

Abstract
The structural changes of bacteriorhodopsin during its photochemical cycle, as revealed by crystal structures of trapped intermediates, have provided insights to the proton translocation mechanism. Because accumulation of the last photointermediate, O, appears to be hindered by lattice forces in the crystals, the only information about the structure of this state is from suggested analogies with the determined structures of the non-illuminated D85S mutant and wild type bacteriorhodopsin at low pH. We used electron paramagnetic resonance spectroscopy of site-directed spin labels at the extracellular protein surface in membranes to test these models. Spin-spin dipolar interactions in the authentic O state compared to the non-illuminated state revealed that the distance between helices C and F increases by ca. 4 Å, there is no distance change between helices D and F, and the distance between helix D and helix B of the adjacent monomer increases. Further, the mobility changes of single labels indicate that helices E and F move outward from the proton channel at the center of the protein, and helix D tilts inward. The overall pattern of movements suggests that the model at acid pH is a better representation of the O state than D85S. However, the mobility analysis of spin-labels on the B-C interhelical loop indicates that the anti-parallel β-sheet maintains its ordered secondary structure in O, instead of the predicted disorder in the two structural models. During decay of the O state, the last step of the photocycle, a proton is transferred from Asp85 to proton release complex in the extracellular proton channel. The structural changes in O suggest the need of large conformational changes to drive the Arg82 side-chain back to its initial orientation towards Asp85, and to rearrange the numerous water molecules in this region in order to conduct the proton away from Asp85.
Keywords: bacteriorhodopsin, electron paramagnetic resonance, site-directed spin labeling, proton translocation, O photointermediate
Introduction
The integral membrane protein, bacteriorhodopsin, is a light-driven proton pump in the purple membrane of Halobacterium salinarum.1 Absorption of a photon by the retinal, bound to Lys216 through a protonated Schiff base, causes its isomerization from all-trans to 13-cis,15-anti, and initiates a series of changes in the conformation of the retinal and the protein (the “photocycle”).2, 3 These changes result in the translocation of the proton across the cell membrane.4 First, the proton from the Schiff base is transferred to Asp85, which is coupled to proton release from an aqueous network to the extracellular surface. Second, the retinal Schiff base is reprotonated by Asp96 that subsequently recruits a proton from the cytoplasmic surface. Third, reprotonation of Asp96 is followed by reisomerization of the retinal to all-trans. Finally, the initial BR state (defined as the non-illuminated form of the protein containing all-trans retinal) is recovered by transfer of a proton from Asp85 to the vacant proton release site.5-7
There are five spectroscopically distinct intermediates (K, L, M, N, and O) identified in this reaction cycle.8 After the N state, the rise of a band at 640 nm in the difference spectrum, which is red-shifted from the BR state at 568 nm, is assigned to the formation of the O intermediate.9 The characteristics of the O state are10, 11: i) the retinal is reisomerized from 13-cis,15-anti in the N state to all-trans, with a twisted configuration indicated by large hydrogen-out-of-plane (HOOP) bands, ii) both the Schiff base and the proton acceptor Asp85 are protonated, iii) the proton release complex at extracellular surface is unprotonated. Thus, the significance of O in the cycle is that it prepares relaxation to the initial state that will require release of the proton from Asp85 to the proton release site, while maintaining a distinctive conformation that provides the barrier to prevent the reverse reaction from BR state to the O intermediate. Although this conformation is not yet known, an earlier study had suggested that the conformation of O enforces an unfavorable ΔpKa for the proton transfer from Asp85 to the proton release site.12 The large dissipation of free energy during the decay of O, as the barrier is removed, will thus ensure that the proton transport across the membrane is unidirectional.
Extensive structural information about the retinal and the protein in the K, L, M, N and BR states has been provided by X-ray diffraction,13-15 electron diffraction,16, 17 and solid-state nuclear magnetic resonance.18 The O intermediate is less studied, and no crystallographic data is available for this state, apparently because accumulation of the O state is hindered by crystal lattice packing.19 This suggests that there might be a large conformational difference between the O and BR states.
Two static diffraction structures, from non-illuminated crystals of the D85S mutant (pdb code:1JV7)20 and wild-type bacteriorhodopsin at pH 2 (pdb code: 1X0I)21 have been suggested to mimic the O state, because the protein in these cases has a red-shifted absorption maximum as it contains a neutral Asp85, either from the serine substitution or from protonation, and the retinal is all-trans (or more precisely, a mixture of all-trans and 13-cis,15-syn). Both structural models show large-scale conformational differences, mainly at the extracellular protein surface, from the BR state. However, there are major discrepancies between the two models, and it is not clear whether the differences arise from differences in crystal packing between the two space groups (C2221 for D85S and P622 for acid bacteriorhodopsin). The D85S structure shows little movement of helices F and G, but large displacements of helices A, B, C, D and E, while in acid bacteriorhodopsin there is little movement of helices A and B, but large movements of helices C, D, E and F. Our purpose was to evaluate the two models through determination of specific structural changes in the authentic O state in the photocycle and compare the results with the predictions of the two models. We used electron paramagnetic resonance spectroscopy of site-directed spin labels22, 23 to detect conformational changes at the extracellular surface in the O state.
EPR has been successfully used in this way in many different systems, e.g., the potassium channel,24 t-SNARE complex,25 Tet repressor,26 T4 lysozyme,27 actin-myosin,28 α-synuclein,29 TonB dependent transporter,30 and annexin B12.31 Since it is not restricted by either conformational hindrances in crystals, or molecular weight limits in solution NMR, EPR spectroscopy has been proved to be a powerful tool for following conformational changes under physiological conditions.
The EPR line-shape of single spin-labels at room temperature contains information about the motions of the nitroxide ring on the nanosecond timescale. The motions include the rotation of the entire protein, the motion of the label from torsional oscillations about bonds of the side-chain, and local backbone fluctuations.32, 33 The latter two modes are modulated by the secondary structure, as well as tertiary contacts. Thus, the EPR line-shape is sensitive to changes in the secondary and tertiary structure, and can identify distinct “fingerprint” characteristics for different sites.33, 34
Spin-spin dipolar interaction leads to spectral broadening to a degree dependent on the inter-spin distance.35-37 At cryogenic temperatures, not only is the rigid lattice condition satisfied, but a possible averaging of anisotropic dipolar interactions can be avoided. Thus, the distance between two different structure elements, and its changes, can be assessed from spin-spin interaction of pairs of such probes in frozen samples.
Our strategy has been to produce photostationary states containing the O intermediate, and analyze line-shape and distance changes of spin-probes covalently linked to engineered cysteine residues on different structural elements. The O state produced in the E204Q mutant should be essentially the authentic O of the photocycle of wild type BR, because the x-ray structures of the unilluminated mutant and the wild type (pdb codes 1F5O and 1C3W, respectively) are very similar except for the Glu204 region, and their O states have the same difference absorption maxima at 640nm. The EPR spectra appear to favor the predictions of the model of the acid form of bacteriorhodopsin over the predictions of D85S. On the other hand, we find that the anti-parallel β-sheet of the BC inter-helical loop is conserved as an ordered structure, in disagreement with both models.
Results
Functional characterization of spin labeled mutants
Because all cysteine mutants constructed contain also the E204Q residue change as background, the decay of the O intermediate is substantially prolonged, from a few milliseconds in the wild-type to hundreds of milliseconds, particularly in D2O buffer at pD 6.38 As shown in Figure 1 as an example, green light caused the accumulation of a red-shifted photoproduct. The unique shape of the spectrum during illumination that has a difference maximum at 640 nm, as in the O state39, rules out significant contributions from other possible photoproducts L, M, or N, all with blue-shifted maxima.39 The accumulation of the O state during continuous illumination was determined from the spectral changes in the photostationary state and its known extinction coefficient relative to the BR state (see Materials and Methods). Table 1 shows that at the available light-intensity, the O state can be accumulated in amounts as high as 50% under illumination. However, according to earlier reports,40 spin labeling may perturb the structure, and thus the photocycle, of bacteriorhodopsin, and the yield of O in the illuminated samples needed to be determined for each spin-labeled mutant.
Figure 1.

Absorption spectra of A196R1 before (grey) and during (black) illumination in D2O buffer (10mM Pi/Na, 100mM NaCl, pD=6.0). A pronounced component with a red-shifted absorption is present in the photostationary state, which is characteristic of the O state. The upward arrow indicates light from the green laser at 532 nm, scattered by the illuminated membrane.
Table 1.
Characteristic absorption properties of bacteriorhodopsin mutants before and after spin labeling. All mutants contained the E204Q residue change as background. The occupancy of O is shown for the spin labeled mutants under continuous illumination in two different buffer systems
| Location of spin label | Abs. max.(nm) before labeling |
Abs. max.(nm) after labeling |
O occupancy at pD 6a % |
O occupancy at pH 9b % |
|---|---|---|---|---|
| --- | 568 | 568 | 50 | N. D. |
| L61R1 | 562 | 559 | 21 | 7 |
| L62R1 | 567 | 566 | 32 | 13 |
| Q75R1 | 568 | 567 | 50 | 19 |
| N76R1 | 566 | 564 | 40 | 8 |
| K129R1 | 567 | 566 | 40 | 7 |
| V130R1 | 566 | 565 | 36 | 12 |
| G195R1 | 568 | 567 | 30 | 8 |
| A196R1 | 568 | 567 | 53 | 28 |
| N76R1/A196R1 | 568 | 564 | 44 | N. D. |
| K129R1/A196R1 | 567 | 566 | 28 | N. D. |
| L61R1/K129R1 | 560 | 552 | 18 | N. D. |
| N78R1 | 564 | 460-570c | <5 | N. D. |
| T128R1 | 566 | 550 | <10 | N. D. |
| Y131R1 | 564 | 555 | <10 | N. D. |
| G192R1 | 564 | 490-570c | <5 | N. D. |
buffer: 10mM Pi/Na, 100mM NaCl, pD=6.0 in D2O
buffer: 10mM Pi/Na, 100mM NaCl, pH=9.0 in H2O
broad peak
N.D.: Not Determined
The results in Table 1 show that the spectra and the photocycle of some of the labeled mutants were considerably perturbed but others less so, as follows. There are three categories: i) Weak or no perturbation (O occupancy > 35 %) was found in Q75R1, N76R1, K129R1, V130R1, A196R1, and N76R1/A196R1, compared to the E204Q background. In these samples, the absorption maxima were shifted (to the blue) by only 1 - 4 nm. This can be understood from the crystal structure of the BR state41 where all of these residues are exposed to the aqueous environment at the protein surface with little contact with nearby residues, precluding the possibility of steric conflicts. ii) Moderate perturbation (O occupancy between 18 and 35%) was found in L61R1, L62R1, G195R1, K129R1/A196R1, and L61R1/K129R1. In these samples the shifts of the absorption maximum were from 1 to 16 nm. Because Leu61 and Leu62 are within helix B and interact with neighboring groups, the addition of a spin label group (ca. 6 Å in length) to L61C and L62C may perturb by interfering with these interactions.41 The same reasoning applies to Gly195. Although at the end of helix F,41 this residue strongly contacts with the neighboring helix E, as confirmed by the EPR line-shape (see results and discussion below). iii) Strong perturbation (O occupancy < 10%) was found in I78R1, T128R1, Y131R1, and G192R1. These locations would have been well suited for constructing spin pairs between helices C, D, E, and F, but they are deeper in the membrane than the others,41 and the labeled samples accumulate too little of the O state to be of utility. They have the largest shifts of the absorption maximum after spin labeling. The very broad peak from 460 nm to 570 nm in I78R1 and G192R1 suggests substantial changes of the structure, or heterogeneous conformations, induced by the spin labeling. When the spin labeled protein was returned to the unlabeled state by reduction with dithiothreitol, the broadened peak disappeared and a maximum at 564 nm, as well as the normal O occupancy, recovered. Thus, the origin of the perturbation is not the cysteine mutation but the addition of the spin label, which creates a longer side-chain than most natural amino acids.
Given these constraints, we used eight single spin-label sites to monitor local mobility changes, and three spin-pairs to monitor distance changes between helices. The locations are all at the extracellular surface of the protein, as shown in Figure 2, where the two models for O show the greatest changes. Leu61 and Leu62 are at the last two turns of helix B near the surface, Gln75 and Asn76 are past the last residue of helix C and on the BC loop. Lys129, Val130, Gly195 and Ala196, are all at the aqueous interface, at the last turns of helices D, E, and F, respectively. .The doubly spin-labeled cysteine mutant N76R1/A196R1 was intended for the distance between helices C and F, and K129R1/A196R1 was for the distance between helices D and F. The K129R1/L61R1 mutant was for measuring the inter-monomer distance between helix D and helix B'. Because it has the highest resolution, we used 1C3W coordinate set to represent the BR state, here and the structure alignment analysis below, instead of the E204Q structure.
Figure 2.

Bacteriorhodopsin model (pdb code: 1C3W) viewed from the extracellular side. Two monomers in a trimer are shown. The amino acids that were replaced by cysteines modified by spin labels are shown as space-filled balls at the coordinates of the side-chain CB atom.
Distance change between helices C and F (N76R1/A196R1)
The D85S model (1JV7)20 and the acid bacteriorhodopsin model (1X0I)21 predict distance increases between helices C and F of ca. 5 Å and ca. 4 Å, respectively, in the O state. When compared to the sum of the spectra of the two single spin-labels, the spin-pair spectra of both non-illuminated and illuminated samples (Figure 3(a)) shows line-shape broadening and large decrease of central line intensity. This indicates strong dipolar interaction (see Materials and Methods), but more in the non-illuminated sample. The decrease of spin-spin interaction, indicated by an increase in the amplitude of the spectrum of the illuminated sample, shows clearly that the distance between helices C and F, increases in the O state relative to the BR state, consistent with both D85S and acid bacteriorhodopsin models.
Figure 3.

EPR spectra at 120K and distance calculation for N76R1/A196R1. (a) Sum of spectra of the N76R1 and A196R1 single mutants without illumination (blue), spectrum of N76R1/A196R1 before illumination (black), and the spectrum of N76R1/A196R1 after illumination (red), superimposed. All spectra are normalized to the same spin number. Panels (b) and (c) show the distance distributions of N76R1/A196R1 in the BR and O states, respectively. The distance distribution in O state was extracted from those in the illuminated samples by subtracting a scaled amount of distribution in the non-illuminated samples according to the occupancy of O in Table 1.The vertical (normalized population) axes of the distributions are arbitrary and selected for convenience of display.
Simulation of the EPR spectra also revealed strong spin-spin dipolar interaction between N76R1 and A196R1. In the non-illuminated state (Figure 3(b)), the distance distribution includes a larger population at ca.13 Å, and a smaller population at ca.10 Å. A mean distance of 12 Å is fully consistent with the distance of 12 Å calculated between the CB atoms of N76C and A196C, based on the BR structure (pdb code: 1C3W)41. In the O state (Figure 3(c)), the distance distribution contains a major population at ca.16 Å. This 4 Å distance increase suggests that either helix C or helix F, or both, move in the O state. The deconvolution also detected a ca. 30% non-interacting component that could have originated from either dipolar interactions outside of the reliable range of the continuous wave EPR method we use or under-labeling (see Materials and Methods). However, the broadened lineshapes are so strong that this non-interacting component can mask neither the spin-spin interaction nor the distance change.
Distance change between helices D and F (K129R1/A196R1)
Both D85S and acid bacteriorhodopsin models exhibit small distance changes, < 1 Å, between helices D and F relative to the BR structure. The spin-pair spectra (Figure 4(a)) show a moderate decrease of the central line intensity compared to the sum of the spectra of the single spin-labels. However, no obvious change of the spin-spin interaction is evident in the amplitude or the line-shape between the non-illuminated and illuminated samples. This suggests that, as predicted by the two models, the distance between helices D and F does not change in the O state.
Figure 4.

EPR spectra at 120K and distance calculation for K129R1/A196R1. (a) Sum of spectra of the K129R1 and A196R1 single mutants without illumination (blue), spectrum of K129R1/A196R1 before illumination (black), and the spectrum of K129R1/A196R1 after illumination (red), superimposed. All spectra are normalized to the same spin number. Panels (b) and (c) show the distance distributions of K129R1/A196R1 in the BR and O states, respectively.
Quantitive analysis detected a medium extent of spin-spin dipolar interaction (Figure 4(b)), with a non-interacting component ca. 34%. In the non-illuminated sample the distance distribution contains one larger population at ca.17 Å, and another smaller population at ca.12 Å. The former is consistent with the distance of ca.16 Å calculated from the structure of the BR state. The latter population may result from a different conformer, which can arise when spin labels are attached to proteins.42 In the O state (Figure 4(c)), this bimodal distance distribution is conserved with only a slight change that is within the 1-2 Å estimated43 uncertainty of the simulation. Thus, we conclude that there is little distance change between helices D and F in the O state. However, this does not necessarily mean that there are no movements of helices D or F, because the same result would be achieved if the two helices were to move in the same direction.
Distance change between helices D and B' (K129R1/L61R1)
The spin-pair mutant K129R1/L61R1 was used to test distance change between helix D and helix B that belongs to the neighboring monomer, labeled B' (Figure 2). According to the BR state structure, the distance between the residues with the labels on helices D and B within the same monomer is ca. 26 Å, which is outside of the upper detection limit of the EPR method used, but the distance between the residues on D and B' is ca. 15 Å. Therefore, any EPR line-shape change modulated by distance changes should be attributed to inter-monomer interaction.
The D85S model predicts a ca. 2 Å decreased distance for this pair, while in acid bacteriorhodopsin the distance is increased by ca. 1 Å relative to the BR state. The EPR line-shape (Figure 5(a)) shows that the non-illuminated sample has much weaker spin-spin interaction than N76R1/A196R1 (Figure 3(a)). The spectrum of the illuminated sample shows an increase of the center line intensity. This suggests that the distance between helices D and B', increases in the O state, supporting the prediction of acid bacteriorhodopsin model.
Figure 5.

EPR spectra at 120K and distance calculation for L61R1/K129R1. (a) Sum of spectra of the L61R1 and K129R1 single mutants without illumination (blue), spectrum of L61/K129R1 before illumination (black), and the spectrum of L61R1/K129R1 after illumination (red), superimposed. All spectra are normalized to the same spin number. Panels (b) and (c) show the deconvoluted distance distributions of L61R1/K129R1 in the BR and O states, respectively.
The simulation analysis reveals one major distance population at ca. 18 Å in the non-illuminated state (Figure 5(b)), while a very broad population from ca. 17 to ca. 22 Å in the O state (Figure 5(c)). The high fraction of non-interacting component of ca. 51% dominates the EPR line-shape so that the simulation yields a greater distance of ca. 18 Å, as compared to ca.15 Å in the BR structural data. Besides, the relatively lower O occupancy at ca.18% makes it difficult to quantify the distance change. Nevertheless, the lesser spin-spin interaction in the raw spectrum of the illuminated sample is at least a qualitative indication of increased distance between the two neighboring helices in the O state, even if the magnitude of this movement cannot be estimated.
BC anti-parallel sheet
In both D85S and acid bacteriorhodopsin the otherwise ordered B-C inter-helical anti-parallel β-sheet could not be modeled from the electron density maps, predicting that it will be disordered in the O state. To test this, we placed two label sites on the B-C loop, but with opposite side-chain orientations relative to the protein surface to detect conformational changes in O state that may occur at either side of the B-C sheet. Backbone motions will increase and tertiary contacts will be absent when ordered secondary structures are transformed to disordered coils.44 Therefore, the overall line-shapes should become sharper and be dominated by a strong isotropic mode from the increased backbone fluctuations and the less restricted side-chain motions.
The label Q75R1 is located on the B-C loop (Figure 2), with the side-chain oriented toward the aqueous environment. There are no obvious tertiary contacts in the BR state. In the absence of constraints imposed by local interactions, the anisotropic internal motion of the spin label may be partially averaged by isotropic backbone fluctuations with nanosecond correlation time. This is apparently the case for the Q75R1 at this exposed site, as the hyperfine spitting is partially resolved (Figure 6). The mobile component at the high-field β region dominants the spectrum, and the narrow center linewidth is ca. 3.7G.
Figure 6.

EPR spectra of singly spin-labeled mutants at room temperature. Spectra in black represent the non-illuminated BR state, the spectra in blue represent the O state. The difference spectra (blue minus black) are shown in red. The low-field α region that identifies the immobile components and the high-field β region that identifies the mobile components are separated by a vertical line. The arrows indicate the changes of immobile and mobile components in the difference spectra (see Results). The EPR spectra of the O state were reconstructed according to the occupancy of O in Table 1 (see materials and methods). The EPR spectra of the non-illuminated BR and O states were normalized to the intensity of the central resonance peak.
Although a large amount (ca. 50%) of the O intermediate accumulates upon illumination, no line-shape change (Figure 6) can be detected between the non-illuminated state and the O state. This appears to rule out a conformational change at this site.
The label N76R1 is located nearer to helix C than Q75R1 (Figure 2). The side-chain orients toward the membrane and may have contacts with neighboring residues, such as Phe71. Consistent with this, the EPR spectra (Figure 6) shows multiple components that may reflect such tertiary contacts. The center linewidth (ca. 4.8G) is greater than in Q75R1, with corresponding wider anisotropic hyperfine spitting. The contribution of the mobile component at the high-field β region is still present but with less amplitude than in Q75R1. These line-shape features show that some tertiary interactions reduce the mobility at this local environment compared to Q75R1. There were no conformational changes observed upon illumination at this site either.
Because there are no significant EPR line-shape and mobility changes at either side of the β sheet as detected by Q75R1 or N76R1, in the O state (Figure 6), conversion of the ordered B-C anti-parallel sheet to disordered coil during the photocycle, as predicted by the two models for O, is doubtful.
Helices D and E
The label K129R1 is located on the extracellular end of helix D, on the D-E interhelical loop. With only four residues, this is the shortest loop in the protein (Figure 2). In Figure 6, a strong isotropic component from the mobile loop structure dominates the line-shape. The narrowest center linewidth ca. 3.2G indicates that K129R1 is the most mobile site of the eight single spin labels tested. A small component at the low field α region can be detected also, which is assigned to tertiary contacts and may originate from the nearest B-C loop elements.
According to the steric packing density analysis (see Materials and Methods), the D85S model predicts increased mobility in O from decreased steric hindrance (ΔN = −5), while the acid bacteriorhodopsin model predicts decreased mobility from increased steric hindrance (ΔN= +3) in Table 2. The EPR spectra (Figure 6) show distinct line-shape changes in the O state. The population of the immobile component at the low-field α region increases (indicated by the upward arrow), while that of the mobile component at the high-field β region decreases (downward arrow). This, and the change of −0.074 G−1 in the mobility parameter (Table 2), suggest slowed motion in the O state. To confirm whether this small mobility and line-shape change can be attributed to the accumulation of O, control samples were prepared at pH 9 in H2O buffer, in which the occupancy of the O state decreased dramatically (Table 1) (see Materials and Methods). If the difference EPR spectra between the non-illuminated and illuminated samples decreases proportionally to the decrease of the O occupancy, the mobility and line-shape changes can be assigned unambiguously to the O state. As shown in Figure 7, this is indeed the case for K129R1. The EPR line-shape of the non-illuminated sample (Figure 6) reveals a small population of tertiary contacts, most likely with Pro70 on the close-lying B-C loop that is within 4 Å ofthe CB of K129R1 (see BR state structure in Figure 8(a)). The increase of this immobile component in the O state (Figure 6) suggests that K129R1 moves closer to the B-C loop. If the conclusion that the anti-parallel sheet structure of the B-C loop is conserved in the O state (see above) is correct, the decreased mobility suggests that helix D moves inward, in the direction of the proton channel. Thus, it is the acid bacteriorhodopsin model that correctly predicts the decrease of the mobility of K129R1 in the O state.
Table 2.
Comparison of the predicted change of steric packing density (ΔN) in 1JV7 and 1X0I with EPR mobility change Δ(ΔH0)−1
| Mutants | ΔN |
Δ(ΔH0)−1,G−1 | |
|---|---|---|---|
| D85S (1JV7) |
Acid BR (1X0I) |
||
| L62R1 | +14 | +6 | −0.008±0.004 |
| K129R1 | −5 | +3 | −0.074±0.021 |
| V130R1 | −2 | −1 | +0.033±0.024 |
| G195R1 | −6 | −11 | +0.037±0.018 |
Figure 7.
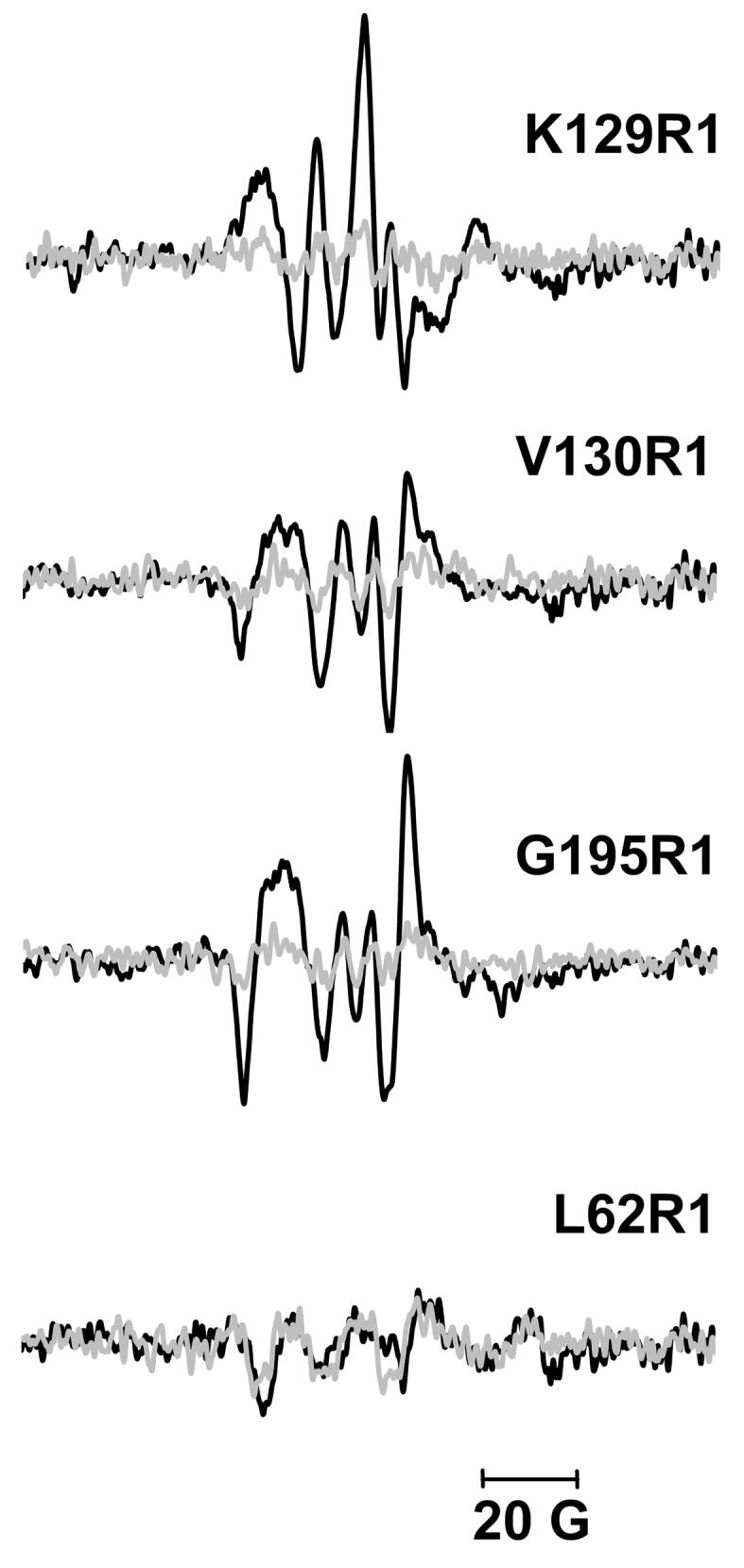
EPR difference spectra at room temperature in D2O buffer (black, 10 mM Pi/Na, 100 mM NaCl, pD=6.0) and in H2O buffer (grey, 10 mM Pi/Na, 100 mM NaCl, pH=9.0). The difference spectra are calculated by subtracting the non-illuminated EPR spectra from the illuminated spectra. At pH=9.0, the amplitude of the difference spectra decreases compared to that at pD=6.0, as the O occupancy decreases (Table 1). Thus, the spectra at pH=9.0 are controls to confirm that the difference spectra at pD=6.0 are attributable to the O state.
Figure 8.

Structure alignments of the BR state (purple, pdb code 1C3W) with the model D85S (blue-green, in (a), (c) and (e), pdb code 1JV7) and acid bacteriorhodopsin model (blue-green, in (b), (d) and (f), pdb code: 1X0I). The spin labels (K129R1 in (a) and (b), G195R1 in (c) and (d), and L62R1 in (e) and (f)) are shown as space-filled balls according to the coordinates of side-chain CB atom. Purple, spin-labels in the BR state; blue, spin-labels in either D85S or acid bacteriorhodopsin. Arrows indicate the directions of the movements of either spin labels or helices.
The label V130R1 is located at the end of helix E, on the D-E loop (Figure 2). In Figure 6, multiple components with resolved hyperfine spitting suggest less backbone fluctuation and more tertiary contacts at this site than at K129R1, consistent with the broader linewidth (ca. 4.7G) for this label. These tertiary contacts may originate from interactions between the two closest structural elements of helices E and F, or helices E and B' on the adjacent monomer (Figure 2).
Both D85S model and acid bacteriorhodopsin model predict increased mobility (ΔN = −2, and −1, respectively) for V130R1. In the O state, we detect a mobility parameter increase of ca. 0.033 G−1 (Table 2), and an increase of the mobile component (upward arrow) and a decrease of the immobile one (downward arrow) in Figure 6. Mollaaghababa45 found, in time-resolved measurements, that an increase of mobility at this location reached its maximum value when M decay was nearly finished. Based on the control experiment (Figure 7) where smaller changes are seen when less O is produced, we conclude that this mobility increase should be assigned to structural changes in the O state. Thus, the mobility increase in the O state suggests that helix E either moves outward, away from the proton channel in order to reduce its steric interaction with helix F, or it moves away from helix B'. In either case, the observed increased mobility is consistent with both structural models for O.
Helix F
The label G195R1 is located at the extracellular end of helix F, facing inward, toward the proton release channel (Figure 2). The EPR line-shape is dominated by the immobile component in the low field α region, with fully resolved hyperfine splitting (Figure 6). Although the label is on the protein surface, the EPR line-shape and the ca. 8.5G center linewidth reveal that this site has the most immobile local environment of the eight sites described. The BR structure in Figure 8(c) shows that the nitroxide side-chain is surrounded by Phe135 on helix E, and Trp189 and Leu190 on helix F. The 36 atoms within 5 Å distance of CB of G195C produce very strong tertiary contacts. Thus, this location provides a sensitive site for detecting local structural changes around helices F and E.
Both D85S and acid bacteriorhodopsin models predict increased mobility (with steric packing factor changes of ΔN= −6 and −11, respectively) in O (Table 2). Upon illumination, there is a 0.037G−1 mobility parameter increase, consistent with a sharp decrease at immobile low-field α region (downward arrow) and an increase at mobile high-field β region (upward arrow) in the difference spectra (Figure 6). The control (Figure 7) confirms that this change originates from O. Therefore, either helix F, or helix E, or both, move away from the proton channel, so that there are fewer tertiary contacts at G195R1. This mobility change is consistent with the predictions of both models.
Helix B
L62R1 is at the last helical turn of helix B, with its side-chain pointing toward helix E' of another monomer (Figure 2). Therefore, the nitroxide side-chain may interact with Tyr133 and Trp137 (BR state model in Figure 8(e)). Consistent with the BR structure, this site shows moderately immobilized line-shape (Figure 6) whose center linewidth is ca. 6.9G, with resolved hyperfine splitting.
Both D85S and acid bacteriorhodopsin models predict decreased mobility in the O state, the first greater than the second (steric packing factor changes ΔN= +14 and +6, respectively). However, at residue 62 we observe less line-shape change upon illumination than in K129R1, V130R1 and G195R1 (Figure 6). This small change is not associated with the O state because the control sample shows a similar change (Figure 7). Thus, the observed small mobility parameter decrease (−0.008 G−1 in Table 2) will have originated either from artifacts induced by illumination, or by low occupancy intermediates, e.g. the M state. Therefore, we conclude that helices B and E' will not approach each other as the D85S model predicts (Figure 8(e)). Although the acid BR model also predicts a decreased mobility, the lesser predicted movement of E' towards B (Figure 8(f)), seems fit EPR data more than D85S.
No line-shape and mobility changes were found at the two other labeling sites, A196R1 and L61R1 (Figure 6). These sites were originally constructed to calculate distances for spin pairs (see above).
Discussion
Spin labeling
Our original intent was to test all structural changes predicted by the two proposed models for the O state, from spin labels at the extracellular ends of each of the seven helices. However, perturbations of the photocycle by many of the spin-labels near the extracellular surface, particularly at helix C, decreased the amount of O that accumulated during illumination, and made this approach unfeasible. A previous report40 showed already that the spin-labeled mutants R82R1 and Y79R1, located inside helix C, were both perturbed in structure and function. We found this to be the case also for I78R1, making it impossible to acquire full motional information for helix C. The sensitivity of this region to labeling is probably due to residues involved in the loss of proton from Asp85 during decay of the O state via the hydrogen-bonding network.46 To avoid substitution of the conserved residue Pro77, the only option left was to label N76C. Thus, instead of I78R1/A196R1, we used N76R1/A196R1 to monitor the distance between helices C and F. Similar problems at the extracellular ends of the other helices severely restricted the location of labeling. Possibly, when the free energy change is small, as in the N to O reaction12, any structural perturbation can cause large changes in the kinetics. In contrast, the cytoplasmic surface was found to be far more permissive to spin-labeling47, 48.
Structural changes of the O state, based on the EPR data
Taken together, the EPR spectra of the unperturbed and moderately perturbed spin-labeled mutants produce a partial but coherent picture of structural changes at the extracellular surface in the O state, as follows.
The secondary structure of the B-C interhelical anti-parallel β sheet is conserved. This conclusion is based on the unchanged line-shapes of N76R1 and Q75R1 (Figure 6) located on the B-C loop, and will need be evaluated by more cysteine substitutions.
Helix F moves outward, away from the proton channel (Figure 9(a)). The increased distance of N76R1 to A196R1, by ca. 4 Å (Figure 3), suggests movement of helix F relative to helix C, either outward or inward. It is very unlikely that helix F moves inward, because decrease of the immobilization of spin-label at G195R1 on helix F (Figure 6 and Table 2) suggests that there are fewer tertiary contacts at this site in the O state.
Figure 9.

(a) Overall structural changes in the O state of bacteriorhodopsin, based on the EPR data. The arrows in (a) indicate the directions of the movements of helices D, E and F. (b) Structural alignment of the M state (pdb code: 1C8S) with acid bacteriorhodopsin (pdb code: 1X0I). Nitrogen and oxygen atoms are shown in purple and red, respectively. Carbon atoms in the M state and acid bacteriorhodopsin are shown in yellow and blue, respectively. Arg82 is oriented to the extracellular surface, toward the proton release site and away from Asp85. However, in the acid bacteriorhodopsin model Glu194 and Glu204 have moved away (indicated by the arrows) from the positively charged Arg82 as compared to the M state, creating a cavity (see Discussion).
An outward tilting of helix F at cytoplasmic surface has been reported, from the distance change between V101R1 and A168R1, with an amplitude of ca. 1 Å,49 and in this time-resolved experiment the change appears in M and decays during the decay of the N and/or the O states. It is not clear from the kinetics when the cytoplasmic tilt of helix F is reversed, but these results, together with our observed outward tilt of helix F at extracellular side, suggest a possible rigid body rocking motion of helix F during the second half of the photocycle.
3. Helix D moves inward, toward the proton channel (Figure 9(a)). The constant distance of K129R1 and A196R1 (Figure 4) suggests that helix D moves in the same direction and amplitude as helix F. This inward movement, away from the center of the trimer and toward the B-C loop, is supported by the decreased mobility of K129R1 (Figure 6 and Table 2). Further, the increased distance between K129R1 and L61R1 (Figure 5) appears to exclude the possibility that helix D moves outward from the proton channel, towards the axis of 3-fold symmetry at the center of the trimer.
4. Helix E tilts outward from the proton channel (Figure 9(a)). This motion may remove some of the rigid contacts of helix E, and the increased backbone fluctuations would be then reflected by the observed increased mobility at V130R1 (Figure 6 and Table 2). The behavior of G195R1 (Figure 6) also contains a clue that there is increased space between helices F and E, from the increased separation of these two helices. However, helix E does not approach helix B' on the neighboring monomer. If the tilt of helix E were to result in stronger tertiary contacts with helix B', the less mobile local environment would be reflected by a decreased mobility at L62R1. Since there is no decreased mobility detected at L62R1 (Figure 6 and 7), the most likely movement of helix E is outward, and sideways from the neighboring helix B'.
However, another model might not be excluded in which helix E does not move. The outward movement of helix F could modify the interface region between helices F and E, or the overall rearrangement of the extracellular side could induce a change of the nitroxide orientation. Either would lead to increase in the mobility at V130R1, as observed, without motion of helix E.
Evaluation of D85S (1JV7) and acid bacteriorhodopsin (1X0I) as models for the O state
The distance changes between helices D and F are very small, less than 1 Å in both D85S and acid bacteriorhodopsin models. They are both consistent with the distance information from K129R1/A196R1 for the O state. However, there is a large discrepancy of the movements of helix D in the two models. In D85S helix D moves sideways relative to the proton channel and away from the B-C loop (Figure 8(a)), while in acid bacteriorhodopsin helix D moves inward to the proton channel and thus approaches the B-C loop, by ca. 2 Å to Phe71 and by ca. 3 Å to Gly72 (Figure 8(b)). The EPR spectra suggest the inward movement of helix D (as in Figure 9(a)) with decreased mobility at K129R1 (Table 2), more as in the acid bacteriorhodopsin model than in D85S.
The distance increase between helices C and F is ca. 5 Å in D85S and ca. 4 Å in acid bacteriorhodopsin. Both agree with the observed distance change of ca. 4 Å in O from N76R1/A196R1, within error. Interestingly however, the directions of the movements of the helices in the two models are different. In D85S (Figure 8(c)), helix F remains unchanged while in acid bacteriorhodopsin (Figure 8(d)), helix F moves outward from the proton channel. Although we could not construct unperturbed singly spin-labeled mutants on helix C, the EPR analysis concludes that the distance change between helices C and F should originate from either the outward tilt of helix F (Figure 9(a)) or the movement of both two helices away from one another, which is nearer to the prediction of the acid bacteriorhodopsin model.
The D85S coordinates predict that the distance between helices D and B' will decrease by ca. 2 Å. In contrast, in acid bacteriorhodopsin this distance increases by ca. 1 Å. The EPR spectra indicate an increased distance of L61R1 and K129R1 in the O state (Figure 5). Because helix D moves inward towards the proton channel (Figure 9(a)), the interaction of helices D and B' between the monomers seems to be reduced, as predicted by the acid bacteriorhodopsin model.
Although both models predict that helix E' will tilt outward, away from the proton channel, helix E' in D85S (Figure 8(e)) moves toward helix B, but in acid bacteriorhodopsin (Figure 8(f)) moves sideways relative to helix B. The tilt of helix E' is supported by the observed increased mobility of V130R1 and G195R1 (Table 2). Because a greater interaction of helices B and E' expected from D85S is not supported by a mobility change of L62R1 (Figure 6 and 7), the conserved line-shape at this site favors the acid bacteriorhodopsin model.
Overall, the structure changes characterized by EPR are consistent with the acid bacteriorhodopsin structure, except at the B-C loop where the absence of electron density suggests increased disorder. However, the disorder may be due to the generally higher temperature factors in 1X0I (average 47 Å2 as compared to 25 Å2 in 1C3W), a less native-like crystal packing in P622 rather than P63, or the shrinkage of the unit cell during acidification (ca. 3.3 Å shortening along the c axis).
How are the movements of helices related to proton translocation at the final step of BR photocycle?
In the M intermediate, Asp85 becomes protonated, and water 402 that connects the Schiff base to Asp85 had moved away or is absent altogether, as are several other water molecules of the hydrogen-bonded network of the extracellular region. For recovery of the initial state through deprotonation of Asp85, the aqueous proton conducting network in the extracellular region must be rebuilt. In acid bacteriorhodopsin, and if it is the correct model for O, in the O state also, the water molecules of the extracellular proton release channel are extensively rearranged. Water 406 moved away from water 401 and Asp212, and water 407 moved away from Tyr57. The water molecules in the network around the Glu194/Glu204 pair is completely rearranged, and are now associated more closely with a sulfate ion rather than the glutamic acids. Water 402 is still absent. These differences break the hydrogen-bonded network that leads from Schiff base region to the extracellular surface and collapse the proton pathway from Asp85 to the proton release site. Thus, the proton pathway is blocked in O and will not recover until the decay of the O state to BR state. In contrast, in D85S the extracellular aqueous network is intact. If the D85S structure were the model for O, the aqueous network between Schiff base and Asp85 would be present in O, i.e., it would have been rebuilt during the N to O transition in the photocycle.
Deprotonation of Asp85 will depend also on recovery of the initial low pKa of Asp85. In the M state the positively charged Arg82 side-chain flips downward50-52 from Asp85 toward the proton release complex that is the aqueous network around Glu194/Glu204.46, 53 This will increase the pKa of Asp85 and cause its protonation, as well as decrease the pKa of the release complex and cause its deprotonation.54 The downward conformation of the Arg82 side-chain is maintained in the acid form of bacteriorhodopsin. This suggests that in the O state the configuration of the Arg82 side-chain continues to support the protonated state of Asp85. Interestingly, because helix F moves outward a cavity appears near the positively charged guanidinium ion of Arg82.21 This would destabilize the downward conformation of Arg82 because not only Glu194 moves ca.4 Å away from Arg82 (Figure 9(b)), but also because the negatively charge at the Glu194/Glu204 pair becomes diffuse as the two acidic groups move away from each other. In acid bacteriorhodopsin, this cavity is partly stabilized by the SO4 anion.21 Therefore, we might expect that in the O state, the energetically costly cavity around the positive charged Arg82 without a counter-ion, like the Glu194/Glu204 pair, may be the driving force to drive the Arg82 back to the upward conformation. Once this occurs, Asp85 will release the proton to the proton release complex, and the cavity is eliminated by return of helix F from its tilted position. Since helix F undergoes little movement in D85S, Arg82 is seen to maintain its downward conformation in this model from stabilization by the Glu194/Glu204 pair, and no clue can be found how the final step of proton translocation is triggered.
Much is still not clear about the final step of bacteriorhodopsin photocycle. What drives the movements of helices D, E, and F in the extracellular region during the decay of N state to O state? Are there conformational changes at the cytoplasmic side in the O state? Is it possible to design a zwitterion to stabilize Arg82 and the Glu194/Glu204 pair by specific binding in the cavity induced by outward movement of helix F, and consequently to lock the O state? The present work tries to understand the final proton translocation from the structural point of view of the O state. By comparing the EPR data with the static structures of D85S and acid bacteriorhodopsin, we find that it is the acid bacteriorhodopsin model that resembles the structure of the O state and provides more insightful information than the former model.
Materials and Methods
Mutagenesis, expression and spin labeling
The site-directed spin labeling method requires substitution of a nitroxide side chain for the native residue at selected sites. This was accomplished by cysteine-substitution mutagenesis, followed by chemical modification of the unique sulfhydryl group with a nitroxide reagent. Other than those introduced for this purpose, bacteriorhodopsin does not contain cysteine. Site-specific mutants of bacteriorhodopsin were prepared according to a method developed by Krebs.55 The shuttle vector PBA2 and the host cell MPK409 were the generous gifts from Dr. Krebs at University of Florida. To aid specific accumulation of the O state during illumination, all cysteine mutants contained also the E204Q mutation.56 The TGC codon for the cysteine was induced into each desired position into E204Q background plasmids by QuikChange method (Stratagene). The mutated PBA2 plasmids were sequence-confirmed, and then transformed into MPK409 host by homologous recombination. After successive selection with mevinolin (4 μg/ml) and (5-fluoroorotic acid 0.25 mg/ml) plates,57 the final recombinants were again confirmed by DNA sequencing. Homologous expression of bacteriorhodopsin in Halobacterium salinarum MPK409 was followed by the standard purification procedure for purple membranes.58 The ratios of absorbance at 280 nm to the retinal chromophore maxima of different mutants were 1.9 - 2.1, which indicated high purity.
The labeling procedure followed a previously reported method,49 with minor modifications. Before labeling, the protein was incubated for 2 hrs at room temperature with 10 mM dithiothreitol solution. After the reducing reagent was removed by five centrifugations in 100 mM NaCl, 10mM phosphate buffer, pH 7.0, ,the purple membranes were resuspended in 1 - 2 ml of the same buffer, to 1 - 2mg/ml concentration, and the spin-label reagent R1 (1-oxyl-2,2,5,5,-tetramethylpyrroline-3-methyl) methanethiosulfonate), in dimethylsulfoxide, was added immediately. The molar ratio of the reagent to the protein was about 10:1. The reaction was carried out for 12 hrs at room temperature. Excess reagent was removed by five centrifugations and washes of the purple membrane with 100 mM NaCl, 10mM phosphate buffer, pH 6.0. The light-dependent accumulation of the O state was increased by replacing H2O with D2O, which slowed the decay of this intermediate. The labeled purple membrane was finally washed and concentrated to 100-150 μM in 100mM NaCl, 10mM phosphate in D2O (pD 6.0) for the EPR measurements. Control samples in which O accumulated to a much lesser extent, were prepared in the same way, but with 100 mM NaCl, 10mM phosphate buffer in H2O (pH 9.0).
The spin labeling reaction was specific to cysteine under these conditions, because the E204Q mutant without cysteine showed no detectable EPR signal after the same labeling procedure. At the high-field region, a sharp EPR signal which is assigned to free spin-label was always present in the spectra of G195R1 and L61R1 (Figure 6). However, the amount of this free label was so small (ca. 5%), that it neither hid the distinct immobile components in the low-field region nor prevented detecting the EPR line-shape changes.
Characterization of spin-labeled mutants
UV-VIS absorption spectra were recorded on a Shimadzu UV 1601 spectrophotometer. Determination of the occupancy of the O state was on the optical spectroscopic multi-channel analyzer (OSMA).59 The spin labeled purple membrane (ca. 5 μl) was loaded into an EPR capillary (1 mm inner diameter), and fixed in a homemade holder to which a green laser was connected. The continuous 532 nm green light (ca. 2 mW/mm2) was passed through the sample from the top of the capillary to produce a photostationary state. The spectra of the illuminated samples showed a pronounced component with a red-shifted absorption. Figure 1 shows an example of such spectra, indicating that a considerable amount of O is accumulated in the illuminated A196R1. To quantify the occupancy of O, the absorption change at 670 nm, which is far from where the L, M, and N intermediates absorb, was used (extinction coefficient 39,000 M−1cm−1).39 With pD 6.0, minor occupancy for M (slight absorption increase at 410 nm in Figure 1) was observed in some spin labeled mutants, but was not considered to significantly affect the calculations of mobility and distances in the O state.
Electron paramagnetic resonance spectroscopy
Continuous-wave EPR experiments were carried out on a Bruker EMX spectrometer with X microwave band. The temperature was maintained by an ER 4112HV controller. For the low temperature (120K ± 0.1K) measurements, the microwave power was 0.2 mW, and the modulation amplitude was 2G. For the room temperature (ca. 298K) measurements, the microwave power was 6 mW and the modulation amplitude was 1.5 G. To avoid possible temperature fluctuations by the illumination, a stream of nitrogen was blown into the cavity. Under these conditions, there was no evidence for power saturation or distortion in the EPR line-shape.
Samples for low-temperature measurements were first tested for occupancy of the O state by recording spectra before and during illumination as described above, and the capillary was plunged into liquid nitrogen while illuminated to freeze-in the photostationary state. Room-temperature samples were directly inserted into the resonance cavity, with the same illumination setup on top of the capillary during data collection.
Structure alignment and steric packing density parameter
To evaluate the two structures 1JV7 and 1X0I (all-trans), both models were aligned with the model for the BR state, 1C3W. First, the 1C3W trimer in the space group P63, was constructed with Swiss-pdb viewer, and then three 1JV7 or 1X0I monomers were loaded into the trimer. Each monomer of 1JV7 or 1X0I was then aligned with a 1C3W monomer on the basis of RMSD minimization of backbone atoms. The final RMSD values relative to 1C3W were 0.91 Å and 0.85 Å for 1JV7 and 1X0I, respectively.
All the distances were measured between CB atoms of interest. To evaluate the distance change between helices C and F, D and F, and D and B' (where the apostrophe indicates that the helix is from a neighboring monomer), measurements of I78C and A196C, K129C and A196C, and K129C and L61C were used respectively. Because the coordinates of Asn76 are absent in both 1JV7 and 1X0I, we used Ile78 instead to measure the distance between helices C and F.
To predict the mobility (ΔH0)−1 change (see below) based on the structure alignment of 1JV7 or 1X0I with 1C3W, the steric packing density parameter (N) was introduced. Because the distance from the CB atom to the ring of the label is about 6 Å, we chose a 5 Å radius to search for close tertiary contacts. Thus, the steric packing density parameter (N) is defined as the number of atoms within a sphere whose center is the CB of spin-labeled side-chain and radius is 5 Å. This parameter evaluates the degree of steric hindrance of the local space surrounding the spin label side-chain, which can modulate the motions of the spin-label. Thus, increase of N suggests stronger steric hindrance that will decrease the mobility, and vice versa. A quantitative relationship between ΔN and mobility change Δ(ΔH0)−1 cannot be established since other complex interactions including Coulombic interaction, van der Waals interaction, and hydrogen bonding may also contribute. The ΔN calculated from the aligned structures exclude the atoms of the spin label side-chain itself. The atoms of lipids are also excluded because the various models contain different coordinates and kinds of lipids.
Mobility parameter
At room temperature, the EPR mobility parameter (ΔH0)−1, a semi-quantitative descriptor of spin motion that implies both rate and order, is the inverse of the width of the central resonance line.32 The EPR spectrum of the O state was calculated from the measured EPR spectrum with illumination by subtracting a scaled amount of the spectrum measured in the dark, and rescaling the result according to the measured occupancy of the O intermediate.
Different from the low temperature EPR experiments, the samples illuminated at room temperature had to be scanned under continuous light for at least 20 mins in order to increase the S/N. Under this condition, the possibility of artifacts induced by the light, increase of temperature, and the possible occupancy of other intermediates had to be considered. Therefore, control samples were prepared at pH 9 in H2O buffer, in which the O occupancy decreased dramatically (Table 1).
Distance calculations
For calculation of inter-spin distances we recorded the EPR spectra of the frozen samples. The spin-spin dipolar interaction can be easily recognized by the decreased amplitude and broadened linewidth from the raw data. Because no line-shape and mobility changes were found at L61R1, A196R1 and N76R1 at both room temperature and in the frozen state, and at K129R1 in the frozen state after the illumination, the center line amplitude change is an unbiased and direct criterion to detect the distance changes in the spin-pair samples.60, 61
The Fourier deconvolution method43 assumes that the dipolar interaction EPR spectrum containing a spin-pair can be represented as the convolution of the non-interacting EPR spectrum with a broadening Pake function. By simulation of the deconvoluted curve, distance distributions from 8-25 Angstroms will be generated (software package written by Dr. Christian Altenbach from UCLA).62 Distances longer than 25 Å, the so called non-interacting components, were not taken into account because they were contributed either by partially labeled cysteine mutants or dipolar interactions outside of the reliable range of this method.43, 60, 62 The distance distribution in O state was extracted from those in the illuminated samples by subtracting a scaled amount of distribution in the non-illuminated samples according to the O occupancy results.
Acknowledgements
We greatly thank Dr. M. P. Krebs at the University of Florida for providing the expression system MPK409, Dr. C. Altenbach at the University of California, Los Angeles and Dr. R. Langen at the University of Southern California for the EPR software package and instructions. We are very grateful for the instructive discussion with B. Schobert, E.S. Imasheva, C. Weihe, S. P. Balashov, A. K. Dioumaev, V. Krishnamani, M. Goldfeld and H. Haigler at University of California, Irvine and with L. S. Brown at University of Guelph. This work was supported by grants from NIH (GM29498) and DOE (DEFG03-86ER13525).
Abbreviations used
- BR
non-illuminated state of bacteriorhodopsin
- NMR
nuclear magnetic resonance
- EPR
electron paramagnetic resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stoeckenius W. Bacterial rhodopsins: evolution of a mechanistic model for the ion pumps. Protein Sci. 1999;8:447–459. doi: 10.1110/ps.8.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanyi JK. Bacteriorhodopsin. Annu. Rev. Physiol. 2004;66:665–688. doi: 10.1146/annurev.physiol.66.032102.150049. [DOI] [PubMed] [Google Scholar]
- 3.Heyn MP, Borucki B, Otto H. Chromophore reorientation during the photocycle of bacteriorhodopsin: experimental methods and functional significance. Biochim. Biophys. Acta. 2000;1460:60–74. doi: 10.1016/s0005-2728(00)00130-4. [DOI] [PubMed] [Google Scholar]
- 4.Khorana HG. Bacteriorhodopsin, a membrane protein that uses light to translocate protons. J. Biol. Chem. 1988;263:7439–7442. [PubMed] [Google Scholar]
- 5.Subramaniam S, Henderson R. Crystallographic analysis of protein conformational changes in the bacteriorhodopsin photocycle. Biochim. Biophys. Acta. 2000;1460:157–165. doi: 10.1016/s0005-2728(00)00136-5. [DOI] [PubMed] [Google Scholar]
- 6.Kandori H. Hydration switch model for the proton transfer in the Schiff base region of bacteriorhodopsin. Biochim. Biophys. Acta. 2004;1658:72–79. doi: 10.1016/j.bbabio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 7.Haupts U, Tittor J, Oesterhelt D. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 1999;28:367–399. doi: 10.1146/annurev.biophys.28.1.367. [DOI] [PubMed] [Google Scholar]
- 8.Balashov SP, Ebrey TG. Trapping and spectroscopic identification of the photointermediates of bacteriorhodopsin at low temperatures. Photochem. Photobiol. 2001;73:453–462. doi: 10.1562/0031-8655(2001)073<0453:tasiot>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 9.Lozier RH, Bogomolni RA, Stoeckenius W. Bacteriorhodopsin: a light-driven proton pump in Halobacterium Halobium. Biophys. J. 1975;15:955–962. doi: 10.1016/S0006-3495(75)85875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith SO, Pardoen JA, Mulder PJ, Mathies R. Chromophore structure in bacteriorhodopsin's O640 photointermediate. Biochemistry. 1983;22:6141–6148. [Google Scholar]
- 11.Kandori H, Yamazaki Y, Hatanaka M, Needleman R, Brown LS, Richter HT, Lanyi JK, Maeda A. Time-resolved fourier transform infrared study of structural changes in the last steps of the photocycles of Glu-204 and Leu-93 mutants of bacteriorhodopsin. Biochemistry. 1997;36:5134–5141. doi: 10.1021/bi9629788. [DOI] [PubMed] [Google Scholar]
- 12.Richter HT, Needleman R, Kandori H, Maeda A, Lanyi JK. Relationship of retinal configuration and internal proton transfer at the end of the bacteriorhodopsin photocycle. Biochemistry. 1996;35:15461–15466. doi: 10.1021/bi9612430. [DOI] [PubMed] [Google Scholar]
- 13.Schobert B, Brown LS, Lanyi JK. Crystallographic structures of the M and N intermediates of bacteriorhodopsin: assembly of a hydrogen-bonded chain of water molecules between Asp-96 and the retinal Schiff base. J. Mol. Biol. 2003;330:553–570. doi: 10.1016/s0022-2836(03)00576-x. [DOI] [PubMed] [Google Scholar]
- 14.Matsui Y, Sakai K, Murakami M, Shiro Y, Adachi S, Okumura H, Kouyama T. Specific damage induced by X-ray radiation and structural changes in the primary photoreaction of bacteriorhodopsin. J. Mol. Biol. 2002;324:469–481. doi: 10.1016/s0022-2836(02)01110-5. [DOI] [PubMed] [Google Scholar]
- 15.Lanyi JK, Schobert B. Mechanism of proton transport in bacteriorhodopsin from crystallographic structures of the K, L, M1, M2, and M2′ intermediates of the photocycle. J. Mol. Biol. 2003;328:439–450. doi: 10.1016/s0022-2836(03)00263-8. [DOI] [PubMed] [Google Scholar]
- 16.Subramaniam S, Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 2000;406:653–657. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 17.Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Surface of bacteriorhodopsin revealed by high-resolution electron crystallography. Nature. 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 18.Herzfeld J, Lansing JC. Magnetic resonance studies of the bacteriorhodopsin pump cycle. Annu. Rev. Biophys. Biomol. Struct. 2002;31:73–95. doi: 10.1146/annurev.biophys.31.082901.134233. [DOI] [PubMed] [Google Scholar]
- 19.Efremov R, Gordeliy VI, Heberle J, Buldt G. Time-resolved microspectroscopy on a single crystal of bacteriorhodopsin reveals lattice-induced differences in the photocycle kinetics. Biophys. J. 2006;91:1441–1451. doi: 10.1529/biophysj.106.083345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rouhani S, Cartailler JP, Facciotti MT, Walian P, Needleman R, Lanyi JK, Glaeser RM, Luecke H. Crystal structure of the D85S mutant of bacteriorhodopsin: model of an O-like photocycle intermediate. J. Mol. Biol. 2001;313:615–628. doi: 10.1006/jmbi.2001.5066. [DOI] [PubMed] [Google Scholar]
- 21.Okumura H, Murakami M, Kouyama T. Crystal structures of acid blue and alkaline purple forms of bacteriorhodopsin. J. Mol. Biol. 2005;351:481–495. doi: 10.1016/j.jmb.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Hubbell WL, Mchaourab HS, Altenbach C, Lietzow MA. Watching proteins move using site-directed spin labeling. Structure. 1996;4:779–783. doi: 10.1016/s0969-2126(96)00085-8. [DOI] [PubMed] [Google Scholar]
- 23.Altenbach C, Marti T, Khorana HG, Hubbell WL. Transmembrane protein structure: spin labeling of bacteriorhodopsin mutants. Science. 1990;248:1088–1092. doi: 10.1126/science.2160734. [DOI] [PubMed] [Google Scholar]
- 24.Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- 25.Xiao W, Poirier MA, Bennett MK, Shin YK. The neuronal t-SNARE complex is a parallel four-helix bundle. Nat. Struct. Biol. 2001;8:308–311. doi: 10.1038/86174. [DOI] [PubMed] [Google Scholar]
- 26.Steinhoff HJ, Suess B. Molecular mechanisms of gene regulation studied by site-directed spin labeling. Methods. 2003;29:188–195. doi: 10.1016/s1046-2023(02)00309-2. [DOI] [PubMed] [Google Scholar]
- 27.Mchaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 28.Korman VL, Anderson SE, Prochniewicz E, Titus MA, Thomas DD. Structural dynamics of the actin-myosin interface by site-directed spectroscopy. J. Mol. Biol. 2006;356:1107–1117. doi: 10.1016/j.jmb.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 29.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merianos HJ, Cadieux N, Lin CH, Kadner RJ, Cafiso DS. Substrate-induced exposure of an energy-coupling motif of a membrane transporter. Nat. Struct. Biol. 2000;7:205–209. doi: 10.1038/73309. [DOI] [PubMed] [Google Scholar]
- 31.Isas JM, Patel DR, Jao C, Jayasinghe S, Cartailler JP, Haigler HT, Langen R. Global structural changes in annexin 12. The roles of phospholipid, Ca2+, and pH. J. Biol. Chem. 2003;278:30227–30234. doi: 10.1074/jbc.M301228200. [DOI] [PubMed] [Google Scholar]
- 32.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem. Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 33.Hubbell WL, Cafiso DS, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 2000;7:735–739. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 34.Kusnetzow AK, Altenbach C, Hubbell WL. Conformational states and dynamics of rhodopsin in micelles and bilayers. Biochemistry. 2006;45:5538–5550. doi: 10.1021/bi060101v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eaton SS, Eaton GR. Distance measurements by CW and pulsed EPR. In: Berliner LJ, Eaton SS, Eaton GR, editors. Biological magnetic resonance: Distance measurements in biological systems by EPR. Kluwer Academic/Plenum; New York: 2000. pp. 2–27. [Google Scholar]
- 36.Hustedt EJ, Beth AH. Nitroxide spin-spin interactions: applications to protein structure and dynamics. Annu. Rev. Biophys. Biomol. Struct. 1999;28:129–153. doi: 10.1146/annurev.biophys.28.1.129. [DOI] [PubMed] [Google Scholar]
- 37.Sale K, Song L, Liu YS, Perozo E, Fajer P. Explicit treatment of spin labels in modeling of distance constraints from dipolar EPR and DEER. J. Am. Chem. Soc. 2005;127:9334–9335. doi: 10.1021/ja051652w. [DOI] [PubMed] [Google Scholar]
- 38.Balashov SP, Lu M, Imasheva ES, Govindjee R, Ebrey TG, Othersen B, III, Chen Y, Crouch RK, Menick DR. The proton release group of bacteriorhodopsin controls the rate of the final step of its photocycle at low pH. Biochemistry. 1999;38:2026–2039. doi: 10.1021/bi981926a. [DOI] [PubMed] [Google Scholar]
- 39.Gergely C, Zimanyi L, Varo G. Bacteriorhodopsin intermediate spectra determined over a wide pH range. Journal of Physical Chemistry B. 1997;101:9390–9395. [Google Scholar]
- 40.Greenhalgh DA, Altenbach C, Hubbell WL, Khorana HG. Locations of Arg-82, Asp-85, and Asp-96 in helix C of bacteriorhodopsin relative to the aqueous boundaries. Proc. Natl. Acad. Sci. U. S. A. 1991;88:8626–8630. doi: 10.1073/pnas.88.19.8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 42.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 43.Rabenstein MD, Shin YK. Determination of the distance between two spin labels attached to a macromolecule. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klug CS, Feix JB. Guanidine hydrochloride unfolding of a transmembrane beta-strand in FepA using site-directed spin labeling. Protein Sci. 1998;7:1469–1476. doi: 10.1002/pro.5560070624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mollaaghababa R, Steinhoff HJ, Hubbell WL, Khorana HG. Time-resolved site-directed spin-labeling studies of bacteriorhodopsin: loop-specific conformational changes in M. Biochemistry. 2000;39:1120–1127. doi: 10.1021/bi991963h. [DOI] [PubMed] [Google Scholar]
- 46.Garczarek F, Brown LS, Lanyi JK, Gerwert K. Proton binding within a membrane protein by a protonated water cluster. Proc. Natl. Acad. Sci. U. S. A. 2005;102:3633–3638. doi: 10.1073/pnas.0500421102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rink T, Pfeiffer M, Oesterhelt D, Gerwert K, Steinhoff HJ. Unraveling photoexcited conformational changes of bacteriorhodopsin by time resolved electron paramagnetic resonance spectroscopy. Biophys. J. 2000;78:1519–1530. doi: 10.1016/S0006-3495(00)76704-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steinhoff HJ, Pfeiffer M, Rink T, Burlon O, Kurz M, Riesle J, Heuberger E, Gerwert K, Oesterhelt D. Azide reduces the hydrophobic barrier of the bacteriorhodopsin proton channel. Biophys. J. 1999;76:2702–2710. doi: 10.1016/S0006-3495(99)77422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Radzwill N, Gerwert K, Steinhoff HJ. Time-resolved detection of transient movement of helices F and G in doubly spin-labeled bacteriorhodopsin. Biophys. J. 2001;80:2856–2866. doi: 10.1016/S0006-3495(01)76252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structural changes in bacteriorhodopsin during ion transport at 2 Angstrom resolution. Science. 1999;286:255–261. doi: 10.1126/science.286.5438.255. [DOI] [PubMed] [Google Scholar]
- 51.Luecke H, Schobert B, Cartailler JP, Richter HT, Rosengarth A, Needleman R, Lanyi JK. Coupling photoisomerization of retinal to directional transport in bacteriorhodopsin. J. Mol. Biol. 2000;300:1237–1255. doi: 10.1006/jmbi.2000.3884. [DOI] [PubMed] [Google Scholar]
- 52.Sass HJ, Buldt G, Gessenich R, Hehn D, Neff D, Schlesinger R, Berendzen J, Ormos P. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature. 2000;406:649–653. doi: 10.1038/35020607. [DOI] [PubMed] [Google Scholar]
- 53.Garczarek F, Gerwert K. Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature. 2006;439:109–112. doi: 10.1038/nature04231. [DOI] [PubMed] [Google Scholar]
- 54.Govindjee R, Misra S, Balashov SP, Ebrey TG, Crouch RK, Menick DR. Arginine-82 regulates the pKa of the group responsible for the light-driven proton release in bacteriorhodopsin. Biophys. J. 1996;71:1011–1023. doi: 10.1016/S0006-3495(96)79302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krebs MP, Mollaaghababa R, Khorana HG. Gene replacement in Halobacterium halobium and expression of bacteriorhodopsin mutants. Proc. Natl. Acad. Sci. U. S. A. 1993;90:1987–1991. doi: 10.1073/pnas.90.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown LS, Sasaki J, Kandori H, Maeda A, Needleman R, Lanyi JK. Glutamic acid 204 is the terminal proton release group at the extracellular surface of bacteriorhodopsin. J. Biol. Chem. 1995;270:27122–27126. doi: 10.1074/jbc.270.45.27122. [DOI] [PubMed] [Google Scholar]
- 57.Peck RF, DasSarma S, Krebs MP. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol. Microbiol. 2000;35:667–676. doi: 10.1046/j.1365-2958.2000.01739.x. [DOI] [PubMed] [Google Scholar]
- 58.Oesterhelt D, Stoeckenius W. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 1974;31:667–678. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 59.Zimanyi L, Keszthelyi L, Lanyi JK. Transient spectroscopy of bacterial rhodopsins with an optical multichannel analyzer. 1. Comparison of the photocycles of bacteriorhodopsin and halorhodopsin. Biochemistry. 1989;28:5165–5172. doi: 10.1021/bi00438a038. [DOI] [PubMed] [Google Scholar]
- 60.Altenbach C, Klein-Seetharaman J, Cai K, Khorana HG, Hubbell WL. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 316 in helix 8 and residues in the sequence 60-75, covering the cytoplasmic end of helices TM1 and TM2 and their connection loop CL1. Biochemistry. 2001;40:15493–15500. doi: 10.1021/bi011545o. [DOI] [PubMed] [Google Scholar]
- 61.Altenbach C, Cai K, Klein-Seetharaman J, Khorana HG, Hubbell WL. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 65 in helix TM1 and residues in the sequence 306-319 at the cytoplasmic end of helix TM7 and in helix H8. Biochemistry. 2001;40:15483–15492. doi: 10.1021/bi011546g. [DOI] [PubMed] [Google Scholar]
- 62.Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]