rate x VTI x aortic cross-sectional area. As a measure of ventricular diastolic function, transmitral flow waveforms were also recorded, using continuous wave Doppler oriented in the parasternal, long axis position, collecting E-wave (passive ventricular filling) and A-wave (component of ventricular filling attributable to atrial contraction) waveforms. Peak transmitral flow, VTI, and acceleration/deceleration slopes were determined for each waveform. Where E and A waveforms were not baseline resolved, E wave slopes were extrapolated from the upper 2/3 of the waveform (from apex toward baseline). Intra- and inter-observer variability for these measurements were 3% and 5% respectively. All systolic and diastolic parameters were analyzed by a blinded observer.
Cardiac immunohistochemistry
Following functional analyses, hearts were rapidly excised and bisected equatorially just distal to the mitral valves, then formalin-fixed and paraffin embedded by standard protocols (Weinstein et al., 2000; Mihm et al., 2002). Left ventricular tissues were prepared as 5μm cross-sections and mounted on slides for histological and immunohistochemical analysis, as we have previously described (Weinstein et al., 2000; Mihm et al., 2002). General cardiac morphology and extent of cardiac fibrosis deposition were assessed using Masson’s Trichrome stain with a kit based approach (Sigma Chemical). Immunohistochemical studies were conducted to measure cardiac levels of β1-adrenergic receptor, protein ubiquitination, protein 3-nitrotyrosine formation, lysine acetylation, and expanded protein-bound polyglutamine repeat domains (>37 repeats recognized as a indirect measure of mutant huntingtin). Antibodies employed for these studies were anti- β1-adrenergic receptor (1:800 dilution, Research Diagnostics); anti-protein-ubiquitin (1:8000 dilution, Dako Corp.); anti-protein-3-nitrotyrosine (1:800 dilution, Upstate Biotechnology); anti-acetyl-lysine (pan-antibody for protein-bound acetylated lysine, 1:4000 dilution, Upstate Biotechnology) and anti-polyglutamine (Trottier et al., 1995) (1C2, 1:2000 dilution, Chemicon International). Diaminobenzidine (0.06% w/v, DAKO) was used to provide visualization of immunoreactivity, with methyl green counterstaining. As staining controls for each antibody, serial 5μm cross-sections were treated with the identical staining protocol, substituting an equivalent concentration of non-immune IgG for the primary antibody to demonstrate antibody specificity.
Cardiac left ventricular tissue samples were also prepared for electron microscopic immunocytochemistry from R6/2 and littermate control animals following echocardiographic assessment at 12 weeks, as we have previously described (Mihm et al., 2002).
Studies to evaluate the heart for development of myocyte DNA fragmentation (preliminary marker of myocyte apoptosis) were conducted by a kit-based histological approach (FragEL DNA Fragmentation Detection Kit, Oncogene Research), as we have previously described (Mihm et al., 2000).
Image capture and digital image analysis
Left ventricular images were captured using a Polaroid digital camera (Polaroid) and transferred into research-based digital image analysis software (Image Pro Plus, Media Cybernetics). Multiple images were captured around each ventricular cross section, under identical lighting and optical settings, so as to encompass the entire cross-section available. Digital imaging approaches for Trichrome staining provided quantitative measures of fibrosis in each cross-section, expressed as percent of total tissue area stained blue. Extent of immunoreactivity for immunohistochemical studies was determined by measuring optical density of diaminobenzidine signal in each tissue. Integrated optical densities were determined for each image as a measure of staining intensity, and values were averaged for each heart to provide a measure of antigen prevalence at each time point. Intra- and inter-observer variability for this procedure were each less than 2% (Mihm et al., 2000; Weinstein et al., 2000; Mihm et al., 2001a; Mihm et al., 2002).
TUNEL-positive nuclear counts were performed using an automated quantification approach. Images were background corrected, and positive nuclei were color segmented and then gated based on size, then automated nuclear counts for TUNEL positive nuclei were performed, and expressed as a percentage of DNAse positive control.
Electron micrographs (55,000x magnification) were scanned using an HP Scanjet 6200c capable of 1290x960 resolution, transferred into research-based digital image analysis software (Image Pro Plus, Media Cybernetics). Images were calibrated and myofibrillar, mitochondrial and nuclear structures were delineated and their areas were calculated. Gold particles were counted and normalized as a function of area. Intra-observer and inter-observer variability were each <2%. Mitochondrial morphology was also evaluated as a measure of mitochondrial health, using a quantitiative digital image analysis approach, as we have previously described (Joshi et al., 2000).
Data Handling
Significant differences determined by two-tailed Student’s t-tests or One-way analyses of variance, with post- hoc Newman-Keuls tests to evaluate significant comparisons. p<0.05 described statistical significance.
RESULTS
HD (R6/2) mice characteristically lose weight as they age, and, as expected, the HD mice used in the present study exhibited less weight gain compared to littermate controls (CTRL) (8 weeks: 27.2±0.5 vs. 23.7±1.1g; 10 weeks: 28.2±1.1 vs. 24.0±1.1g; 12 weeks: 30.0±1.3 vs. 22.1±1.2g, CTRL vs. HD, p<0.05 at each time point studied), but tibial lengths were not significantly different at sacrifice (17.5±0.8 vs. 17.7±1.3mm at 12 weeks, CTRL vs. HD, p = NS). R6/2 and littermate control mice were studied longitudinally at 8, 10, and 12 weeks using echocardiography, under light inhalation anesthesia. Left ventricular wall motion during systole was severely diminished in HD mice compared to littermate controls (Figure 1A, representative M-mode images shown from HD and control mice at 12 weeks). Fractional shortening was significantly impaired at 10 and 12 weeks of age in HD mice compared to controls (Figure 1B). Cardiac output was reduced by 50% by 12 weeks (Figure 1B). Alterations in cardiac output were caused by the progressive decrement in stroke volume (determined from aortic flow per beat) observed in HD mice, as there were no differences in heart rate in HD versus control mice at any time point studied (p=NS, Table 1). Transmitral flow velocities were also altered in HD hearts, as E/A ratio was significantly decreased at all time points studied (Figure 1B), resulting from significant decreases in passive ventricular filling (E wave flow and E wave VTI, Table 1) and significant increases in atrial-mediated LV filling (A wave flow and A wave VTI, Table 1). These changes were not explained by age-related effects, as littermate controls did not show age-dependent decreases in any parameter studied (8 vs. 12 week data, p=NS, Table 1).
Figure 1.
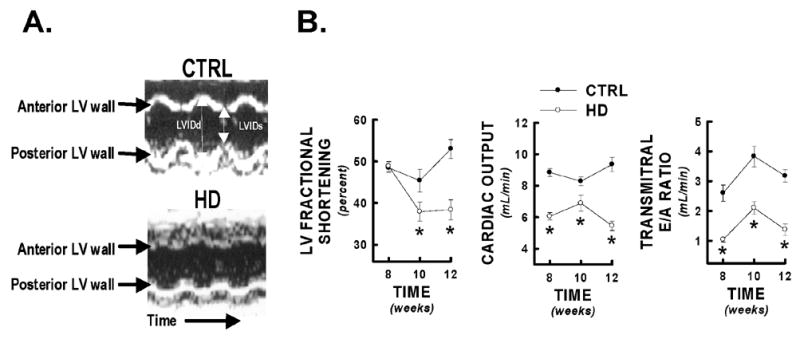
Echocardiographic evidence of decreased cardiac performance in HD mice. HD mice and littermate controls (CTRL) were serially assessed for systolic and diastolic cardiac performance at 8, 10, and 12 weeks of age by non-invasive echocardiography. Panel A) Representative M-mode images of control and HD cardiac left ventricles at 12 weeks of age. HD mice demonstrated significantly diminished anterior and posterior wall motion compared to littermate controls. Panel B) Selected systolic (LV fractional shortening, cardiac output) and diastolic (transmitral E/A ratio) cardiac performance indicators from HD mice and littermate controls at 8, 10, and 12 weeks. * = p<0.05 from control values at equivalent time point.
Table 1.
Echocardiographic cardiac performance parameters for R6/2 mice and littermate controls
| 8 WEEKS | 10 WEEKS | 12 WEEKS | ||||
|---|---|---|---|---|---|---|
| CTRL | HD | CTRL | HD | CTRL | HD | |
| Heart Rate (beats per minute) | 498±43 | 458±26 | 500±34 | 482±22 | 488±27 | 467±15 |
| LVIDs (cm) | 0.161±0.005 | 0.158±0.004 | 0.165±0.009 | 0.189±0.007* | 0.142±0.008 | 0.176±.0.008* |
| LVIDd (cm) | 0.312±0.007 | 0.310±0.006 | 0.304±0.010 | 0.305±0.007 | 0.303±0.013 | 0.286±0.008 |
| Peak aortic flow (cm/s) | 118.8±4.2 | 106.1±4.8 | 117.2±3.9 | 104.5±4.4* | 126.2±7.7 | 78.9±2.3* |
| Aortic VTI (cm) | 6.2±0.18 | 4.55±0.18 | 5.55±0.18 | 4.60±0.34* | 6.26±0.30 | 3.86±0.20* |
| Aortic acceleration slope (cm/s2) | 5060.5±417.8 | 5082.8±464.6 | 4378.9±351.9 | 4190.0±449.3 | 5652.7±514.0 | 3493.7±295.3* |
| Aortic deceleration slope (cm/s2) | 2530.0±331.9 | 2375.3±287.7 | 2415.2±177.9 | 2153.0±164.2 | 2910.3±200.2 | 1405.2±69.7* |
| E wave VTI (cm) | 3.04±0.20 | 2.09±0.10* | 3.21±0.18 | 2.20±0.25* | 3.80±0.39 | 2.30±0.07* |
| E wave acceleration slope (cm/s2) | 5914.5±378.0 | 4770.0±452.2* | 6282.2±388.2 | 3581.6±482.1* | 7397.2±271.4 | 5577.2±194.7* |
| E wave deceleration slope (cm/s2) | 3306.6±257.3 | 3997.3±224.7 | 3246.5±235.7 | 1767.0±329.8* | 3672.7±409.1 | 2521.3±249.6* |
| A wave VTI (cm) | 1.21±0.07 | 2.04±0.07* | 0.86±0.05 | 1.18±0.11* | 1.23±0.15 | 1.88±0.22* |
| A wave acceleration slope (cm/s2) | 1958.3±332.1 | 1400.0±91.3 | 1391.0±164.4 | 929.0±117.7* | 1841.4±310.6 | 1794.1±218.9 |
| A wave deceleration slope (cm/s2) | 973.0±68.6 | 1088.3±32.2 | 875.8±73.2 | 616.3±54.1* | 1053.6±81.0 | 1278.3±192.5 |
= p<0.05 versus littermate control
Heart weights were significantly reduced at 12 weeks in HD mice compared to littermate controls (Figure 2B), and exhibited LV dilation compared to CTRL (Figure 2B). LV dilation was not accompanied by changes in relative Trichrome staining for cardiac fibrosis (Figure 2B), immunohistochemical staining for β1-adrenoreceptor (Figure 3A, p=NS), or TUNEL positive nuclei (Figure 3B).
Figure 2.
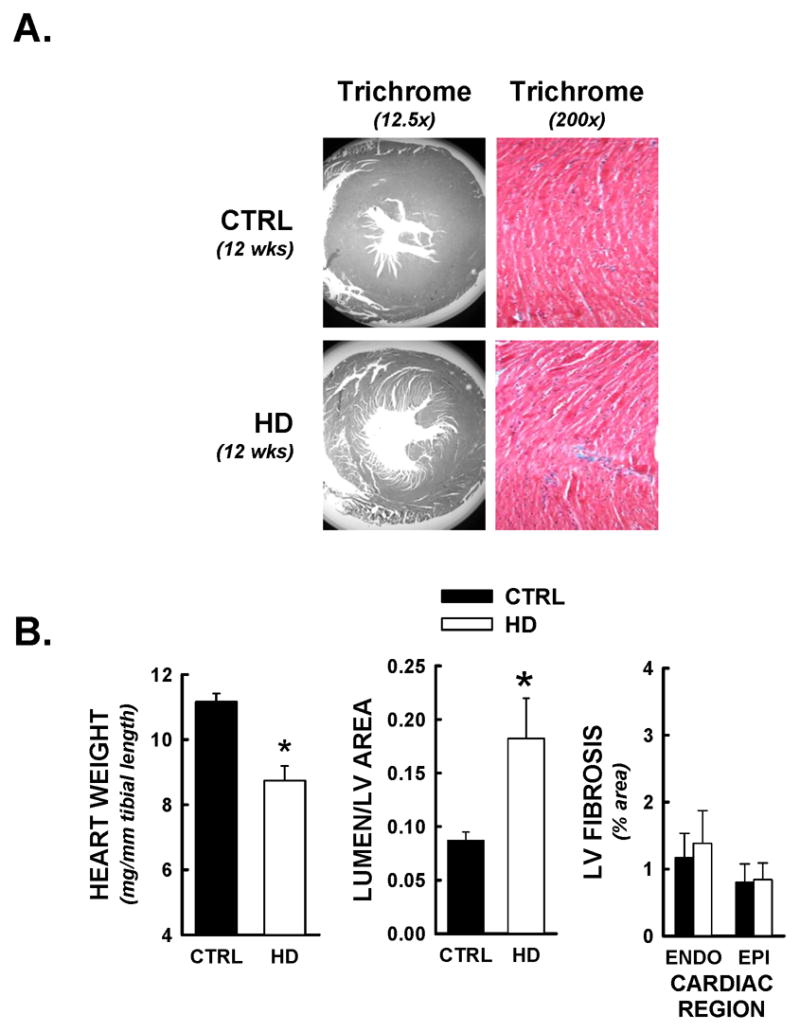
Limited evidence of cardiac structural remodeling in HD mice. Following functional analyses, hearts were prepared for histopathological studies, and stained with Masson’s Trichrome (Myocardium = red, nuclei = black, collagen/fibrosis = blue). All data are from animals sacrificed at 12 weeks of age. Panel A) Representative photomicrographs of Masson’s Trichrome staining at 12.5 and 200x magnifications. Myocardial cross-sectional areas from HD mice were smaller than littermate control (CTRL) hearts, and demonstrated equivalent and limited evidence of interstitial fibrosis. Panel B) Heart weights (normalized to tibial length) for HD mice were significantly decreased relative to littermate controls. HD hearts demonstrated significant LV chamber dilation, determined by lumenal/myocardial area ratios. No increases in interstitial fibrosis was detected, as both endocardial (inner 50% of myocardium) and epicardial (outer 50% of myocardium) Trichrome staining were under 2% of total tissue area. *, p<0.05 versus control values at equivalent time point.
Figure 3.
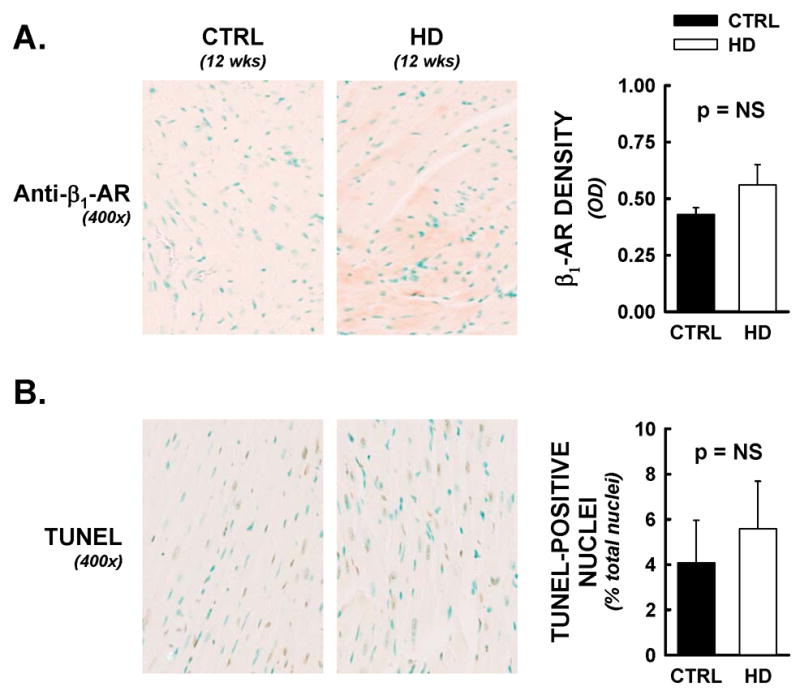
Cardiac β1-adrenergic receptor densities and myocyte apoptotic events were not altered in HD mice. Following functional analyses, hearts were assessed for β1-adrenergic receptor densities and DNA nicks by immunohistochemical methods. All data are from animals sacrificed at 12 weeks of age. Panel A) Representative photomicrographs from anti- β1 adrenergic receptor labelling (400x). No significant differences were observed between HD hearts and littermate controls, by optical density analysis. Panel B) TUNEL staining was employed as an indirect marker of apoptotic events. TUNEL positive nuclei (brown nuclei) were counted by an automated digital imaging approach, and expressed as a percentage of DNAse positive control (maximal nuclear staining) from serial sections. Percentages of total nuclei that were TUNEL-positive were not significantly different between treatment groups.
An antibody raised against an expanded poly-glutamine repeat sequence was used as an indirect marker of the mutant huntingtin protein (Trottier et al., 1995), and immunohistochemical studies were conducted at the light microscopy (400x) and the electron microscopy (42,500–55,000x) levels. Control tissues exhibited negligible staining, but positive staining was significantly elevated in HD hearts (Figure 4A, 400x). Both cytosolic and nuclear staining was evident, and staining prevalence was equivalent throughout the epicardial and endocardial areas of each cardiac cross-section. Representative electron photomicrographs are shown from longitudinal sections of cardiomyocytes from a control and HD mouse at 12 weeks of age (Figure 4B, 42,500x). Digital image analysis demonstrated the nucleus to be the predominant site of poly-glutamine staining, with staining density increased 5-fold in HD hearts versus CTRL (Figure 4B). Mitochondrial localization was significantly increased in HD hearts as well (p<0.05 at 12 weeks), with negligible staining in the cardiac myofibrils.
Figure 4.
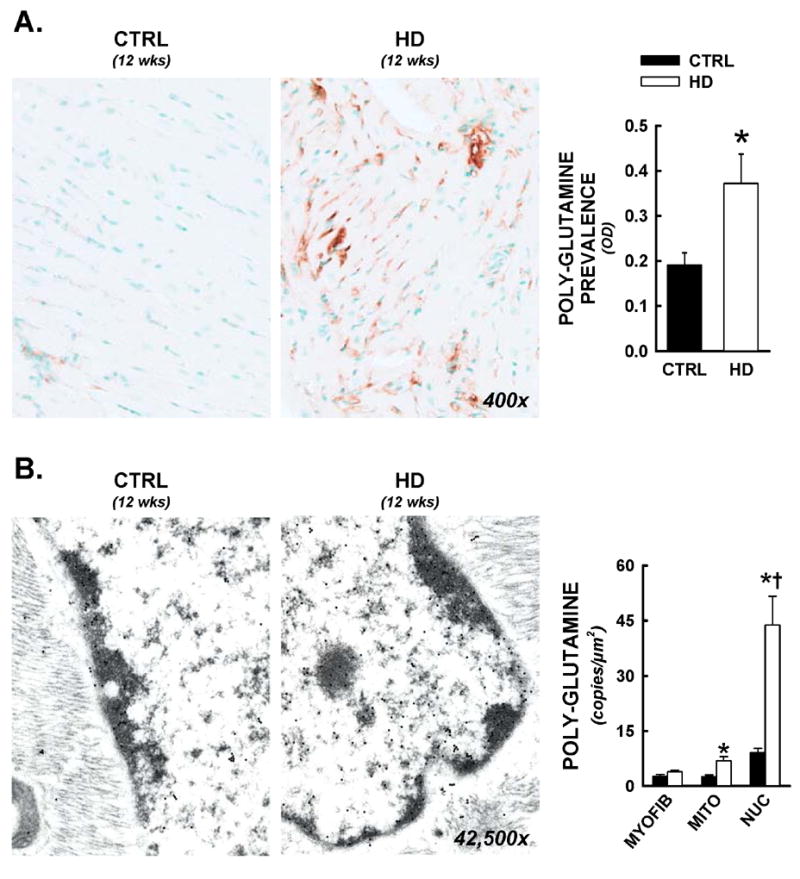
Nuclear and mitochondrial residence of mutant huntingtin in R6/2 mice. Following functional analyses, cardiac tissues were prepared for both light microscopy and electron microscopy immunohistochemistry. All data are from animals sacrificed at 12 weeks of age. Panel A) Representative photomicrographs from anti-polyglutamine staining (antibody raised against poly-glutamine repeats >37, 400x). Increased prevalence of the expanded polyglutamine sequence was visualized in both cytosolic and nuclear spaces. Total cardiac polyglutamine prevalence was significantly elevated in HD hearts by optical density analysis. Panel B) Immunogold electron microscopy for intracellular distributions of mutant huntingtin. Immunohistochemistry was conducted as above, with a gold-colloid secondary antibody (10nm particles, visualized as electron dense circles under tunneling electron microscopic examination). Images of myocytes with longitudinal orientation were captured and nuclear, mitochondrial, and myofibrillar areas were delineated and calibrated, then positive copies for the expanded polyglutamine sequence were counted, and expressed per nuclear, mitochondrial, or myofibrillar area. Mean data is represented in histogram format. *, p<0.05 versus littermate control.
Cardiac cross-sections were also assessed for evidence of myocyte post-translational protein modifications that have been implicated in HD neuronal pathogenesis. HD hearts exhibited significant increases in both cytosolic and nuclear prevalence of protein-ubiquitin, which has been shown to associate with intranuclear mutant huntingtin aggregates in HD neurons (Figure 5A). These increases did not functionally correlate to left ventricular fractional shortening measures (Spearman’s non-parametric correlation, p = NS). Protein acetylation was also assessed in situ, using a pan-antibody for protein-acetylation (i.e. not selective for single histone or p53 acetylation sites) (Figure 5B). Protein acetylation was significantly increased in HD hearts at 12 weeks, and extent of lysine acetylation was significantly correlated to fractional shortening (Figure 5B, left panel). Similarly, protein tyrosine nitration was increased in HD hearts relative to controls, and a significant negative correlation was observed between cardiac protein nitration and left ventricular fractional shortening (Figure 5C, left panel).
Figure 5.
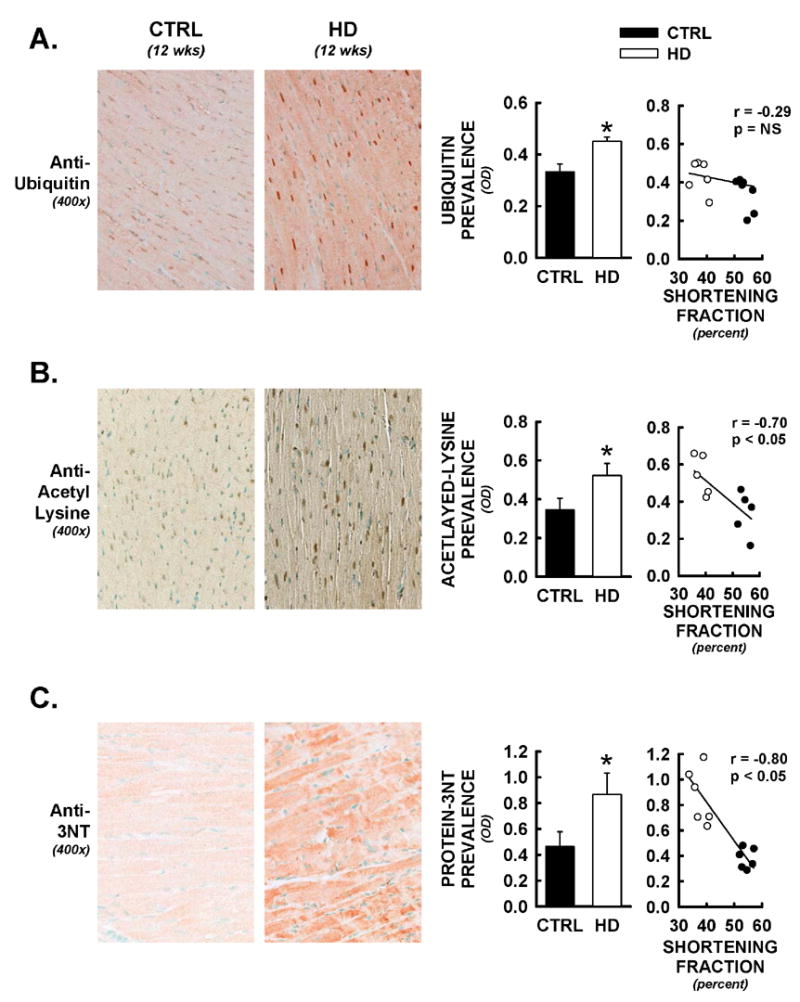
Alterations in protein post-translational modifications in HD hearts. Following functional analyses, hearts were assessed for protein-ubiquitin, lysine acetylation, and protein-nitrotyrosine formation by immunohistochemical methods. All data are from animals sacrificed at 12 weeks of age. Panel A) Representative photomicrographs from anti-ubiquitin staining (400x). Increases in cytosolic and nuclear staining were observed in HD hearts. These changes were not significantly correlated to alterations in fractional shortening in these same mice (Spearman’s non-parametric correlation). Panel B) Protein lysine-acetylation was assessed using a pan-antibody to acetylated-lysine. Nuclear as well as cytosolic increases in acetylated-lysine were observed in HD hearts, and optical densities for cardiac lysine-acetylation were significantly negatively correlated to LV fractional shortening in these same mice. Panel C) Protein-3-nitrotyrosine (protein-3NT) was also increased in cardiac cross-sections from HD mice relative to controls. Staining was predominantly observed in cardiac myocytes themselves and was widespread throughout the myocardium. Extent of cardiac protein-3NT formation was significantly correlated to alterations in fractional shortening in these same mice. *, p<0.05 versus littermate control.
Mitochondrial areas and roundness were assessed in the same electron micrographs that were used to determine mutant huntingtin distributions, representative micrographs of mitochondria from control and HD cardiac myocytes at 12 weeks of age are shown in Figure 6. Healthy normal mitochondria generally present as elliptical, with uniform optical densities under electron microscopic observation. HD mitochondria demonstrated a more circular appearance, with a disruption of their uniform densities (Figure 6, left panels). Although mitochondrial areas were not significantly different between groups, digital imaging parameters that describe the elliptical versus circular shape of the mitochondria were significantly different between control and HD hearts. Both mitochondrial aspect ratio (major axis/minor axis, collapses to 1.0 for a perfect circle) and mitochondrial roundness (perimeter2/2π*area, collapses to 1.0 for a perfect circle) were significantly decreased in HD hearts relative to controls at 12 weeks, consistent with the loss of normal mitochondrial appearance in situ.
Figure 6.
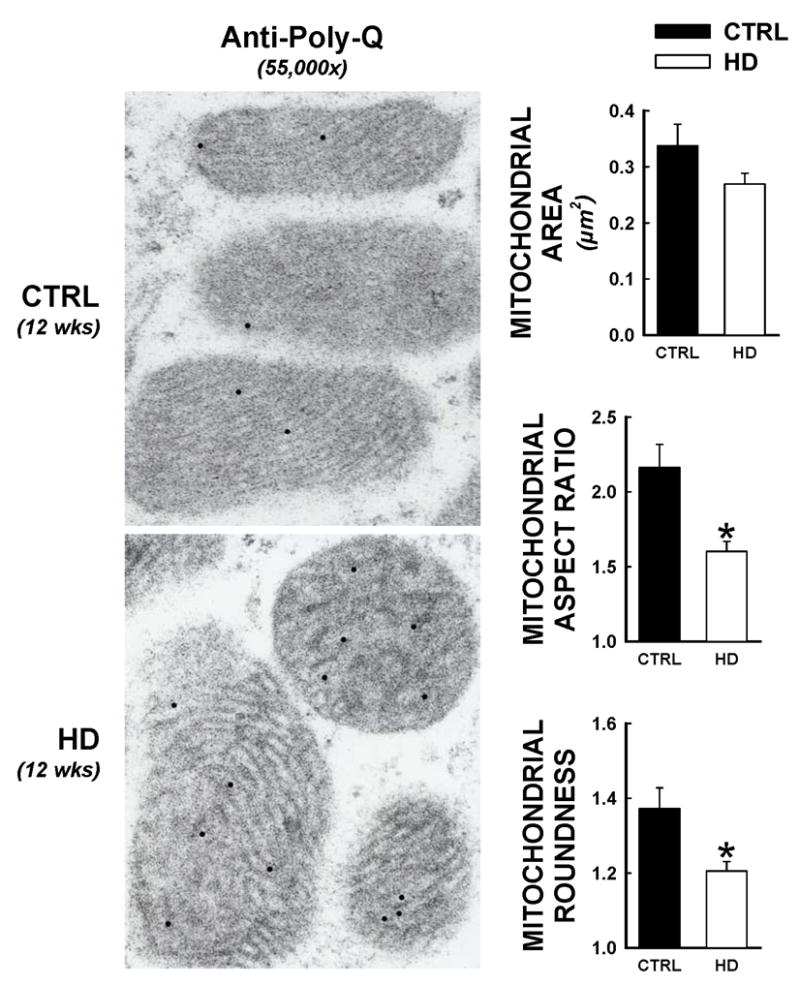
Altered mitochondrial ultrastructure in HD hearts. Electron micrographs from HD and control hearts were assessed using quantitative measures for mitochondrial morphology, at 12 weeks of age. These assessments were made in the same electron micrographs as immunogold studies for the expanded polyglutamine sequence, such that electron dense circles indicate mutant huntingtin presence. Over 300 individual mitochondrion were assessed for areas, aspect ratio (defined as the length of the major mitochondrial axis divided by the minor mitochondrial axis, which converges to 1.0 for a perfect circle), and roundness (defined as: [(mitochondrial perimeter)2]/[2π*(mitochondrial area)], which converges to 1.0 for a perfect circle). *, p<0.05 versus littermate control.
DISCUSSION
Over 30,000 Americans are currently diagnosed with HD, and at present, no fully effective symptomatic treatment or cure exists. Although its most prominent effects manifest as neurodegenerative events in striatal and cortical neurons, mutant huntingtin protein is expressed throughout the body (Strong et al., 1993), and the systemic effects of mutant huntingtin remain largely unstudied. While broncho-pneumonia is the leading recognized cause of death in HD patients, a review of available epidemiological studies indicate that cardiac failure is a commonly cited cause of mortality in this patient population as well (Chiu and Alexander, 1982; Lanska et al., 1988; Sorensen and Fenger, 1992). A recent report has shown that the mitochondrial complex II inhibitor, 3-nitropropionic acid, causes a dramatic cardiac failure phenotype, at doses that are also used to produce neurological deficits modeling HD (Gabrielson et al., 2001). These data suggest that mutant huntingtin may have cardiotropic effects, however, the cardiospecific activities of mutant huntingtin remain completely undefined.
Here we used a well-established mouse model of HD, the R6/2 transgenic mouse to test the hypothesis that expression of the mutant huntingtin protein can cause altered cardiac performance in vivo. The R6/2 afforded several advantages in these studies, including a well-described neuropathology and the direct evaluation of the cardiac response to only the (human) mutant huntingtin protein itself (Mangiarini et al., 1996; Carter et al., 1999). We used echocardiography to study cardiac function over the same time course in which neurological deficits typically become manifest in this mouse model (Carter et al., 1999). The use of this important diagnostic tool provided an array of clinically relevant performance indicators of systolic and diastolic function. HD mice presented with multiple systolic and diastolic performance deficits, detectable as early as 8 weeks, and progressing to 50% reduction in cardiac output by 12 weeks of age. The magnitude of these changes was consistent with moderate to severe cardiac failure in mice—expression of mutant huntingtin resulted in the rapid development of significant and substantial cardiac contractility impairments, over the same time course in which neurological impairments have been described in this model. The magnitude of these changes match or surpass those that we have observed with these same methods in other murine models of severe cardiac disease, including retrovirus induced cardiac injury, doxorubicin-related cardiac toxicity, and cocaine-induced cardiac injury, such that HD-related cardiac disease may be a significant source of mortality in this animal model (Weinstein et al., 2000; Mihm et al., 2002). This is an important consideration, given that mortality is often employed as a therapeutic endpoint for neurological benefit in the R6/2 mouse (Ferrante et al., 2000; Andreassen et al., 2001a; Schiefer et al., 2002). These data provide first-time evidence that mutant huntingtin can have significant cardiotoxic effects, and are consistent with the hypothesis that HD-related cardiac failure may be directly related to the ubiquitous expression of the mutated gene product.
Given the parallel time courses of these cardiac changes and the development of neurological deficits, the use of cardiovascular performance indicators as “state biomarkers” of disease progression may warrant consideration and study. The absence of convenient or reliable biomarkers of disease progression represents a significant unmet need with respect to HD patient care and the rational development of therapeutic strategies. The highly quantitative nature of cardiac performance indicators and their convenient and routine clinical application may represent significant advantages as potential state biomarkers of HD progression, relative to currently employed methodologies. Evidence that HD patients exhibit changes in heart rate variability that correlate to some aspects of the Unified Huntington’s Disease Rating Scale is consistent with the potential value of this approach (Sharma et al., 1999).
These functional investigations were followed by pilot histopathological investigations, determining the localization of mutant huntingtin in the cardiac myocyte, and assessing changes that are traditionally associated with classical settings of heart failure or with neuronal changes reported to occur in HD. The trafficking of mutant huntingtin in HD neurons has been extensively studied, and the presence of intranuclear poly-glutamine-rich aggregates has been described in a variety of cell types, in both experimental models as well as HD patients (Davies et al., 1997; DiFiglia et al., 1997). The possibility for differential mutant huntingtin localizations in myocytes versus neurons is a potentially important consideration, since myofibrils constitute the majority of cardiac myocyte volume (60–70%), and these structures are absent in neurons. We found that the predominant localization of mutant huntingtin in the R6/2 mouse was nuclear, approximately 6- and 12-fold higher than mitochondrial and myofibrillar densities, respectively. Increased nuclear and mitochondrial polyglutamine densities in HD hearts are consistent with findings in R6/2 neurons. The localization of mutant huntingtin in a “minority” of the total myocyte area suggests that mutant huntingtin may be selectively trafficked in cardiac myocytes, and implicates cardiac mitochondria and nuclei as potential sites of mutant huntingtin-mediated toxicities. Interestingly, although immunoprevalence of ubiquitination was elevated in HD myocytes relative to control, this presence was not correlated to changes in cardiac function. Since protein ubiquitination can be useful as an indirect marker of aggregate formation in HD (Davies et al., 1998; Kuemmerle et al., 1999; Martin-Aparicio et al., 2001), cardiac functional changes in this model may not be directly caused by the formation of mutant huntingtin aggregates.
Although HD was associated with significant LV chamber dilation, HD hearts did not demonstrate evidence of hypertrophic remodeling, fibrosis deposition, changes in β1-adrenergic receptor densities, or TUNEL positive nuclei (as an indirect marker of increased apoptosis), which are commonly seen in more traditional (particularly ischemic) settings of cardiac disease (Francis et al., 1995; Braunwald et al., 2000). Therefore, myocardial apoptosis and overt cardiac remodeling are apparently not operable mechanisms of cardiac dysfunction in the HD mouse. These data are consistent with contractility impairments at the level of the cardiac myocyte driving cardiac functional impairment, rather than global pump performance alterations secondary to perturbations in cardiac structure—the absence of these commonly seen cardiac structural and biochemical alterations is unusual with this degree of cardiac functional impairment, although the rapid time course of functional decline in this model may be a contributing factor.
In addition to defining the intracellular distributions of mutant huntingtin in the cardiac myocyte, our electron microscopy studies allowed us to examine the effect of mutant huntingtin expression on mitochondrial morphology. We have previously developed a digital imaging approach to quantitatively assess mitochondrial morphology, as a histological measure of mitochondrial health (Joshi et al., 2000). Mitochondria from HD myocytes exhibited morphological changes that are indicative of mitochondrial dysfunction, including a loss of elongated shape (critical for the maintenance of high surface area/volume ratios) and a general diffusion of mitochondrial densities under electron microscopic examination. These changes represent the first indication that cardiac mitochondrial health and functionality may be impaired in the R6/2 mouse, and are consistent with previous reports describing energetic deficiencies in neurons from both patients and the R6/2 mouse (Bowling and Beal, 1995; Gu et al., 1996; Sawa et al., 1999; Panov et al., 2002; Panov et al., 2003; Brustovetsky et al., 2005; Milakovic and Johnson, 2005; Panov et al., 2005; Saft et al., 2005; Benchoua et al., 2006; Gizatullina et al., 2006).
Post-translational protein modifications have been implicated in the pathogenesis of HD, as well as many other neurological and cardiac diseases. Mutant huntingtin has been shown to modulate histone acetylation patterns in the Drosophila model of HD and this may play a mechanistic role in the neuronal degeneration observed (Steffan et al., 2001). We observed significant increases in nuclear protein acetylation in HD hearts, and the extent of cardiac protein acetylation was correlated to the extent of cardiac dysfunction (in contrast to the result seen with protein ubiquitination). A limitation to this approach is that the pan-antibody did not identify the specific protein targets involved; as a result, it is difficult to directly relate these observations in cardiac myocytes to those that have been described in HD models for histone tails H3 and H4 in neurons. However, these data demonstrate that protein acetylation patterns are dramatically altered in the HD heart, that these changes predominate in the myocyte nucleus, and that these changes are strongly associated with the functional deficits that occur. HD hearts also exhibited significant increases in cardiac protein nitration. Protein nitration is a selective oxidative modification of tyrosine residues mediated by reactive nitrogen species, a particularly reactive sub-family of biological oxidants (Beckman and Koppenol, 1996), and has been shown to occur in a number of neurodegenerative disease states, including HD (Bowling and Beal, 1995; Browne et al., 1999; Tabrizi et al., 1999; Tabrizi et al., 2000). Additionally, we and others have shown that protein nitration occurs in a wide array of acute and chronic cardiovascular disease states, and often correlates to the severity of cardiac disease (Weinstein et al., 2000; Mihm et al., 2001b; Mihm and Bauer, 2002; Mihm et al., 2002; Turko and Murad, 2002). As we have observed in other settings of decompensated cardiac failure, HD hearts had significantly elevated protein nitration, which was negatively correlated to cardiac performance, suggesting that RNS formation and attendant protein oxidation may have deleterious effects on cardiac contractility in this setting. We have shown that increases in cardiac protein oxidation often occur secondarily to energetic dysfunction, which is consistent with the metabolic hypothesis of HD (Bowling and Beal, 1995). Taken in total, these preliminary data suggest that select protein modifications are influencing the rapid and severe cardiac functional deficits seen in the HD heart—this hypothesis is also currently active in HD neurological research as well. The specific myocyte protein targets involved, and whether these activities are resulting in changes in cardiac energetics and/or gene expression are currently not known, and these experiments are currently underway in our laboratories.
The R6/2 mouse is generally considered one of the more aggressive models of HD, given the truncated fragment of huntingtin that is expressed, the high number of glutamine repeats, and the rapid onset of symptoms. Additionally, a previously recognized aspect of the R6/2 model is the tendency of these mice to develop symptoms of diabetes mellitus, including elevations in fasting plasma glucose, decreased insulin production, and increased circulating glucagon levels (Hurlbert et al., 1999) (interestingly, an increased incidence of diabetes has been suggested to occur in the human HD population as well (Podolsky et al., 1972; Farrer, 1985)). Because diabetes mellitus is well-established as an independent risk factor for cardiovascular disease, this aspect of the R6/2 model is a limitation to the current study design. However, hyperglycemia associated with the R6/2 mouse is likely to have only a limited influence on the alterations in cardiac performance observed, for the following reasons: 1) The development of detectable hyperglycemia occurs late (after detectable deficits in cardiac performance) in the time course of the R6/2 mouse, and only moderate hyperglycemia (fasting plasma glucose = 211±19mg/dl) (Hurlbert et al., 1999) has been described. 2) The time course of HD related cardiac failure is much more rapidly progressive (2–4 weeks) than that generally observed in chemically and genetically induced rodent models of diabetic cardiomyopathy. 3) The limited degree of hyperglycemia observed in the HD mouse is generally associated with only mild cardiac impairment in rodent models of diabetic cardiomyopathy. More moderate models of HD are currently available—cardiac functional studies in some of these models would add important insights with respect to the broader implications of these findings and time course relative to neurological deficits, as well as the impact of hyperglycemia on the potential cardiac complications of HD.
In summary, expression of the mutated huntingtin protein is sufficient to cause the rapid development of severe cardiac systolic and diastolic impairments in mice, by mechanisms that more closely resemble mutant huntingtin-related cellular dysfunction than hallmark cardiac failure pathways. Although further experimental studies are clearly required, these data provide first-time evidence that the cardiotropic effects of mutant huntingtin must be considered in the assessment of experimental models of HD, and that further consideration of this phenomenon in humans may be warranted.
Acknowledgments
We appreciate the expert technical assistance of Jessica Buescher, Mandar Joshi, PhD, and Kathy Wolken, MS. This work was supported in part by grants from the National Institutes of Health (HL59791, HL63067, NS41003), The Hereditary Disease Foundation, and the American Heart Association, Ohio Valley Affiliate.
Abbreviations
- HD
Huntington’s disease
- TUNEL
terminal deoxy nucleotidyl fragment end labeling
- LV
left ventricle
- FS
fractional shortening
- LVID
LV internal dimension at diastole
- VTI
velocity-time integrals
- CO
cardiac output
- β1-AR
β1-adrenergic receptor
- 3NT
3-nitrotyrosine
- RNS
reactive nitrogen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreassen OA, Ferrante RJ, Dedeoglu A, Beal MF. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport. 2001a;12:3371–3373. doi: 10.1097/00001756-200110290-00044. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington’s disease. Neurobiol Dis. 2001b;8:479–491. doi: 10.1006/nbdi.2001.0406. [DOI] [PubMed] [Google Scholar]
- Arenas J, Campos Y, Ribacoba R, Martin MA, Rubio JC, Ablanedo P, Cabello A. Complex I defect in muscle from patients with Huntington’s disease. Annals of Neurology. 1998;43:397–400. doi: 10.1002/ana.410430321. [DOI] [PubMed] [Google Scholar]
- Bates G, Harper P, Jones L, editors. Huntington’s Disease. 3. Oxford ; New York: Oxford University Press; 2002. [Google Scholar]
- Bates GP, Mangiarini L, Davies SW. Transgenic mice in the study of polyglutamine repeat expansion diseases. Brain Pathol. 1998;8:699–714. doi: 10.1111/j.1750-3639.1998.tb00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. American Journal of Physiology. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling AC, Beal MF. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 1995;56:1151–1171. doi: 10.1016/0024-3205(95)00055-b. [DOI] [PubMed] [Google Scholar]
- Braunwald E, Zipes D, Libby P, editors. Heart Disease-A textbook of cardiovascular medicine. 6. Portland, OR: Book News, Inc; 2000. [Google Scholar]
- Brennan WA, Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington’s disease brain. Journal of Neurochemistry. 1985;44:1948–1950. doi: 10.1111/j.1471-4159.1985.tb07192.x. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington’s disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington’s disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Annals of Neurology. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, Dubinsky JM. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington’s Disease. J Neurochem. 2005;93:1361–1370. doi: 10.1111/j.1471-4159.2005.03036.x. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC/NCHS, AHA. National Health and Nutrition Examination Study (NHANES III) 1988–94. CDC/NCHS and the American Heart Association. [Google Scholar]
- Chiu E, Alexander L. Causes of death in Huntington’s disease. Med J Aust. 1982;1:153. doi: 10.5694/j.1326-5377.1982.tb132224.x. [DOI] [PubMed] [Google Scholar]
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- Davies SW, Beardsall K, Turmaine M, DiFiglia M, Aronin N, Bates GP. Are neuronal intranuclear inclusions the common neuropathology of triplet-repeat disorders with polyglutamine-repeat expansions? Lancet. 1998;351:131–133. doi: 10.1016/S0140-6736(97)08360-8. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, Raza AS, Mahal A, Mangiarini L, Bates GP. From neuronal inclusions to neurodegeneration: neuropathological investigation of a transgenic mouse model of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:981–989. doi: 10.1098/rstb.1999.0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Kubilus JK, Yang L, Ferrante KL, Hersch SM, Beal MF, Ferrante RJ. Creatine therapy provides neuroprotection after onset of clinical symptoms in Huntington’s disease transgenic mice. J Neurochem. 2003;85:1359–1367. doi: 10.1046/j.1471-4159.2003.01706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Djousse L, Knowlton B, Cupples LA, Marder K, Shoulson I, Myers RH. Weight loss in early stage of Huntington’s disease. Neurology. 2002;59:1325–1330. doi: 10.1212/01.wnl.0000031791.10922.cf. [DOI] [PubMed] [Google Scholar]
- Farrer LA. Diabetes mellitus in Huntington disease. Clin Genet. 1985;27:62–67. doi: 10.1111/j.1399-0004.1985.tb00185.x. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J Neurosci. 2000;20:4389–4397. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis GS, McDonald K, Chu C, Cohn JN. Pathophysiologic aspects of end-stage heart failure. Am J Cardiol. 1995;75:11A–16A. doi: 10.1016/s0002-9149(99)80378-6. [DOI] [PubMed] [Google Scholar]
- Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington’s disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielson KL, Hogue BA, Bohr VA, Cardounel AJ, Nakajima W, Kofler J, Zweier JL, Rodriguez ER, Martin LJ, de Souza-Pinto NC, Bressler J. Mitochondrial toxin 3-nitropropionic acid induces cardiac and neurotoxicity differentially in mice. Am J Pathol. 2001;159:1507–1520. doi: 10.1016/S0002-9440(10)62536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizatullina ZZ, Lindenberg KS, Harjes P, Chen Y, Kosinski CM, Landwehrmeyer BG, Ludolph AC, Striggow F, Zierz S, Gellerich FN. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann Neurol. 2006;59:407–411. doi: 10.1002/ana.20754. [DOI] [PubMed] [Google Scholar]
- Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington’s disease caudate nucleus. Annals of Neurology. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- Hamilton JM, Wolfson T, Peavy GM, Jacobson MW, Corey-Bloom J. Rate and correlates of weight change in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2004;75:209–212. doi: 10.1136/jnnp.2003.017822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Gevorkian S, Marder K, Moskowitz C, Feigin A, Cox M, Como P, Zimmerman C, Lin M, Zhang L, Ulug AM, Beal MF, Matson W, Bogdanov M, Ebbel E, Zaleta A, Kaneko Y, Jenkins B, Hevelone N, Zhang H, Yu H, Schoenfeld D, Ferrante R, Rosas HD. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology. 2006;66:250–252. doi: 10.1212/01.wnl.0000194318.74946.b6. [DOI] [PubMed] [Google Scholar]
- Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Huntington’s Disease Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology. 2001;57:397–404. doi: 10.1212/wnl.57.3.397. [DOI] [PubMed] [Google Scholar]
- Hurlbert MS, Zhou W, Wasmeier C, Kaddis FG, Hutton JC, Freed CR. Mice transgenic for an expanded CAG repeat in the Huntington’s disease gene develop diabetes. Diabetes. 1999;48:649–651. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- Ingwall J. On the hypothesis: cardiac failure is due to decreased energy reserve. Circulation. 1993;87:VII.58–VII.62. [Google Scholar]
- Jenkins BG, Klivenyi P, Kustermann E, Andreassen OA, Ferrante RJ, Rosen BR, Beal MF. Nonlinear decrease over time in N-acetyl aspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington’s disease mice. J Neurochem. 2000;74:2108–2119. doi: 10.1046/j.1471-4159.2000.0742108.x. [DOI] [PubMed] [Google Scholar]
- Jenkins BG, Andreassen OA, Dedeoglu A, Leavitt B, Hayden M, Borchelt D, Ross CA, Ferrante RJ, Beal MF. Effects of CAG repeat length, HTT protein length and protein context on cerebral metabolism measured using magnetic resonance spectroscopy in transgenic mouse models of Huntington’s disease. J Neurochem. 2005;95:553–562. doi: 10.1111/j.1471-4159.2005.03411.x. [DOI] [PubMed] [Google Scholar]
- Joshi MS, Crouser ED, Julian MW, Schanbacher BL, Bauer JA. Digital imaging analysis for the study of endotoxin-induced mitochondrial ultrastructure injury. Anal Cell Pathol. 2000;21:41–48. doi: 10.1155/2000/201406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz AM. Cardiol Clin. Vol. 16. 1998. Is the failing heart energy depleted? pp. 633–644. viii. [DOI] [PubMed] [Google Scholar]
- Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- Lanska DJ, Lavine L, Lanska MJ, Schoenberg BS. Huntington’s disease mortality in the United States. Neurology. 1988;38:769–772. doi: 10.1212/wnl.38.5.769. [DOI] [PubMed] [Google Scholar]
- Li JY, Popovic N, Brundin P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRx. 2005;2:447–464. doi: 10.1602/neurorx.2.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi R, Schapira AH, Manners D, Styles P, Wood NW, Taylor DJ, Warner TT. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann Neurol. 2000;48:72–76. [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Martin-Aparicio E, Yamamoto A, Hernandez F, Hen R, Avila J, Lucas JJ. Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington’s disease. J Neurosci. 2001;21:8772–8781. doi: 10.1523/JNEUROSCI.21-22-08772.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Yang L, Jenkins BG, Ferrante RJ, Rosen BR, Kaddurah-Daouk R, Beal MF. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. Journal of Neuroscience. 1998;18:156–163. doi: 10.1523/JNEUROSCI.18-01-00156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihm MJ, Bauer JA. Peroxynitrite-induced inhibition and nitration of cardiac myofibrillar creatine kinase. Biochimie. 2002;84:1013–1019. doi: 10.1016/s0300-9084(02)00005-6. [DOI] [PubMed] [Google Scholar]
- Mihm MJ, Jing L, Bauer JA. Nitrotyrosine causes selective vascular endothelial dysfunction and DNA damage. J Cardiovasc Pharmacol. 2000;36:182–187. doi: 10.1097/00005344-200008000-00007. [DOI] [PubMed] [Google Scholar]
- Mihm MJ, Seifert JL, Coyle CM, Bauer JA. Diabetes related cardiomyopathy time dependent echocardiographic evaluation in an experimental rat model. Life Sci. 2001a;69:527–542. doi: 10.1016/s0024-3205(01)01141-9. [DOI] [PubMed] [Google Scholar]
- Mihm MJ, Yu F, Weinstein DM, Reiser PJ, Bauer JA. Intracellular distribution of peroxynitrite during doxorubicin cardiomyopathy: evidence for selective impairment of myofibrillar creatine kinase. Br J Pharmacol. 2002;135:581–588. doi: 10.1038/sj.bjp.0704495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihm MJ, Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, Bauer JA. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation. 2001b;104:174–180. doi: 10.1161/01.cir.104.2.174. [DOI] [PubMed] [Google Scholar]
- Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280:30773–30782. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- Orth M, Cooper JM, Bates GP, Schapira AH. Inclusion formation in Huntington’s disease R6/2 mouse muscle cultures. J Neurochem. 2003;87:1–6. doi: 10.1046/j.1471-4159.2003.02009.x. [DOI] [PubMed] [Google Scholar]
- Panov AV, Lund S, Greenamyre JT. Ca2+-induced permeability transition in human lymphoblastoid cell mitochondria from normal and Huntington’s disease individuals. Mol Cell Biochem. 2005;269:143–152. doi: 10.1007/s11010-005-3454-9. [DOI] [PubMed] [Google Scholar]
- Panov AV, Burke JR, Strittmatter WJ, Greenamyre JT. In vitro effects of polyglutamine tracts on Ca2+-dependent depolarization of rat and human mitochondria: relevance to Huntington’s disease. Arch Biochem Biophys. 2003;410:1–6. doi: 10.1016/s0003-9861(02)00585-4. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Paolisso G, Gambardella A, Galzerano D, D’Amore A, Rubino P, Verza M, Teasuro P, Varricchio M, D’Onofrio F. Total-body and myocardial substrate oxidation in congestive heart failure. Metabolism. 1994;43:174–179. doi: 10.1016/0026-0495(94)90241-0. [DOI] [PubMed] [Google Scholar]
- Podolsky S, Leopold NA, Sax DS. Increased frequency of diabetes mellitus in patients with Huntington’s chorea. Lancet. 1972;1:1356–1358. doi: 10.1016/s0140-6736(72)91092-6. [DOI] [PubMed] [Google Scholar]
- Reynolds NC, Jr, Prost RW, Mark LP. Heterogeneity in 1H-MRS profiles of presymptomatic and early manifest Huntington’s disease. Brain Res. 2005;1031:82–89. doi: 10.1016/j.brainres.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Ribchester RR, Thomson D, Wood NI, Hinks T, Gillingwater TH, Wishart TM, Court FA, Morton AJ. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur J Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- Ryu H, Rosas HD, Hersch SM, Ferrante RJ. The therapeutic role of creatine in Huntington’s disease. Pharmacol Ther. 2005;108:193–207. doi: 10.1016/j.pharmthera.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Saft C, Zange J, Andrich J, Muller K, Lindenberg K, Landwehrmeyer B, Vorgerd M, Kraus PH, Przuntek H, Schols L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov Disord. 2005;20:674–679. doi: 10.1002/mds.20373. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pernaute R, Garcia-Segura JM, del Barrio Alba A, Viano J, de Yebenes JG. Clinical correlation of striatal 1H MRS changes in Huntington’s disease. Neurology. 1999;53:806–812. doi: 10.1212/wnl.53.4.806. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F, Wanker EE, Doherty P, Davies SW, Bates GP. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet. 1999;8:813–822. doi: 10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Jr, Greenamyre JT, Snyder SH, Ross CA. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- Schapira A, Lodi R. Assessment of in vitro and in vivo mitochondrial function in Friedreich’s ataxia and Huntington’s disease. Methods Mol Biol. 2004;277:293–307. doi: 10.1385/1-59259-804-8:293. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial involvement in Parkinson’s disease, Huntington’s disease, hereditary spastic paraplegia and Friedreich’s ataxia. Biochim Biophys Acta. 1999;1410:159–170. doi: 10.1016/s0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- Schapiro M, Cecil KM, Doescher J, Kiefer AM, Jones BV. MR imaging and spectroscopy in juvenile Huntington disease. Pediatr Radiol. 2004;34:640–643. doi: 10.1007/s00247-004-1159-y. [DOI] [PubMed] [Google Scholar]
- Schiefer J, Landwehrmeyer GB, Luesse HG, Sprunken A, Puls C, Milkereit A, Milkereit E, Kosinski CM. Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington’s disease. Mov Disord. 2002;17:748–757. doi: 10.1002/mds.10229. [DOI] [PubMed] [Google Scholar]
- Schilling G, Coonfield ML, Ross CA, Borchelt DR. Coenzyme Q10 and remacemide hydrochloride ameliorate motor deficits in a Huntington’s disease transgenic mouse model. Neurosci Lett. 2001;315:149–153. doi: 10.1016/s0304-3940(01)02326-6. [DOI] [PubMed] [Google Scholar]
- Seong IS, Ivanova E, Lee JM, Choo YS, Fossale E, Anderson M, Gusella JF, Laramie JM, Myers RH, Lesort M, MacDonald ME. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- Sharma KR, Romano JG, Ayyar DR, Rotta FT, Facca A, Sanchez-Ramos J. Sympathetic skin response and heart rate variability in patients with Huntington disease. Arch Neurol. 1999;56:1248–1252. doi: 10.1001/archneur.56.10.1248. [DOI] [PubMed] [Google Scholar]
- Smith KM, Matson S, Matson WR, Cormier K, Del Signore SJ, Hagerty SW, Stack EC, Ryu H, Ferrante RJ. Dose ranging and efficacy study of high-dose coenzyme Q(10) formulations in Huntington’s disease mice. Biochim Biophys Acta. 2006;1762:616–626. doi: 10.1016/j.bbadis.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Sorensen SA, Fenger K. Causes of death in patients with Huntington’s disease and in unaffected first degree relatives. J Med Genet. 1992;29:911–914. doi: 10.1136/jmg.29.12.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squitieri F, Cannella M, Sgarbi G, Maglione V, Falleni A, Lenzi P, Baracca A, Cislaghi G, Saft C, Ragona G, Russo MA, Thompson LM, Solaini G, Fornai F. Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech Ageing Dev. 2006;127:217–220. doi: 10.1016/j.mad.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington’s disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- Stack EC, Smith KM, Ryu H, Cormier K, Chen M, Hagerty SW, Del Signore SJ, Cudkowicz ME, Friedlander RM, Ferrante RJ. Combination therapy using minocycline and coenzyme Q10 in R6/2 transgenic Huntington’s disease mice. Biochim Biophys Acta. 2006;1762:373–380. doi: 10.1016/j.bbadis.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Strand AD, Aragaki AK, Shaw D, Bird T, Holton J, Turner C, Tapscott SJ, Tabrizi SJ, Schapira AH, Kooperberg C, Olson JM. Gene expression in Huntington’s disease skeletal muscle: a potential biomarker. Hum Mol Genet. 2005;14:1863–1876. doi: 10.1093/hmg/ddi192. [DOI] [PubMed] [Google Scholar]
- Strong TV, Tagle DA, Valdes JM, Elmer LW, Boehm K, Swaroop M, Kaatz KW, Collins FS, Albin RL. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nature Genetics. 1993;5:259–265. doi: 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH. Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol. 1999;45:25–32. doi: 10.1002/1531-8249(199901)45:1<25::aid-art6>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Blamire AM, Manners DN, Rajagopalan B, Styles P, Schapira AH, Warner TT. Creatine therapy for Huntington’s disease: clinical and MRS findings in a 1-year pilot study. Neurology. 2003;61:141–142. doi: 10.1212/01.wnl.0000070186.97463.a7. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Blamire AM, Manners DN, Rajagopalan B, Styles P, Schapira AH, Warner TT. High-dose creatine therapy for Huntington disease: a 2-year clinical and MRS study. Neurology. 2005;64:1655–1656. doi: 10.1212/01.WNL.0000160388.96242.77. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. 2000;47:80–86. doi: 10.1002/1531-8249(200001)47:1<80::aid-ana13>3.3.co;2-b. [DOI] [PubMed] [Google Scholar]
- Trejo A, Tarrats RM, Alonso ME, Boll MC, Ochoa A, Velasquez L. Assessment of the nutrition status of patients with Huntington’s disease. Nutrition. 2004;20:192–196. doi: 10.1016/j.nut.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, Agid Y, Brice A, Mandel J-L. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- Tsang TM, Woodman B, McLoughlin GA, Griffin JL, Tabrizi SJ, Bates GP, Holmes E. Metabolic characterization of the R6/2 transgenic mouse model of Huntington’s disease by high-resolution MAS 1H NMR spectroscopy. J Proteome Res. 2006;5:483–492. doi: 10.1021/pr050244o. [DOI] [PubMed] [Google Scholar]
- Turko IV, Murad F. Protein nitration in cardiovascular diseases. Pharmacol Rev. 2002;54:619–634. doi: 10.1124/pr.54.4.619. [DOI] [PubMed] [Google Scholar]
- Verbessem P, Lemiere J, Eijnde BO, Swinnen S, Vanhees L, Van Leemputte M, Hespel P, Dom R. Creatine supplementation in Huntington’s disease: a placebo-controlled pilot trial. Neurology. 2003;61:925–930. doi: 10.1212/01.wnl.0000090629.40891.4b. [DOI] [PubMed] [Google Scholar]
- Vogt A, Kubler W. Cardiac energy metabolism-a historical perspective. Heart Failure Reviews. 1999;4:211–219. [Google Scholar]
- Weinstein DM, Mihm MJ, Bauer JA. Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exp Ther. 2000;294:396–401. [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]