

Abstract
Neurodegeneration with brain iron accumulation, type 1 (NBIA 1), or Hallervorden-Spatz syndrome, is a rare neurodegenerative disorder characterized clinically by Parkinsonism, cognitive impairment, pseudobulbar features, as well as cerebellar ataxia, and neuropathologically by neuronal loss, gliosis, and iron deposition in the globus pallidus, red nucleus, and substantia nigra. The hallmark pathological lesions of NBIA 1 are axonal spheroids, but Lewy body (LB)-like intraneuronal inclusions, glial inclusions, and rare neurofibrillary tangles also occur. Here we show that there is an accumulation of α-synuclein (αS) in LB-like inclusions, glial inclusions, and spheroids in the brains of three NBIA 1 patients. Further, β-synuclein (βS) and γ-synuclein (γS) immunoreactivity was detected in spheroids but not in LB-like or glial inclusions. Western blot analysis demonstrated high-molecular weight αS aggregates in the high-salt-soluble and Triton X-100-insoluble/sodium dodecyl sulfate-soluble fraction of the NBIA 1 brain. Significantly, the levels of αS were markedly reduced in the Triton X-100-soluble fractions compared to control brain, and unlike other synucleinopathies, insoluble αS did not accumulate in the formic acid-soluble fraction. These findings expand the concept of neurodegenerative synucleinopathies by implicating αS, βS, and γS in the pathogenesis of NBIA 1.
Neurodegeneration with brain iron accumulation (NBIA 1), also known as Hallervorden-Spatz syndrome or adult neuroaxonal dystrophy, is a rare familial and sporadic neurodegenerative disorder. 1-3 Symptoms usually present in late adolescence or early adult life 2-5 and this disease is relentlessly progressive. NBIA 1 is characterized clinically by rigidity, dystonia, dyskinesia, and choreoathetosis, 5-8 as well as dysarthria, dysphagia, ataxia, and dementia. 2-4,8 NBIA 1 also may be associated with other clinical manifestations such as spasticity and extensor plantar responses, 1-4 optic atrophy, 9 retinitis pigmentosa, 8,10 as well as seizures and myoclonic jerks. 8
Pathologically, NBIA 1 is characterized by cerebral atrophy, symmetrical partially destructive lesions of the globus pallidus with iron deposition in the medial globus pallidus, red nucleus, substantia nigra pars reticularis, and dentate nucleus. 1-4,10,11 The iron deposition results in a characteristic magnetic resonance image with a hypointense center surrounded by a hyperintense area in the pallidum (“eye of the tiger”). 8,12 The designation of NBIA 1 is increasingly used instead of the term Hallervorden-Spatz syndrome to describe this disorder. 13 Associated with neuronal loss is an intense gliosis of the medial globus pallidus 3,8,14 the external segment is typically spared. 3 Muscle pathology includes myeloid structures, dense bodies, and fiber splitting. 15 Nonnervous tissue may also be involved as liver and pituitary abnormalities have been described. 16 Bone marrow biopsy has demonstrated sea-blue histiocytes and osmophilic inclusions 8,17 have been described in lymphocytes suggesting that NBIA 1 is a systemic disorder.
The major histopathological hallmarks of NBIA 1 are axonal spheroids, 3,4,11,14 which have been shown to contain immunoreactive (IR) neurofilament (NF) proteins, 7,18,19 superoxide dismutase, 11 amyloid precursor protein (APP), 20 and α-synuclein (αS). 18,19,21 In addition to spheroids, other characteristic lesions include glial cytoplasmic inclusions (GCIs), 18 Lewy body (LB)-like intraneuronal inclusions (NCIs), 7,13,18,19,22 and dystrophic neurites (DNs), 7,18,19 whereas in late onset NBIA 1, tau pathology has been demonstrated 14,22,23 consisting of both paired helical filaments and straight filaments without amyloid β-protein (Aβ) deposition.
In this study, we examined three cases of NBIA 1 immunohistochemically with antibodies to NFs, purified LBs, tau, Aβ, APP, αS, as well as β-synuclein (βS) and γ-synuclein (γS). Moreover, we also mapped topographically-distinct epitopes extending throughout αS in lesions that were immunostained by anti-αS antibodies, and we demonstrated species of normal and abnormal αS in the NBIA 1 brain by Western blot analysis.
Materials and Methods
Case Materials
Three autopsy proven cases (one infantile, one adult, one late-onset) of NBIA 1 were obtained from the brain banks at the Center for Neurodegenerative Disease Research, Case Western Reserve University, and the Universities of Miami and Maryland (see Table 1 ▶ for clinical demographics). Tissue was fixed in either 10% neutral buffered formalin or 70% ethanol/150 mmol/L NaCl and paraffin-embedded. The following brain regions were examined: motor cortex, sensory cortex, midfrontal lobe, orbitofrontal lobe, cingulate gyrus, superior temporal gyrus, amygdala, hippocampus (anterior and posterior), visual cortex, insula, caudate, putamen, globus pallidus, nucleus accumbens, basal forebrain, thalamus, hypothalamus, midbrain/substantia nigra, pons/locus ceruleus, medulla, cervical spinal cord, olfactory bulb, and trigeminal ganglion when available. Serial sections of 6 μm thickness were analyzed.
Table 1.
Clinical Demographics and Neuropathology of NBIA 1 Cases
| Case | Age at onset | Age at death (years) | Cause of death | Family history | Presenting symptom | Other features | PMI (hrs) | Brain wt (grams) | Macroscopic and microscopic neuropathology |
|---|---|---|---|---|---|---|---|---|---|
| Univ. of Miami | 14 months | 8 | Aspiration pneumonia | NO | Loss of developmental milestones | Flaccidity, hearing loss, muscle cramps, dementia, dysphagia, seizures | 5 | 1024 | Severe atrophy, mild ventricular enlargement, severe neuronal loss in spinal cord, iron deposition, spheroids, NCIs, GCIs |
| CNDR | 18 years | 29 | Inanition | NO | Tremor | Parkinsonism, optic atrophy, dementia, dysphagia, dysarthria, spasticity, pes cavus | 6 | 1080 | Severe atrophy, mild ventricular enlargement, depigmentation of locus ceruleus, iron deposition, spheroids, NCIs, GCIs, glial tangles |
| Case Western Reserve | 75 years | 77 | Aspiration pneumonia | NO | Confusion | Dementia, behavioral abnormalities | 60 | 1530 | No atrophy, severe ventricular enlargement, small cortical infarct, iron deposition, spheroids, NCIs, GCIs |
PMI, postmortem interval; NCI, neuronal cytoplasmic inclusions; GCI, glial cytoplasmic inclusion; CNDR, Center for Neurodegenerative Disease Research.
Immunohistochemistry
Histochemical studies were performed using hematoxylin and eosin (H&E) and silver stains whereas immunohistochemistry was performed using well-characterized antibodies as described. 24,25 The antibodies used here included those specific for NF subunits (RMdO20, NFL), 26 ubiquitin (mAb 1510, Chemicon, Temecula, CA), 27 purified cortical LBs (LB48), 24 αS (LB509, Syn204, Syn208, SNL1), 28,29 αS/βS (Syn202, Syn205, Syn214, SNL4), 29 βS (Syn207), 18 γS (antisera20), 18 tau (17026), 30 APP (LN39), 31 and Aβ (2332). 32 Sections were probed by immunohistochemistry, developed using the avidin-biotin-peroxidase complex method (Vector Labs, Burlingame, CA) and then lightly counterstained with hematoxylin.
Sections were examined and graded semiquantitatively using the following system: 0 = no pathology, 1 = rare pathology, 2 = mild pathology, 3 = moderate pathology, and 4 = severe pathology. Each section was analyzed for synuclein IR spheroids, LBs, NCIs, GCIs, and DNs as well as for tau and amyloid pathologies.
Western Blot Analysis
Western blotting and biochemical fractionation were performed as described previously. 29,33 Briefly, gray matter (0.3 gram) from the midfrontal cortex of NBIA 1 (Case Western Reserve) and a normal age-matched control brain was homogenized in 2 ml/gram of tissue of high-salt (HS) buffer (50 mmol/L Tris, pH 7.4, 750 mmol/L NaCl, 10 mmol/L NaF, 5 mmol/L ethylenediaminetetraacetic acid with protease inhibitors) and centrifuged at 100,000 × g for 30 minutes. The pellets were re-extracted and the supernatants were pooled. The pellets were sequentially extracted twice with 2 ml/gram of HS buffer/1% Triton X-100 (HS/T) and once with 1 ml/gram sodium dodecyl sulfate (SDS) sample buffer (1% SDS, 10% sucrose, 10 mmol/L Tris, pH 6.8, 1 mmol/L ethylenediaminetetraacetic acid, 40 mmol/L dithiothreitol). The pellets were extracted with 0.67 ml/gram 70% formic acid (FA) and disrupted with two sequential 2-second sonication bursts. FA was evaporated in an Automatic Environment SpeedVAc System (Savant Instruments, Holbrook, NY). SDS sample buffer (0.67 ml/gram) was added to the dried pellets, followed by vigorous vortex, and the pH was adjusted with NaOH. SDS sample buffer was added to the HS and HS/Triton fraction and all of the samples were boiled for 5 minutes. The FA samples were centrifuged at 13,000 × g for 5 minutes to remove insoluble debris. Five μl of each fraction was loaded in separate lanes for SDS-polyacrylamide gel electrophoresis followed by Western blot analysis. 29,33
Results
Macroscopic and Microscopic Neuropathology
Two of the three brains exhibited severe atrophy with mildly dilated ventricles. The third brain (late onset) had marked ventricular dilatation without significant atrophy (see Table 1 ▶ ). All of the cases examined had iron accumulation in the globus pallidus, substantia nigra, red nucleus, and dentate nucleus whereas microscopic evaluation revealed neuronal loss with gliosis. The infantile onset case also displayed marked loss of anterior horn cells in cervical and lumbar spinal cord. Dystrophic axon spheroids were noted with H&E and silver stains in the basal ganglia and brainstem in all three cases.
Synuclein Pathology
αS pathology was seen throughout all three NBIA 1 brains (Figure 1) ▶ . For example, αS IR NCIs and spheroids were abundant in the globus pallidus, putamen, thalamus, midfrontal gyrus, precentral gyrus, postcentral gyrus, amygdala, and entorhinal cortex (Figure 1, A–H) ▶ . In addition, most cortical regions displayed numerous αS-positive DNs, but they were most prominent in the CA2/3 region of the hippocampus (Figure 1I) ▶ . Antibodies specific for βS and γS labeled axonal spheroids but not NCIs or DNs.
Figure 1.
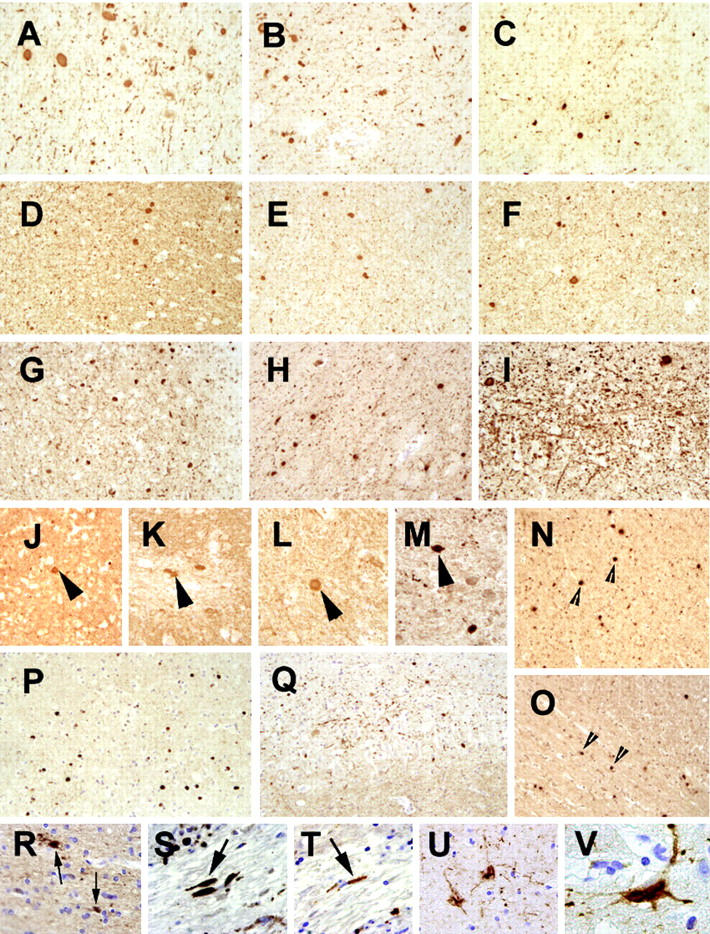
Neuropathology of NBIA 1. A–I: Panels demonstrate abundant αS pathology in the NBIA 1 brain. There are numerous NCIs, DNs, and spheroids distributed in all regions examined including globus pallidus (A), putamen (B), thalamus (C), midfrontal gyrus (D), motor cortex (E), sensory cortex (F), amygdala (G), and entorhinal cortex (H). Note the extensive αS IR DNs in the CA3 region of the hippocampus (I). J-M: Panels demonstrate βS IR in spheroids (large arrowheads) but not in NCIs or DNs. These spheroids are less numerous than the αS IR spheroids, but they are also found throughout the NBIA 1 brain including cingulate gyrus (J), inferior olive (K), cervical spinal cord (L), and putamen (M). Spheroids, but not NCIs or DNs are also demonstrated with antibodies against γS (small arrowheads) in midfrontal gyrus (N) and cingulate gyrus (O). NCIs and spheroids are labeled in cingulate gyrus (P) by the antibody, LB48, which recognizes the mid-sized NF subunit. DNs are also seen with NF antibodies in the CA3 region of the hippocampus (Q), but to a lesser degree than with the αS antibodies. GCIs in oligodendrocytes were detected throughout the white matter in NBIA 1 with antibodies to αS (small arrows in R). No SPs were seen, however axonal dilatations (large arrows) were detected in the basal ganglia with antibodies against Aβ (S) and tau (T). In the hippocampus, glial tangles were seen with antibodies against tau in (U) with a higher power view in (V).
Panels J to M in Figure 1 ▶ illustrate representative βS IR spheroids in the cingulate gyrus, inferior olive, cervical spinal cord, and putamen, respectively, whereas panels N to O in Figure 1 ▶ show typical γS IR spheroids in the cingulate and midfrontal cortices. However, similar NCI, DNs and spheroids also were detected with antibodies to NF proteins and ubiquitin (data not shown) as well as with antibodies raised against purified cortical LBs (LB48) that recognize the mid-sized NF subunit 24 as shown in Figure 1, P–Q ▶ . Finally, GCIs in white matter oligodendrocytes were best demonstrated with antibodies specific for αS (Figure 1R) ▶ but not by antibodies to βS or γS (data not shown).
Tau and Aβ Pathology
Senile plaques were not detected in any of the three cases by immunohistochemistry with antibodies to Aβ or by silver-staining methods. However, many dystrophic processes were labeled by antibodies to Aβ (Figure 1S) ▶ and to APP (data not shown). Dystrophic neurites also were labeled with antibodies to tau proteins (Figure 1T) ▶ . Rare neurofibrillary tangles were noted in the entorhinal cortex of the late onset case whereas the brain of the Center for Neurodegenerative Disease Research case contained tau IR glial tangles in the hippocampus and entorhinal cortex (Figure 1, U–V) ▶ .
Epitope Analysis
To define the extent to which αS was incorporated in neuropathological lesions in the NBIA 1 brain, we used antibodies specific for defined epitopes in different domains spanning the αS molecule in immunohistochemical epitope mapping studies (Figure 2) ▶ . Remarkably, we noted variations in staining intensity and pattern of distinct lesions with different epitope-specific anti-αS antibodies. For example, NCIs, DNs, and spheroids were seen best with LB509, which recognizes amino acids 115 to 122 in αS 28 and the αS amino-terminal antibody SNL4 (amino acids 2 to 12). 29 In contrast, NCIs, DNs, and spheroids were less immunoreactive with antibodies directed against the carboxy-terminal region of αS (ie, Syn202, Syn205, and Syn214 to epitopes within amino acids 130 to 140). 29 The antibodies directed against the hydrophobic-middle region of αS, (ie, Syn204 and Syn208 to epitopes in amino acids 87 to 110 and SNL1 to amino acids 104 to 119) 29 poorly stained NCIs and DNs. Almost no spheroids were detectable with Syn208 and none were labled with Syn204 or SNL1. Moreover, LB509, SNL4 and the carboxy-terminal antibodies (Syn202, Syn205, and Syn214) stained NCIs in a dense homogenous pattern whereas the antibodies to the middle hydrophobic region of αS (Syn204, Syn208, and SNL1) displayed a less intense puntate pattern of immunoreactivity. Finally, GCIs were seen best by LB509 and SNL4 but they were difficult to recognize with the other antibodies against αS (data not shown).
Figure 2.

Epitope mapping of NBIA 1 pathology with αS antibodies. A schematic representation of the αS protein is shown with the six KTKEGV repeat motifs depicted in red. The NAC peptide (amino acids 61–95) is demonstrated in the hydrophobic-middle region and the two familial PD mutations (A30P and A53T) are shown. Epitope mapping of eight αS antibodies is shown with their amino acid sequences. Antibodies were used at equivalent titers as assayed by enzyme-linked immunosorbent assays as previously described. 29 NCIs are recognized by all αS antibodies, however there are differences in staining intensity. Antibodies directed against the middle hydrophobic region (Syn204, Syn208, SNL1) do not stain NCIs as intensely as amino-terminal antibodies (SNL4) or carboxy-terminal antibodies (Syn202, Syn205, Syn214). Interestingly, antibody LB509, which recognizes a polypeptide stretch between the middle and carboxy-terminal regions, intensely stains NCIs. DNs are best recognized by SNL4 and LB509 and to a lesser extent by the carboxy-terminal antibodies. Middle hydrophobic region antibodies either weakly stained (Syn204, Syn208) or did not recognize (SNL1) DNs. Spheroids were also best recognized by SNL4 and LB509. Middle region antibodies weakly stained (Syn208) or did not recognize (Syn204, SNL1) spheroids at all. See text for complete description of epitope mapping.
Biochemistry
Western blot analysis demonstrated the presence of αS in the HS-soluble fraction in the control and NBIA 1 brains (Figure 3 ▶ , lanes 1–2), however there was a marked increase in high-molecular weight aggregates in the NBIA 1. In contrast to the control brain (Figure 3 ▶ , lane 3), however, there was a significant reduction in the levels of αS in the HS/T-soluble fraction of the NBIA 1 brain (Figure 3 ▶ , lane 4, small arrow). In the SDS-soluble fraction from the NBIA 1 brain, there was an accumulation of high-molecular weight aggregates of αS (Figure 3 ▶ , lane 6, large arrow), but not in the control case (Figure 3 ▶ , lane 5). In comparison to other synucleinopathies, ie, Parkinson’s disease (PD), dementia with LBs (DLB), and multiple system atrophy, αS did not aggregate or accumulate in the FA-soluble fraction of the control or NBIA 1 brains (Figure 3 ▶ , lanes 7 and 8, respectively).
Figure 3.

Western blot analysis of the NBIA 1 brain. Control normal (C) and NBIA 1 (D) brains were sequentially extracted with HS, HS/T, SDS, and FA as described in Materials and Methods. Five μl of each fraction were loaded in separate lanes of a 12% SDS-polyacrylamide gel electrophoresis gel and after electrophoretic transfer to nitrocellulose membranes, the anti-αS antibody Syn208 was used to probe the membranes. All of the blots were probed at the same time and developed for the same amount of time with ECL reagents (NEN Life Sciences, Bedford, MA). αS was present in the HS soluble fraction in both the control (lane 1) and NBIA 1 brain (lane 2) but there was a marked increase in high-molecular weight aggregates in the NBIA 1 brain. Compared to the HS/T extract of the control brain (lane 3), there was a marked reduction in the level of αS in the NBIA 1 brain (lane 4, arrowhead). Also note the presence of higher molecular weight aggregates of αS in the SDS fractions of the NBIA 1 brain (arrow in lane 6) but not in the same fraction from the control brain (lane 5). Finally, there were no αS aggregates in the FA extracts of either the control (lane 7) or the NBIA 1 brain (lane 8), although αS aggregates are present in the FA extracts of other synucleinopathies (see text for further discussion).
Discussion
The synucleins are a family of soluble presynaptic proteins that are widely expressed in neurons of the central nervous system. Two missense mutations in the αS gene have been described in rare familial PD kindreds. 34,35 LBs of sporadic and familial PD, DLB, the LB variant of Alzheimer’s disease (AD), 36,37 familial AD, 33 and Down’s syndrome 38 are also composed primarily of αS, but other proteins, such as NF subunits, are found consistently in these inclusions. 24,37 In addition, αS is a major component of GCIs in multiple system atrophy. 18,39 NBIA 1 is a rare neurodegenerative disorder with neuronal, axonal, and glial pathology that may be widely distributed or relatively restricted in the central nervous system. Whereas many cases begin in the late juvenile to early adult years, there have been reports of late onset cases, 4,14 including one of the cases described here. The histopathological hallmark of NBIA 1 is the axonal spheroid that seems to be primarily comprised of insoluble αS.
In addition to αS-rich spheroids, we show here that αS IR NCIs, GCIs, and DNs are dispersed throughout the NBIA 1 brain. These pathological lesions, together with iron deposition, distinguish NBIA 1 from other synucleinopathies. Significantly, AD pathology may be absent from NBIA 1 brains even when patients with this disorder become demented. Although our cases showed no evidence of Aβ IR plaques and tau IR neurofibrillary tangles were very rare in the late onset case, the Center for Neurodegenerative Disease Research brain had abundant tau IR glial tangles that also were weakly immunostained by antibodies to αS.
Recently, it was reported that NCIs but not spheroids in NBIA 1 brains are αS IR. 19 The discordance between the results in this study and the data described here may be because of a number of factors including differences in the specificities of the anti-αS antibodies used in each study. Indeed, our epitope mapping study, using antibodies to topographically separate antigenic sites throughout αS, suggests that SNL4 (amino terminal, amino acids 2 to 12) and LB509 (amino acids 115 to 122) are the most sensitive probes for detecting axon spheroids. Moreover, consistent with a previous report, 22 we show here carboxy-terminal-specific antibodies (Syn202, Syn205, Syn214) as well as antibodies to epitopes in the hydrophobic central region of αS (Syn204, Syn208, and SNL1) label these spheroids weakly or not at all, and are less sensitive for detecting NCIs and DNs. Notably, LB509, which recognizes an epitope between the hydrophobic-middle and the carboxy-terminal regions of αS, labels αS IR pathology in the NBIA 1 brains more intensely than antibodies to the two flanking regions. Thus, the LB509 epitope may be more accessible for antibody binding than the flanking epitopes detected by our other anti-αS antibodies, and this may reflect pathological alterations of αS specific to NBIA 1. Nonetheless, our study confirms and extends previous reports that axonal spheroids in the NBIA 1 brain are robustly labeled by antibodies to αS. 18,21
Although initially, βS and γS had not been implicated in neurodegenerative disease, 25 we reported recently that the hippocampus of patients with PD and DLB contain pathological aggregates of αS and βS in mossy fiber projections as well as γS IR spheroids in the molecular layer of the dentate gyrus. 25 γS IR spheroids were not seen in other disorders with axonal dilatations such as Pick’s disease. 25 Here, we extend these findings by demonstrating βS and γS IR spheroids in another neurodegenerative disease, ie, NBIA 1. However, the βS IR vesicular pathology that we detected in PD and DLB 25 was not seen in NBIA 1.
Significantly, our Western blot analysis demonstrated αS in the HS-soluble fraction of both the control and NBIA 1 brains, but multiple higher molecular weight species of αS were more prominent in this fraction of the NBIA 1 brain. Moreover, there also was a dramatic reduction of αS in the HS/T fraction of the NBIA 1 brain compared to control, similar to that reported in familial AD. 33 Because this fraction may represent a membrane-bound αS component, the loss of membrane integrity, perhaps including synaptic vesicle membranes may be a common feature of several different synucleinopathies. A significant pool of high-molecular weight species of αS accumulated in the SDS-soluble fraction only in the NBIA 1 brain, some of which failed to enter the gel. Interestingly, little or no αS was detected in the FA fraction. Although this distinguishes NBIA 1 from other synucleinopathies, ie, multiple system atrophy and familial AD with LBs, 33 where αS has been detected in the FA fraction, the reasons for this remain to be elucidated.
The aggregation of normally soluble proteins into fibrillar lesions is the neuropathological hallmark of many diseases. The involvement of αS, βS, and γS in the neuronal, glial, and axonal pathology in NBIA 1 continues to expand the concept of neurodegenerative synucleiopathies. 37,39-42 Although it is not known if the conversion of soluble normal proteins into insoluble variants that form pathological aggregates (ie, LBs, GCIs, and spheroids) causes neurodegeneration, it is likely that these alterations impair the long-term viability of neurons. For example, we have previously proposed 24,25,37,41 that LB-containing neurons could undergo a dying-back phenomenon caused by LBs that block axonal transport, thereby disconnecting one brain region from another. Similarly, αS-rich axonal spheroids of NBIA 1 could interfere with the transport of important structural or functional proteins to distal intracellular targets. Continued efforts to elucidate the pathological mechanisms leading to the conversion of soluble proteins, such as αS, into insoluble aggregates as NCIs, as well as the accumulation of proteins such as αS, βS, and γS in axonal spheroids could lead to novel diagnostic and therapeutic strategies to treat the large number of patients with clinically diverse synucleinopathies such as PD, DLB, and multiple system atrophy as well as the rare patients with NBIA 1.
Acknowledgments
We thank Ms. Theresa Schuck and Ms. Danielle Lavalla for their expert technical assistance. We also thank the Brain and Tissue Bank for Developmental Disorders at the University of Maryland and University of Miami through National Institute of Child Health and Human Development contract N01-HD-8–3284 and the Brain Bank at the Alzheimer Disease Research Center at Case Western Reserve University for tissue.
Footnotes
Address reprint requests to John Q. Trojanowski, M.D., Ph.D., Center for Neurodegenerative Disease Research, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, 3rd floor Maloney Building, 3600 Spruce St., Philadelphia, PA 19104. E-mail: trojanow@mail.med.upenn.edu.
Supported by grants from the National Institute on Aging and a Pioneer Award from the Alzheimer’s Association. B. G. is a recipient of a fellowship from the Human Frontier Science Program Organization.
J. E. G. and B. G. contributed equally to this manuscript
References
- 1.Halliday W: The nosology of Hallervorden-Spatz disease. J Neurol Sci 1995, 134:84-91 [DOI] [PubMed] [Google Scholar]
- 2.Jellinger K: Neuroaxonal dystrophy: its natural history and related disorders. Zimmerman HM eds. Progress in Neuropathology, 1973, vol 2.:pp 129-180 Grune and Stratton, Inc., New York [Google Scholar]
- 3.Dooling EC, Schoene WC, Richardson EP: Hallervorden-Spatz syndrome. Arch Neurol 1974, 30:70-83 [DOI] [PubMed] [Google Scholar]
- 4.Jankovic J, Kirkpatrick JB, Blomquist KA, Langlias PJ, Bird ED: Late onset Hallervorden-Spatz disease presenting as familial parkinsonism. Neurology 1985, 35:227-234 [DOI] [PubMed] [Google Scholar]
- 5.Taylor TD, Litt M, Kramer P, Pandolfo M, Angelini L, Nardocci N, Davis S, Pineda M, Hattori H, Flett PJ, Cilio MR, Bertini E, Hayflick SJ: Homozygosity mapping of Hallervorden-Spatz syndrome to chromosome 20p12.3-p13. Nat Genet 1996, 14:479-481 [DOI] [PubMed] [Google Scholar]
- 6.Maladrini A, Cesaretti S, Mulinari M, Palmeri S, Fabrizi GM, Villanova M, Parrotta E, Matagnani A, Montagnani M, Anichini M, Guazzi GC: Acanthosytosis, retinitis pigmentosa, pallidal degeneration. Report of two cases without serum lipid abnormalities. J Neurol Sci 1996, 140:129-131 [DOI] [PubMed] [Google Scholar]
- 7.Sugiyama H, Hainfellner JA, Schid-Siegal B, Budka H: Neuroaxonal dystrophy combined with diffuse Lewy body disease in a young adult. Clin Neuropathol 1993, 12:147-153 [PubMed] [Google Scholar]
- 8.Swaiman KF: Hallervorden-Spatz syndrome and brain iron metabolism. Arch Neurol 1991, 48:1285-1293 [DOI] [PubMed] [Google Scholar]
- 9.Battistella PA, Midena E, Suppiej A, Carollo C: Optic atrophy as the first symptom in Hallervorden-Spatz syndrome. Childs Nerv Syst 1998, 14:135-138 [DOI] [PubMed] [Google Scholar]
- 10.Tripathi RC, Tripathi BJ, Bauserman SC, Park JK: Clinicopathologic correlation and pathogenesis of ocular and central nervous system manifestations in Hallervorden-Spatz syndrome. Acta Neuropathol 1992, 83:113-119 [DOI] [PubMed] [Google Scholar]
- 11.Nishiyama K, Murayama S, Nishimura Y, Asayama K, Kanazawa I: Superoxide dismutase-like immunoreactivity in spheroids in Hallervorden-Spatz disease. Acta Neuropathol 1997, 93:19-23 [DOI] [PubMed] [Google Scholar]
- 12.Savoiardo M, Halliday WC, Nardocci N, Strada L, D’Incerti L, Angelini L, Rumi V, Tesoro-Tess JD: Hallervorden-Spatz disease: MR and pathologic findings. Am J Neuroradiol 1993, 14:155-162 [PMC free article] [PubMed] [Google Scholar]
- 13.Arawaka S, Saito Y, Murayama S, Mori H: Lewy body in neurodegeneration with brain iron accumulation type 1 is immunoreactive for alpha-synuclein. Neurology 1998, 51:887-889 [DOI] [PubMed] [Google Scholar]
- 14.Eidelberg D, Sotrel A, Joachim C, Selkoe D, Forman A, Pendlebury WW, Perl DP: Adult onset Hallervorden-Spatz disease with neurofibrillary pathology. Brain 1987, 110:993-1013 [DOI] [PubMed] [Google Scholar]
- 15.Maladrini A, Bonucelli U, Parrotta E, Ceravolo R, Berti G, Guazzi GC: Myopathic involvement in two cases of Hallervorden-Spatz disease. Brain Dev 1995, 17:286-290 [DOI] [PubMed] [Google Scholar]
- 16.Williams DJ, Ironside JW: Liver and pituitary abnormalities in Hallervorden-Spatz disease. J Neurol Neursurg Psychiatry 1989, 52:1410-1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zupanc ML, Chun RW, Gilbert-Barness EF: Osmiophilic deposits in cytosomes in Hallervorden-Spatz syndrome. Pediatr Neurol 1990, 6:349-352 [DOI] [PubMed] [Google Scholar]
- 18.Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM-Y: Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple systems atrophy brains contain insoluble α-synuclein. Ann Neurol 1998, 44:415-422 [DOI] [PubMed] [Google Scholar]
- 19.Wakabayashi K, Yoshimoto M, Fukushima T, Koide R, Horikawa Y, Morita T, Takahashi H: Widespread occurrence of α-synucelin/NACP-immunoreactive neuronal inclusions in juvenile and adult-onset Hallervorden-Spatz disease with Lewy bodies. Neuropathol Appl Neurobiol 1999, 25:363-368 [DOI] [PubMed] [Google Scholar]
- 20.Ohgami T, Kitamoto T, Taeishi J: Alzheimer’s amyloid precursor protein accumulates within axonal swelling in human brain lesions. Neurosci Lett 1992, 136:75-78 [DOI] [PubMed] [Google Scholar]
- 21.Newell KL, Boyer P, Gomez-Tortosa E, Hobbs W, Hedley-Whyte ET, Vonsattel JP, Hyman BT: Alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. J Neuropathol Exp Neurol 1999, 58:1263-1268 [DOI] [PubMed] [Google Scholar]
- 22.Wakabayashi K, Fukushima T, Koide R, Horikawa Y, Hasegawa M, Watanabe Y, Noda T, Eguchi I, Morita T, Yoshimoto M, Iwatsubo T, Takahasi H: Juvenile-onset generalized neuroaxonal dystrophy (Hallervorden-Spatz disease) with diffuse neurofibrillary and Lewy body pathology. Acta Neuropathol 2000, 99:331-336 [DOI] [PubMed] [Google Scholar]
- 23.Wisniewski K, Jervis GA, Moretz RC, Wisniewski HM: Alzheimer neurofibrillary tangles in diseases other than senile and presenile dementia. Ann Neurol 1979, 5:288-294 [DOI] [PubMed] [Google Scholar]
- 24.Galvin JE, Lee VMY, Baba M, Mann DMA, Dickson DW, Yamaguchi H, Schmidt ML, Iwatsubo T, Trojanowski JQ: Monoclonal antibodies to purified cortical Lewy bodies recognize the mid-size neurofilament subunit. Ann Neurol 1997, 42:595-603 [DOI] [PubMed] [Google Scholar]
- 25.Galvin JE, Uryu K, Lee VMY, Trojanowski JQ: Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β- and γ-synuclein. Proc Natl Acad Sci USA 1999, 96:13450-13455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt ML, Murray J, Lee VMY, Hill WD, Werkin A, Trojanowski JQ: Epitope map of neurofilament protein domains in cortical and peripheral nervous system Lewy bodies. Am J Pathol 1991, 139:53-65 [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw G, Chau V: Ubiquitin and microtubule-associated protein tau immunoreactivity each define distinct structures with differing distributions and solubility properties in Alzheimer brain. Proc Natl Acad Sci USA 1988, 85:2854-2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jakes R, Crowther A, Lee VMY, Trojanowski JQ, Iwatsubo T, Goedert M: Epitope mapping of LB509, a monoclonal antibody directed against human α-synuclein. Neurosci Lett 1999, 269:13-16 [DOI] [PubMed] [Google Scholar]
- 29.Giasson BI, Ross J, Goedert M, Duda JE, Leight S, Trojanowski JQ, Lee VMY: A panel of epitope-specific antibodies detect protein domains distributed throughout human α-synuclein in Lewy bodies of Parkinson’s disease. J Neurosci Res 2000, 59:528-533 [DOI] [PubMed] [Google Scholar]
- 30.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VMY: Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 82:1914-1917 [DOI] [PubMed] [Google Scholar]
- 31.Arai H, Lee VMY, Messinger ML, Greenberg BD, Lowery DE, Trojanowski JQ: Expression patterns of β-amyloid precursor protein (β-APP) in neural and non-neural human tissues from Alzheimer’s disease and control subjects. Ann Neurol 1991, 30:686-693 [DOI] [PubMed] [Google Scholar]
- 32.Schmidt ML, DiDario AG, Otvos L, Hoshi N, Kant A, Lee VMY, Trojanowski JQ: Plaque associated neuronal proteins: a recurrent motif in neuritic amyloid deposits throughout diverse cortical areas of the Alzheimer’s disease brain. Exp Neurol 1994, 130:311-322 [DOI] [PubMed] [Google Scholar]
- 33.Lippa CF, Fujiwara H, Mann DMA, Giasson B, Baba M, Schmidt ML, Nee LE, O’Connell B, Pollen DA, George-Hyslop P, Ghetti B, Nochlin D, Bird TD, Cairns NJ, Lee VMY, Iwatsubo T, Trojanowski JQ: Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 1998, 153:1365-1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL: Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276:2045-2047 [DOI] [PubMed] [Google Scholar]
- 35.Kruger R, Kuhn W, Muller T, Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O: Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998, 18:106-108 [DOI] [PubMed] [Google Scholar]
- 36.Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VMY, Trojanowski JQ, Iwatsubo T: Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol 1998, 152:879-884 [PMC free article] [PubMed] [Google Scholar]
- 37.Giasson BI, Galvin JE, Lee VMY, Trojanowski JQ: The cellular and molecular pathology of Parkinson’s disease. Clark CM Trojanowski JQ eds. Neurodegenerative Dementias: Clinical Features and Pathological Mechanisms. 2000, :pp 219-228 McGraw-Hill, New York [Google Scholar]
- 38.Lippa CF, Schmidt ML, Lee VMY, Trojanowski JQ: Antibodies to α-synuclein detect Lewy bodies in many Down’s syndrome brains with Alzheimer’s disease. Ann Neurol 1999, 45:353-357 [DOI] [PubMed] [Google Scholar]
- 39.Clayton DF, George JM: Synucleins in synaptic plasticity and neurodegenerative disorders. J Neurosci Res 1999, 58:120-129 [PubMed] [Google Scholar]
- 40.Hardy J, Gwinn-Hardy K: Genetic classification of primary neurodegenerative disease. Science 1999, 282:1075-1079 [DOI] [PubMed] [Google Scholar]
- 41.Trojanowski JQ, Goedert M, Iwatsubo T, Lee VMY: Fatal attractions: abnormal protein aggregation and neuron death in Parkinson’s disease and Lewy body dementia. Cell Death Diff 1998, 5:832-837 [DOI] [PubMed] [Google Scholar]
- 42.Dickson DW, Liu WK, Hardy J, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Sima A, D’Amato C, Albin R, Gilman S, Yen SH: Widespread alterations of α-synuclein in multiple system atrophy. Am J Pathol 1999, 155:1241-1251 [DOI] [PMC free article] [PubMed] [Google Scholar]