

Abstract
Several animal models manifesting lesions resembling neointimal hyperplasia of human vein grafts have been developed, but no spontaneous atheromatous lesions in their vein grafts have been observed. We developed and here characterize a new animal model of vein graft atheroma, a maturated atherosclerotic plaque, in apoE-deficient mice. The lesion displayed classical complex morphological features and heterogeneous cellular compositions and consisted of a fibrous cap, infiltrated mononuclear cells, foam cells, cholesterol crystal structure, necrotic core with calcification, and neovasculature. Cell component analysis revealed smooth muscle cells (SMCs) localized in the cap region, macrophages which made up a large portion of the lesions, and CD4+ T cells scattered under the cap. Importantly, apoptotic/necrotic cells determined by TUNEL assay in vein grafts into apoE−/− mice were significantly higher than wild-type mice, although a similar number of proliferating cell nuclear antigen-positive cells in both types of lesions was found. Interestingly, vascular SMCs cultivated from aortas of apoE-deficient mice showed a high rate of spontaneous apoptosis/necrosis and a higher rate of cell death stimulated by a nitric oxide donor, sodium nitroprusside, H2O2, and oxidized low density lipoprotein (LDL), although no difference in proliferation of both SMCs incubated with platelet-derived growth factor, angiotensin II, LDL, and oxidized LDL was seen. Thus, the pathogenic mechanisms of vein graft atheroma involve increased intimal cell death initiated by biomechanical stress and amplified by hypercholesterolemia, which leads to continuous recruitment of blood mononuclear cells to constitute atheromatous lesions. This mouse model resembling human vein graft disease has many advantages over other animal models.
Autogenous vein grafts are commonly used to replace arteries rendered useless by severe atherosclerosis. 1 Although the treatment is highly successful in relieving symptoms and prolonging survival, nearly all veins implanted into the arterial circulation develop neointimal hyperplasia within 4 to 6 weeks, proceeding to atheroma and consequent serious clinical problems. The pathogenic mechanisms of atheroma remain elusive and few effective techniques are available to prevent this event. 1-3
Hypercholesterolemia has been shown to be a significant risk factor for the development of vein graft atheroma. 4 Evidence indicates that rates of obstructive atheroma in grafted veins were highly correlated to preoperative serum cholesterol levels, 5 and that the lesion development was predicted by higher levels of plasma very low density lipoprotein (VLDL) and low density lipoprotein (LDL). 6 Accordingly, patients with familial hypercholesterolemia exhibit a high incidence of late graft occlusion after bypass surgery. 7 However, the molecular mechanism of vein graft atherosclerosis by which hypercholesterolemia initiates, promotes, or perpetuates the lesion remains largely unknown.
Several animal models manifesting lesions resembling neointimal hyperplasia of human vein grafts have been developed 8-12 and have helped address specific interventional issues, but no spontaneous atheromatous lesions in their vein grafts have been observed. The apolipoprotein E-deficient (apoE−/−) mouse generated by gene targeting 13 provides an appropriate animal model for such a study. In apoE−/− mice, blood cholesterol is markedly elevated because of increased levels of VLDL and LDL. 13
In our previous study, we established a new model for the study of neointima formation of venous bypass grafts in mice, and demonstrated the presence of abundant MAC-1-positive monocytes/macrophages and smooth muscle cells (SMCs) in the early stages of lesions in vein grafts. 14 In the present study, we developed and characterized an animal model of vein graft atheroma in apoE knockout mice, and demonstrated that hypercholesterolemia promotes the formation of typical atherosclerotic plaques, ie, atheroma in vein bypass grafts.
Materials and Methods
Mice and Vein Graft Procedure
All animal experiments were performed according to protocols approved by the institutional committee for use and care of laboratory animals. ApoE-deficient mice of the C57BL/6J strain 13 were purchased from The Jackson Laboratory (Bar Harbor, ME). Three genotypes of apoE−/−, apoE+/−, and apoE+/+ mice were identified using The Jackson Laboratory’s polymerase chain reaction (PCR) protocol (primers: oIMR180 5′-GCC TAG CCG AGG GAG AGC CG-3′, oIMR181 5′-TGT GAC TTG GGA GCT CTG CAG C-3′ and oIMR182 5′-GCC GCC CCG ACT GCA TCT-3′). The mice were maintained on a 12 hours light/12 hours dark cycle at 22°C, receiving food and water ad libitum.
The procedure used for vein grafts was similar to that described previously. 14 Briefly, 3-month-old mice were anesthetized with pentobarbital sodium (50 mg/kg body weight, i.p.). The vena cava vein was harvested and washed with saline solution containing 100 U/ml of heparin. The right common carotid artery was mobilized free from the bifurcation at the distal end toward the proximal, cut in the middle, and a cuff was placed at the end. The cuff was made of an autoclavable nylon tubing 0.63 mm in diameter outside and 0.5 mm inside (cat. no. 800/200/100/200, Portex Ltd., Hythe-Kent, UK). The artery was turned inside out over the cuff and ligated. The vein segment was grafted between the two ends of the carotid artery by sleeving the ends of the vein over the artery-cuff and ligating them together with the 8–0 suture. Vigorous pulsations in the grafted vessel confirmed successful engraftment.
Tissue Preparation
Vein grafts were harvested at 2, 4, and 8 weeks postoperatively (4 to 8 mice at each time point/group) by cutting the implanted segments from the native vessels at the cuff end. For histological analysis, perfusion with 4% phosphate-buffered formaldehyde was performed, as described previously. 14 For frozen section preparation, vein grafts were harvested without perfusion and immediately frozen in liquid nitrogen.
Histology and Lesion Quantification
The grafts were dehydrated in graded ethanol baths, cleared in xylol, embedded in paraffin, and sectioned. The thickness of the vessel wall was determined by measuring 4 regions of a section along a cross, and recorded in micrometers (means ± SEM). The procedure for lesional area measurement is similar to that described elsewhere. 15 Briefly, the lesion was defined as the region between the lumen and the adventitia. Using a transmission scanning microscope (LSM 510, Zeiss, Jena, Germany) vessels were scanned, saved, and then overlaid by different linings to trace the lumen and adventitia. The lesion area was determined by subtracting the area of the lumen from the area enclosed by the line inside of adventitia. Six to eight cross-sections were obtained by selecting the first of every three sections from each graft. Areas were measured and recorded in square micrometers.
Immunohistochemical Staining
The procedure used in the present study was similar to that described previously. 14,15 Briefly, serial 5-μm-thick frozen sections were overlaid with rat monoclonal antibodies against mouse MAC-1 leukocytes (CD11b/18), CD4, or CD8 (PharMingen, San Diego, CA). Sections were visualized with alkaline phosphatase-anti-alkaline phosphatase complex (Dakopatts, Copenhagen, Denmark) and developed using a substrate solution containing 9.8 ml Tris buffer (0.1 mol/L, pH 8.2), 0.2 ml dimethylformamide, 8 mg naphthol AS-MX phosphate, 3 mg levamisole, and 10 mg Fast Red TR salt (Sigma). For SMC staining, a mouse monoclonal antibody against α-actin (Sigma) labeled with phosphatase was used. Semiquantitive evaluation was performed at 10 × 40 magnification. Positive stained cells in the intima and media were counted on two regions of each section and expressed as the range of the cell number or the percentage of total nuclei per 100 micrometers of the vessel wall.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) Assay
Accumulated internucleosomal DNA fragments (apoptosis) were detected using an in situ apoptosis detection kit (Boehringer Mannheim, Mannheim, Germany) as described previously. 16 Sections were developed with Fast Red substrate and counterstained with hematoxylin. Percentages of positive-stained cells were determined by counting the numbers of labeled and total cells using a light microscope. Positive cells of two regions of each section of 4- and 8-week grafts were counted. Morphological features of apoptosis were assessed on light microscope.
Immunofluorescence Double Staining
For double staining, sections were incubated with the rat monoclonal antibody against MAC-1 and visualized with swine anti-rat Ig conjugated with fluorescein isothiocyanate (FITC, Dakopatts). Sections were rinsed and stained with rabbit anti-proliferating cell nuclear antigen (PCNA) antibody (Santa Cruz Biotechnology, Santa Cruz, CA) developed with swine anti-rabbit Ig-TRITC (Dakopatts). For SMC and PCNA staining, a mouse monoclonal antibody against α-actin (Sigma) labeled with FITC was used. For visualization of nuclei, sections were counterstained with the DNA stain Hoechst 33258.
LDL Isolation and Oxidation
EDTA plasma was pooled from normolipemic, fasting (12–14 hours) humans, aged 20 to 30 years. Lipoproteins were prepared by differential centrifugation using solid KBr to adjust the density, as described previously. 17,18 LDL were obtained in fractions between 1.020 and 1.050 g/ml. Concentrations of LDL were determined gravimetrically by aliquot weight after drying, and quantities of lipoproteins were expressed as total weights. 17,18 LDL oxidation was performed by incubation of LDL (1 mg/ml phosphate-buffered saline) with 10 μmol/L CuCl2 at 37°C for 18 hours. 19 The extent of oxidation was assessed by measurement of thiobarbituric acid reactive substances (9.8 ± 1.3 nmol/mg). 20
SMC Culture
Vascular SMCs from apoE−/− and apoE+/+ mice were cultivated from their aortas, as described elsewhere. 21,22 Cells were incubated at 37°C for 7 to 10 days and passaged by treatment with 0.05% trypsin/0.02% EDTA solution. The purity of SMCs was routinely confirmed by immunostaining with antibodies against α-actin. Experiments were conducted on SMCs that had just achieved confluence.
Cell Proliferation and Viability Assays
For proliferation assays, SMCs cultured in 96-well plates in medium containing 10% fetal calf serum at 37°C for 24 hours were serum-starved for 2 days. Platelet-derived growth factor-AB, angiotensin II, LDL, oxidized LDL, and fetal calf serum were added, respectively, and incubated at 37°C for 24 hours. For cell viability assay, SMCs were plated at a density of 2 × 10 3 cells/well (96-well plate) in the medium containing 10% fetal calf serum and incubated at 37°C for 48 hours. Oxidized LDL, LDL, H2O2, and sodium nitroprusside (SNP) were added to the culture, respectively, and incubated at 37°C for 24 hours. A solution from a proliferation/apoptosis kit (Promega) was added 4 hours before measurement. The optical density at 490 nm was recorded with a photometer.
Annexin V/Propidium Iodide Double Staining and Fluorescence-Activated Cell Sorter (FACS) Analysis
Annexin staining was performed according to the manufacturer’s instructions (PharMingen) and as described previously. 16 The cellular fluorescence signal was recorded on the FL1 and FL2 channel of a FACS scan flow cytometer (Program Cell Quest, Becton Dickinson, Mountain View, CA) and expressed on a logarithmic scale. After appropriate markings for negative and positive populations, the percentage of annexin V+/PI− or V+/PI + cells was determined and compared with unstressed controls.
Statistical Analysis
Statistical analyses were performed using the Mann-Whitney U test and analysis of variance, respectively. Results are given as mean ± SEM or ± SD. A P value < 0.05 was considered significant.
Results
Vein Graft Atheroma in ApoE−/− Mice
The venous wall of apoE−/− and wild-type mice is composed of intima, a monolayer of endothelium, media, 1- or 2-layered SMCs, and adventitia, a small amount of connective tissue. Vein grafts of wild-type mice at 8 weeks (Figure 1a) ▶ showed neointimal hyperplasia, ie, thickening of the vessel wall up to 10 or 20 layers of cells. Interestingly, intimal lesions of vein grafts in apoE−/− mice showed a marked increase at 8 weeks (Figure 1b) ▶ . The lumen of vein grafts reduced up to 70% compared to that of wild-types.
Figure 1.

Hematoxylin-eosin (HE)-stained sections of mouse vein grafts. Under anesthesia, vena cava veins of apoE+/+ (a) or apoE−/− (b) mice were removed and isografted into carotid arteries of apoE +/+ or apoE−/−, respectively. Animals were sacrificed 8 weeks after surgery, and the grafted tissue fragments were fixed in 4% phosphate-buffered formaldehyde, pH 7.2, embedded in paraffin, sectioned, and stained with HE. Upper arrow indicates necrosis, and lower arrow indicates loss of tissues possibly due to necrosis. Original magnification, ×40.
Figure 2 ▶ summarizes statistical data of lesion thickness and areas measured with a laser microscope in transmission scan mode. A significant difference between groups of apoE−/− and wild-type mice was found at 4 and 8 weeks (P < 0.05 and P < 0.0001, respectively). Likewise, two- to threefold increases in lesion areas of vein grafts in apoE−/− mice were observed (Figure 2 ▶ , low panel).
Figure 2.
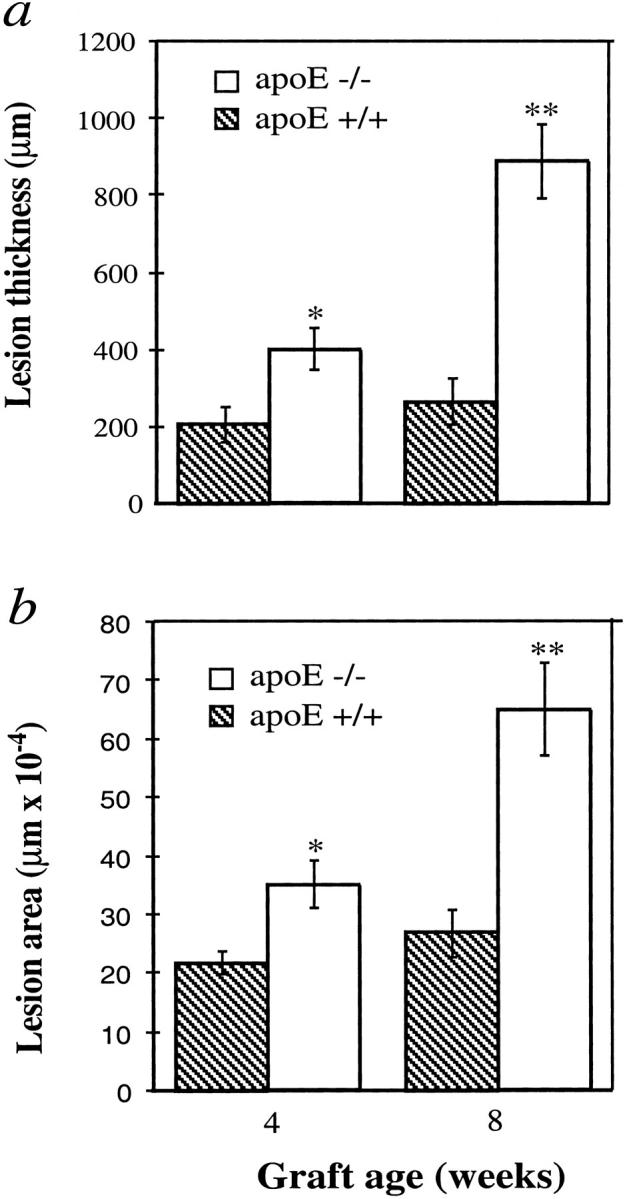
Increased lesion thickness and area of vein grafts in apoE−/− mice. The procedure for animal models and the preparation of HE-stained sections are the same as those described in the legend to Figure 1 ▶ . Thickness was measured microscopically. Four regions of each section along a cross were measured and 5 sections per animal selected. Lesional areas were determined with a laser microscope, as described in Materials and Methods. Data shown as graphs of neointima or lesion thickness (a) and area (b) (means ± SEM) obtained from 6 or 8 animals per group at each time point. Significant difference between apoE−/− and apoE+/+ groups were seen. *P < 0.05, **P < 0.0001.
Neointimal hyperplasia in vein grafts in wild-type mice developed 4 or 8 weeks after engraftment and was characterized by mononuclear cell infiltration, SMC accumulation, and matrix protein deposition (Figure 3a) ▶ , but no fully maturated atheroma could be found even at 16 weeks. 14 In apoE−/− mice, typical atheroma in 8-week vein grafts displayed the classical complex morphological features and heterogeneous cellular compositions. Atheromatous lesions consisted of the fibrous cap (Figure 3, b and c) ▶ , infiltrated mononuclear cells (Figure 3b) ▶ , foam cells (Figure 3c) ▶ , cholesterol crystal structure (Figure 3d) ▶ , necrotic core with calcification (Figure 3e) ▶ , and neovasculature (Figure 3f) ▶ . In addition, the cell density in apoE-null vein graft atheroma was much lower than that of apoE+/+ neointimal lesions. These complicated lesions closely parallel those characteristic of human vein graft atherosclerosis.
Figure 3.
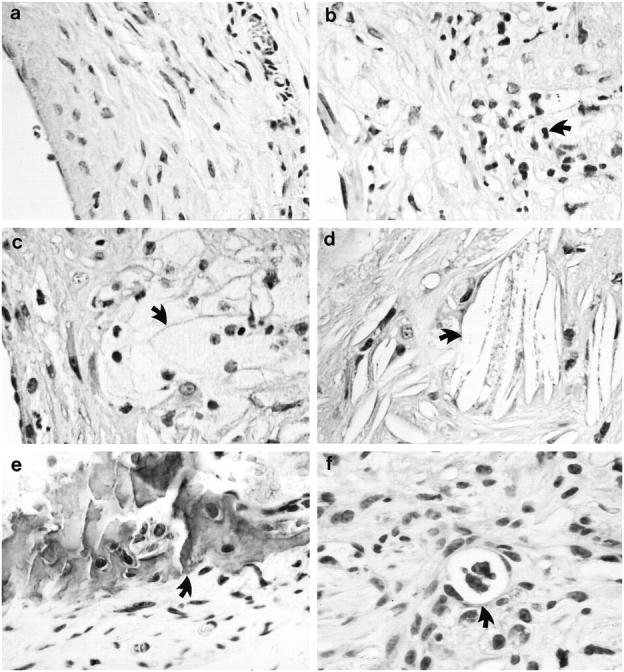
Morphological characteristics of vein graft atheroma. HE-stained sections of 8-week neointima (a) of vein grafts from apoE+/+ mice and vein graft atheroma (b-f) from apoE−/− mice. Arrows indicate typical examples of infiltrated mononuclear cell (b), foam cell (c), cholesterol crystal (d), calcified necrotic core (e), and neovasculature (f).
Cell Compositions in Vein Graft Atheroma
We have provided evidence that lesions of vein graft-induced neointima from wild-type mice contain mainly macrophages and SMCs. 14 Figure 4 ▶ and Table 1 ▶ demonstrate the presence of abundant SMCs in venous bypass graft lesions 4 and 8 weeks after surgery. Initially, a monolayer of SMCs in normal vein walls stained strongly positive (data not shown), and no staining was seen in vein segments stained with normal rat serum as a negative control (Figure 4a) ▶ . Importantly, α-actin-positive SMCs distributed in the most area of neointima of 4- and 8-week vein grafts from wild-type mice (Table 1) ▶ , whereas a majority of these positive cells localized in the cap region of vein graft atheroma in apoE−/− mice (Figure 4b ▶ and Table 1 ▶ ). A significant difference in SMC numbers in 8-week graft lesions between apoE−/− and apoE+/+ was found (33 vs. 60%; Table 1 ▶ ). Abundant infiltration of MAC-1-positive cells was found in intima and/or adventitia of 4-week vein grafts of both types of mice (Table 1) ▶ . MAC-1-positive cells remained predominant in atheroma of 8-week grafts (Figure 4c) ▶ , whereas they were decreased in neointima of wild-type mice 8 weeks after the operation (Table 1) ▶ . Most foam cells displayed MAC-1 positivity, indicating a mononuclear cell origin. MAC-1-positive cells could be found around the necrotic core and the most area under the fibrous cap. Thus, mononuclear cells constituted a large portion of atheroma in vein grafts of apoE-deficient mice.
Figure 4.
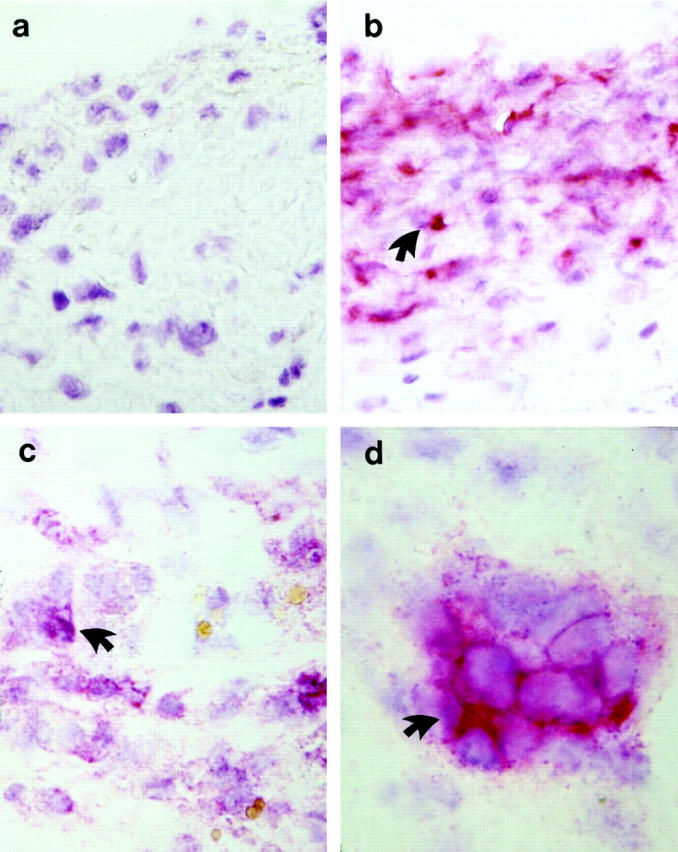
Immunohistochemical staining demonstrates the presence of α-actin-, MAC-1-, and CD4-positive cells in vein grafts. Sections derived from vein grafts at 8 weeks were labeled with normal rat serum (a), a mouse monoclonal antibody against α-actin conjugated with alkaline phosphatase (b), rat monoclonal antibodies against MAC-1-positive leukocytes (CD11b/18; c) or against CD+ T-cells (d) and developed with alkaline phosphatase-anti-alkaline phosphatase techniques. A counterstaining (blue) with hematoxylin was performed. Arrows indicate examples of typical positive cells (red). Original magnifications, ×400 (a-c) and ×1000 (d).
Table 1.
Cell Compositions in Neointima or Atheroma
| Mouse group | MAC-1+ cells | α-actin+ cells | CD4+ cells |
|---|---|---|---|
| 4 weeks | |||
| apoE+/+ | 47 ± 23% | 11 ± 7% | 0–5 |
| apoE−/− | 58 ± 31% | 14 ± 10% | 30–50 |
| 8 weeks | |||
| apoE+/+ | 23 ± 15% | 60 ± 19% | 0–3 |
| apoE−/− | 63 ± 21%* | 33 ± 17%* | 10–30 |
The positive stained cells and total number of nuclei were counted. In each section, two fields (10 × 40) were evaluated. Data from CD4+ cells represent ranges of the number of positive cells, since total numbers per field were less than 5 in neointima of apoE+/+ mice. The values are ranges of means ± SD from 2 to 4 sections/animal, n = 4.
* Significant difference between apoE−/− and +/+ mice, P < 0.05.
T lymphocytes are prominent components of human atherosclerotic lesions, 23,24 but their presence in animal vein graft models has not been confirmed. T lymphocytes, as determined by CD4 and CD8 immunoreactivities, were present in vein grafts from both apoE−/− and apoE+/+ mice. The density of CD4+ T cells was greatest in 4- and 8-week graft lesions of apoE−/− mice, but much lower in neointima from control mice (Table 1 ▶ , Figure 4d ▶ ). Interestingly, the distribution of CD4+ lymphocytes was patchy, with small foci of cells generally located beneath the fibrous cap of atheroma (Figure 4d) ▶ . Few CD8+ T cells were found in neointima or atheromatous lesions.
Increased Cell Death in Vein Graft Atheroma
Because of rapid development of atheromatous lesions, it would be interesting to determine cell turnover in vein grafts. PCNA is widely used as a cell replicating marker, and was also used for labeling of vein grafts from both groups. Data summarized in Figure 5 ▶ show a similarity in the percentage of PCNA+ cells in neointima and atheromatous lesions of apoE+/+ and apoE−/− mice. Double-labeling studies revealed that most PCNA+ cells were α-actin-positive (82 ± 15%), whereas MAC-1 and PCNA double-positive cells were rare (<5%). These results indicate that SMC replications in situ in the intima of vein grafts occur, and that there is no significant difference between apoE−/− and apoE+/+ mice.
Figure 5.

Immunofluorescent labeling for PCNA-positive cells in vein grafts. Sections derived from vein grafts of apoE+/+ and apoE−/− mice at 4 and 8 weeks were labeled with a rabbit anti-PCNA antibody and visualized by swine anti-rabbit Ig conjugated with FITC. Cell nuclei were stained with Hoechst 33258 for 1 minute. Positive cells and total nuclei were counted in a 400× field. Two fields of each section were enumerated, and 3 sections per vein graft were selected. Data are means ± SD of 4 animals per time point in each group.
Next, we performed the experiments with TUNEL assays to determine cell death in vein grafts. Figure 6 ▶ shows typical examples of TUNEL-positive cells in both neointima and atherosclerotic lesions of vein grafts. Although TUNEL-positive cells were found throughout the lesions, they were more abundant around the necrotic core (Figure 6e) ▶ . Quantitative analysis revealed that apoptotic/necrotic cells in atheroma of apoE−/− mice were significantly higher than those in neointima from the wild-types (Figure 7 ▶ ; 11 vs. 5.6%; P < 0.05). To study the mechanism of higher rate of cell death in atheroma, vein isografts between apoE−/− and apoE+/+ mice were performed. Figure 8 ▶ shows data indicating that hyperlipidemia is largely responsible for increased numbers of cell death as well as atheroma formation. However, apoE−/− veins grafted into apoE+/+ mice showed that numbers of apoptotic/necrotic cells were not significantly different from veins grafted among apoE+/+ mice (Figure 8b) ▶ .
Figure 6.
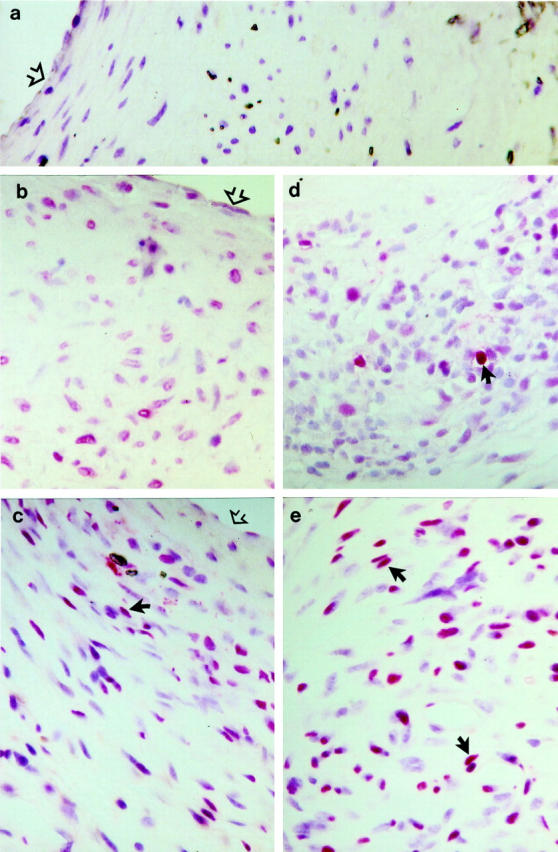
TUNEL-stained sections of vein grafts of apoE+/+ (b and d) and apoE−/− (a, c, and e) mice. Vein grafts were harvested 4 (b and c) and 8 (a, d, and e) weeks after surgery, fixed in 4% phosphate-buffered formaldehyde, pH 7.2, embedded in paraffin, sectioned, and stained with TUNEL as described in Materials and Methods. a: Negative control. Open arrows indicate the surface of the intima; filled arrows indicate typical examples of TUNEL-positive cells in lesions.
Figure 7.

Statistical data of TUNEL-positive cells in vein grafts. TUNEL-stained sections were prepared as described in the legend to Figure 6 ▶ . TUNEL-positive and total nuclei in neointima or atheroma of vein grafts were enumerated under the microscope. Data are means ± SD of 6 to 8 animals per time point each group. *Significant difference between apoE−/− and apoE+/+ mice, P < 0.05.
Figure 8.
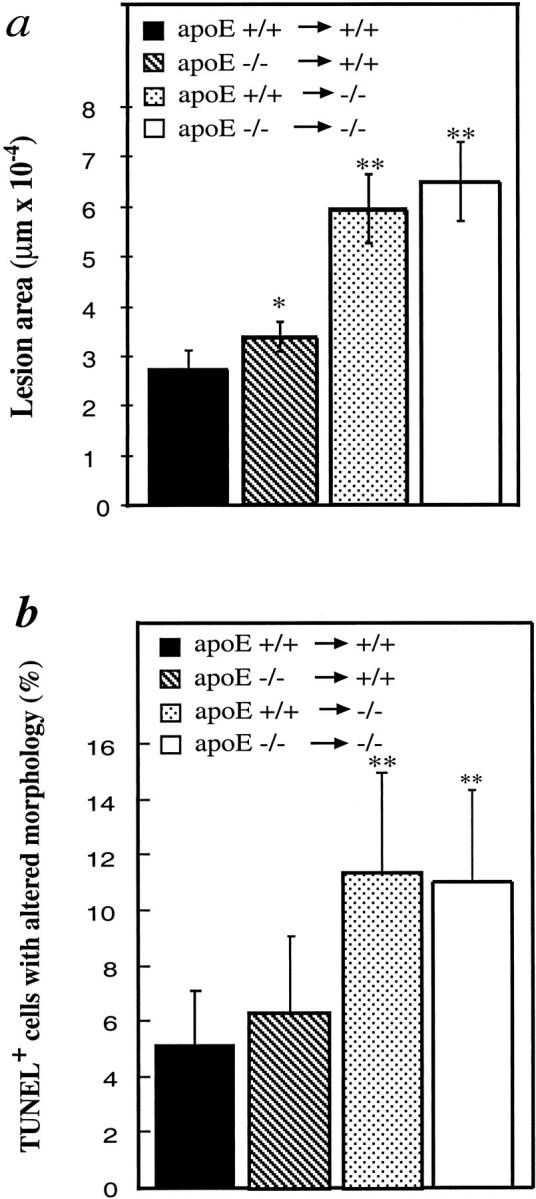
Lesions and TUNEL-positive cells in vein isografts between apoE−/− and apoE+/+ mice. Vein grafts were performed as described in the legend to Figure 1 ▶ and the Materials and Methods. TUNEL labeling was performed using the procedure same as for Figure 6 ▶ . Data are means ± SEM (a) or SD (b) of 6 animals per group. *, Significant difference from apoE+/+ veins grafted to apoE+/+ mice; **, significant difference from apoE+/+ or apoE−/− veins grafted to apoE+/+ mice. P < 0.05.
ApoE−/− SMCs Sensitive for Death in Vitro
Because of increased cell death in apoE−/− grafts, we compared the proliferative ability and viability of cultivated arterial SMCs from apoE−/− and apoE+/+ mice. In response to serum, platelet-derived growth factor-AB, angiotensin II, LDL, and oxidized LDL, SMCs from both types of mice replicated markedly, but no significant differences in cell numbers were observed (Figure 9a) ▶ . Interestingly, apoE-deficient SMCs revealed a significant decrease in cell viability when stimulated by a NO donor sodium nitroprusside (SNP), free radical H2O2, and oxidized LDL (Figure 9b) ▶ . Low concentrations (up to 50 μg/ml) of oxidized LDL-induced SMC replication were observed, whereas higher concentrations (>100 μg/ml) resulted in cell death (Figure 9, a and b) ▶ , similar to the findings obtained in SMCs from other species. To further determine whether oxidized LDL stimulates SMC apoptosis or necrosis, FACS analysis of double-stained SMCs was performed. The data shown in Figure 8, c ▶ -f, indicate that the main population of dying SMCs underwent apoptosis. ApoE-deficient SMCs had a higher rate of both spontaneous and oxidized LDL-stimulated apoptosis/death (Figure 9, c ▶ -f).
Figure 9.
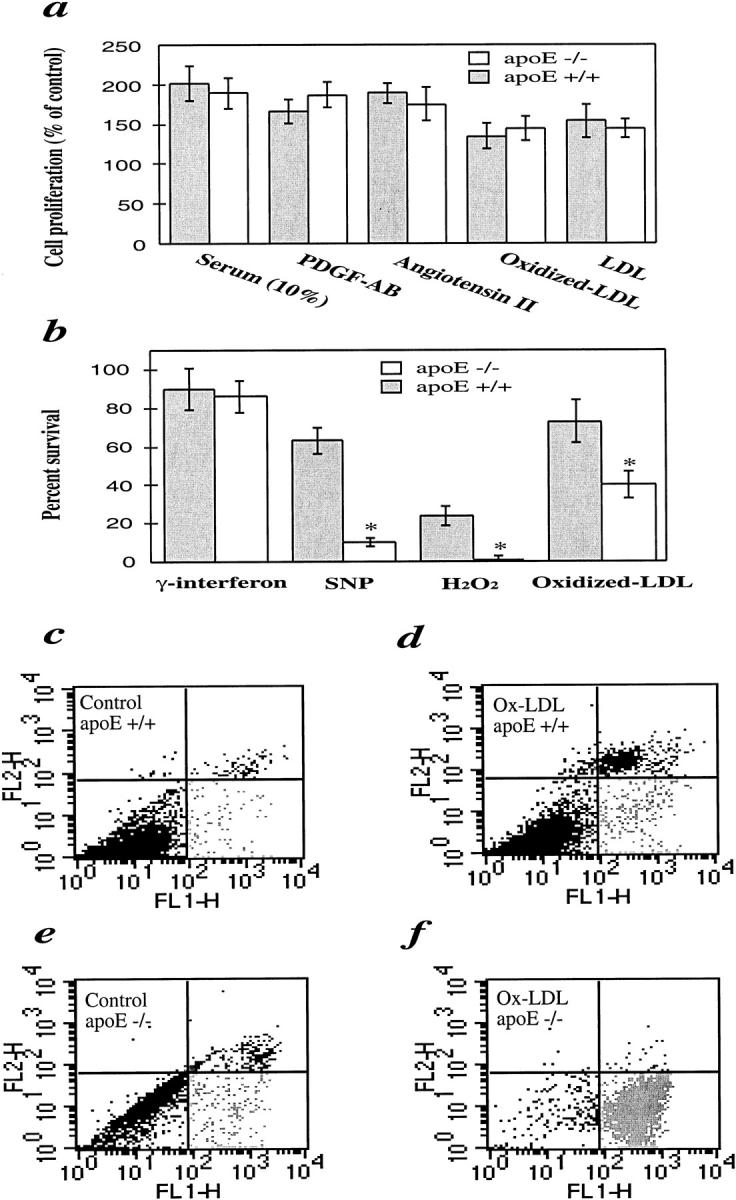
Comparison of SMC proliferation (a), viability (b), and apoptosis/necrosis (c-f) between apoE−/− and apoE+/+ mice. a: Serum-starved SMCs were treated with platelet-derived growth factor-AB (50 ng/ml), angiotensin II (50 nmol/L), fetal calf serum (10%), LDL (100 μg/ml), and oxidized LDL (10 μg/ml), respectively, at 37°C for 24 hours. b: SMCs were incubated at 37°C for 24 hours with oxidized LDL (200 μg/ml), H2O2 (50 μmol/L), sodium nitroprusside (SNP; 0.5 mmol/L), and γ-interferon (100 ng/ml). Solution of proliferation/apoptosis kit was added 4 hours before measurement. The optical density at 490 nm was recorded with a photometry. Data are means ± SD of three experiments. *Significant difference between apoE−/− and apoE+/+ SMCs. c–f: FACS analysis of annexin V/propidium iodide double-stained SMCs. SMCs were treated with oxidized LDL (d and f) for 24 hours and labeled with annexin V and propidium iodide. Cellular fluorescence signal was recorded on FL1 and FL2 channels of a FACS scan flow cytometer and expressed on a logarithmic scale. Note the higher number of spontaneous cell death (c and e) and oxidized LDL-induced apoptosis (d and f) in apoE−/− SMCs.
Discussion
We have recently established a new mouse model for the study of neointima hyperplasia of venous bypass grafts. 14 In the present study, we developed and characterized an animal model of vein graft atheroma in apoE-deficient mice. The morphological features of this mouse vascular graft model resemble those of human venous bypass graft atheroma. 1-3,25 First, the human lesions have an inflammatory nature characterized by mononuclear cell infiltration in the intima of vein bypass grafts, and the lesions seen in our mouse models contain abundant MAC-1-positive monocytes/macrophages 26 (Figure 4 ▶ and Table 1 ▶ ). Second, both the human and mouse atheromas are characterized by concentric intimal proliferation, with a dominance of SMCs in the cap region 27 (Figure 4 ▶ and Table 1 ▶ ). Third, lipid deposition, including foam cells and cholesterol crystal, can be found in atherosclerotic lesions of both species. Finally, apoE-knockout mice develop complicated atheromatous lesions, including necrotic core, calcification, and lumen occlusion that also occur in vein bypass grafts in humans. Thus, our model could be useful in the study of human venous bypass graft atherosclerosis. This model offers the potential for significant progress in the near future in understanding the pathogenesis and treatment of the vein graft disease.
The rabbit is the first and most frequently used animal at all points of atherosclerosis research, including vein bypass grafts. 28,29 Atheromatous lesions analogous to those in humans have been induced in vein grafts of rabbits by administration of a cholesterol-enriched diet. 30 However, our animal model of vein graft atheroma in apoE-deficient mice has several advantages. First, the number of animals used for the graft experiments can be reduced, since vein graft lesions show very low viability among apoE−/− inbred mice. Second, there is a large disparity in blood cholesterol levels of rabbits fed with a cholesterol-enriched diet, whereas blood lipid concentrations are similar in apoE-deficient mice of the same age. Third, when vein isografts between apoE−/− and other knockout mice are performed, the role of the specific gene or protein can be clarified. For example, if vein segments from ICAM-1-deficient C57BL/6J mice are grafted to carotid arteries of apoE−/− C57BL/6J mice, the role of ICAM-1 in vein grafts can be identified. 21
What are the initial factors stimulating the development of vein graft lesions? We hypothesize that biomechanical stress is an initiator for altered gene expression in the early stage of vein grafts, followed by vascular cell apoptosis/death, inflammation, SMC proliferation, and lipid deposition. 16,21,31,32 Support for this hypothesis includes the fact that neointima hyperplasia in vein segments grafted to veins does not occur in mice 14 and that atherosclerotic lesions in arteries grafted to arteries are very rare in apoE-null mice 8 weeks after surgery (data not shown). In grafted veins, mechanical force on the vessel segment suddenly increases more than 10-fold (arterial versus venous blood pressure), which provides a strong stimulus to vascular endothelial and SMCs. 32 We previously demonstrated that acutely elevated blood pressure and mechanical stress activate growth factor receptor-MAPK signal pathways, 33-36 which are critical in mediating mechanical stress-induced cell apoptosis and proliferation. Hyperlipidemia may be an amplifying factor that exerts its effects on vein graft atherosclerosis in the presence of elevated biomechanical stress.
One of the most important observations made in the current study is that cell death in vein graft atheroma of apoE-deficient mice was markedly increased and that increased cell death in apoE+/+ veins grafted into apoE−/− mice could also be observed. SMCs from apoE−/− mice had a higher rate of spontaneous apoptosis/death and were sensitive to a variety of stimuli leading to cell death. A possible explanation for this phenomenon is that high blood cholesterol levels may lead to SMC differentiation to a phenotype sensitive to stress stimuli. Such alterations in SMCs can be maintained for a period of time, because the rate of cell death in apoE−/− veins grafted into apoE+/+ mice and cultured apoE−/− SMCs is still higher (Figures 8 and 9) ▶ ▶ . Concomitantly, Bennett et al 37,38 provided evidence of increased sensitivity of human vascular SMCs from atherosclerotic plaques. These findings indicate that sustained exposure of SMCs to a hypercholesterolemic environment alters the cell phenotype and promotes cell differentiation. For example, they found elevated p53 protein levels in SMCs from atherosclerotic lesions, which may be responsible for increased sensitivity to apoptosis. 37,38 Importantly, increased cell death coincides with rapid development of atheroma in apoE-null mice. Although apoptosis is believed to evoke inflammation responses rarely in other tissues, 39,40 apoptotic cells release the S19 ribosomal protein dimer, 41 a chemokine for macrophages/monocytes, by molecular mimicry to complement C5a. 42 Necrotic cells in atheroma may release oxidized lipids, resulting in continuous recruitment of blood monocytes, ie, inflammatory responses. We found that the number of monocytes/macrophages in 8-week atheroma is significantly higher than in neointima (Table 1) ▶ , suggesting that cell recruitment from blood is a key event in maintaining the enlargement of atheromatous lesions.
In summary, we have developed and characterized a new animal model of vein graft atheroma in apoE-deficient mice. Atheromatous lesions bear many similarities to vein graft disease in humans. The mechanism of the pathogenesis of vein graft atheroma involves increased cell death and continuous recruitment of blood cell, by which biomechanical stress may serve as an initiator, and hypercholesterolemia synergistically enhances or amplifies the progress of lesions. This mouse model has several advantages over other animal models and, therefore, is highly recommended for use in the study of the pathogenesis and therapeutic interventions in vein graft disease.
Acknowledgments
We are grateful to Dr. G. Wick for his continuous support. We thank A. Strübl and A. Jenewein for excellent technical assistance.
Footnotes
Address reprint requests to Dr. Qingbo Xu, Institute for Biomedical Aging Research, Austrian Academy of Sciences, Rennweg 10, A-6020 Innsbruck, Austria. E-mail: Qingbo.Xu@oeaw.ac.at.
Supported by grants P12847-MED from the Austrian Science Fund, and P7917 from the Jubiläumsfonds of the Austrian National Bank. Dr. Hu is a recipient of APART Award from the Austrian Academy of Sciences.
References
- 1.Ip JH, Fuster V, Badimon FL, Badimon J, Taubman MB, Chesebro JH: Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol 1990, 15:1667-1687 [DOI] [PubMed] [Google Scholar]
- 2.Davies MG, Hagen P-O: Pathobiology of intimal hyperplasia. Br J Surg 1994, 81:1254-1269 [DOI] [PubMed] [Google Scholar]
- 3.Motwani JG, Topol EJ: Aortocoronary saphenous vein graft disease: Pathogenesis, predisposition, and prevention. Circulation 1998, 97:916-931 [DOI] [PubMed] [Google Scholar]
- 4.Hunninghake DB: Is aggressive cholesterol control justified? Review of the post-coronary artery bypass graft trial. Am J Cardiol 1998, 82:45T-48T [DOI] [PubMed] [Google Scholar]
- 5.Daida H, Yokoi H, Miyano H, Mokuno H, Satoh H, Kottke TE, Hosoda Y, Yamaguchi H: Relation of saphenous vein graft obstruction to serum cholesterol levels. J Am Coll Cardiol 1995, 25:193-197 [DOI] [PubMed] [Google Scholar]
- 6.Campeau L, Enjalbert M, Lesperance J, Bourassa MG, Kwiterovich P, Jr, Wacholder S, Sniderman A: The relation of risk factors to the development of atherosclerosis in saphenous-vein bypass grafts and the progression of disease in the native circulation: a study 10 years after aortocoronary bypass surgery. N Engl J Med 1984, 311:1329-1332 [DOI] [PubMed] [Google Scholar]
- 7.Kawasuji M, Sakakibara N, Takemura H, Matsumoto Y, Mabuchi H, Watanabe Y: Coronary artery bypass grafting in familial hypercholesterolemia. J Thorac Cardiovasc Surg 1995, 109:364-369 [DOI] [PubMed] [Google Scholar]
- 8.Angelini GD, Bryan AJ, Williams HMJ, Morgan R, Newby AC: Distention promotes platelet and leukocyte adhesion and reduces short-term patency in pig arteriovenous bypass grafts. J Thorac Cardiovasc Surg 1990, 99:433-439 [PubMed] [Google Scholar]
- 9.Davies MG, Klyachkin ML, Dalen H, Massey MF, Svendsen E, Hagen PO: The integrity of experimental vein graft endothelium: implications on the etiology of early vein graft failure. Eur J Vasc Surg 1993, 7:156-165 [DOI] [PubMed] [Google Scholar]
- 10.Boerboom LE, Olinger GN, Liu TZ, Rodriguez ER, Ferrans VJ, Kissebah AH: Histologic, morphometric, and biochemical evaluation of vein bypass grafts in a nonhuman primate model. I. Sequential changes within the first three months. J Thorac Cardiovasc Surg 1990, 99:97-106 [PubMed] [Google Scholar]
- 11.Landymore RW, Kinley CE, Cameron CA: Intimal hyperplasia in autogenous vein grafts used for arterial bypass: a canine model. Cardiovasc Res 1985, 19:589-592 [DOI] [PubMed] [Google Scholar]
- 12.Norman PE, House AK: The influence of nifedipine on microvascular vein graft intimal thickening. Aust N Z J Surg 1993, 63:294-298 [DOI] [PubMed] [Google Scholar]
- 13.Zhang SH, Reddick RL, Piedrahita JA, Maeda N: Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258:468-471 [DOI] [PubMed] [Google Scholar]
- 14.Zou Y, Dietrich H, Hu Y, Metzler B, Wick G, Xu Q: Mouse model of venous bypass graft arteriosclerosis. Am J Pathol 1998, 153:1301-1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietrich H, Hu Y, Zou Y, Dirnhofer S, Kleindienst R, Wick G, Xu Q: Mouse model of transplant arteriosclerosis: role of intercellular adhesion molecule-1. Arterioscler Thromb Vasc Biol 2000, 20:343-352 [DOI] [PubMed] [Google Scholar]
- 16.Mayr M, Li C, Zou Y, Huemer U, Hu Y, Xu Q: Biomechanical stress-induced apoptosis in vein grafts involves p38 mitogen-activated protein kinases. FASEB J 2000, 14:261-270 [DOI] [PubMed] [Google Scholar]
- 17.Jürgens G, Xu Q, Huber LA, Böck G, Howanietz H, Wick G, Traill KN: Promotion of lymphocyte growth by high density lipoproteins (HDL): physiological significance of the HDL binding site. J Biol Chem 1989, 264:8549-8556 [PubMed] [Google Scholar]
- 18.Xu Q, Jürgens G, Huber LA, Böck G, Wolf H, Wick G: Lipid utilization by human lymphocytes is correlated with high-density-lipoprotein binding site activity. Biochem J 1992, 285:105-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amberger A, Maczek C, Jürgens G, Michaelis D, Schett G, Xu Q, Wick G: Co-expression of ICAM-1, VCAM-1, ELAM-1 and Hsp60 in human arterial venous endothelial cells in response to cytokines and oxidized low density lipoproteins. Cell Stress Chaperones 1997, 2:94-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buege JA, August SD: Microsomal lipid peroxidation. Methods Enzymol 1978, 52:302-310 [DOI] [PubMed] [Google Scholar]
- 21.Zou Y, Hu Y, Mayr M, Dietrich H, Wick G, Xu Q: Reduced neointima hyperplasia of vein bypass grafts in intercellular adhesion molecule-1-deficient mice. Circ Res 2000, 86:434-440 [DOI] [PubMed] [Google Scholar]
- 22.Metzler B, Li C, Hu Y, Stum G, Ghaffari-Tabrizi N, Xu Q: LDL stimulates mitogen-activated protein kinase phosphatase-1 expression independent of LDL receptors in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1999, 19:1862-1871 [DOI] [PubMed] [Google Scholar]
- 23.Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK: Regional accumulations of T cells, macrophages and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis 1986, 6:131-138 [DOI] [PubMed] [Google Scholar]
- 24.Xu Q, Oberhuber G, Gruschwitz M, Wick G: Immunology of atherosclerosis: cellular composition and major histocompatibility complex class II antigen expression in aortic intima, fatty streaks, and atherosclerotic plaques in young and aged human specimens. Clin Immunol Immunopathol 1990, 56:344-359 [DOI] [PubMed] [Google Scholar]
- 25.Kockx MM, Cambier BA, Bortier HE, De Meyer GR, Declercq SC, van Cauwelaert PA, Bultinck J: Foam cell replication and smooth muscle cell apoptosis in human saphenous vein grafts. Histopathol 1994, 25:365-371 [DOI] [PubMed] [Google Scholar]
- 26.Tsukada T, Tejima T, Amano J, Suzuki A, Numano F: Immunocytochemical investigation of atherosclerotic changes observed in aortocoronary bypass vein grafts using monoclonal antibodies. Exp Mol Pathol 1988, 48:193-205 [DOI] [PubMed] [Google Scholar]
- 27.Westerband A, Mills JL, Marek JM, Heimark RL, Hunter GC, Williams SK: Immunocytochemical determination of cell type and proliferation rate in human vein graft stenoses. J Vasc Surg 1997, 25:64-73 [DOI] [PubMed] [Google Scholar]
- 28.Zwolak RM, Kirkman TR, Clowes AW: Atherosclerosis in rabbit vein grafts. Arteriosclerosis 1989, 9:374-379 [DOI] [PubMed] [Google Scholar]
- 29.Davies MG, Huynh TT, Fulton GJ, Barber L, Svendsen E, Hagen PO: Early morphology of accelerated vein graft atheroma in experimental vein grafts. Ann Vasc Surg 1999, 13:378-385 [DOI] [PubMed] [Google Scholar]
- 30.Klyachkin ML, Davies MG, Svendsen E, Kim JH, Massey MF, Barber L, McCann RL, Hagen PO: Hypercholesterolemia and experimental vein grafts: accelerated development of intimal hyperplasia and an increase in abnormal vasomotor function. J Surg Res 1993, 54:451-468 [DOI] [PubMed] [Google Scholar]
- 31.Hu Y, Zou Y, Dietrich H, Wick G, Xu Q: Inhibition of neointima hyperplasia of mouse vein grafts by locally applied suramin. Circulation 1999, 100:861-868 [DOI] [PubMed] [Google Scholar]
- 32.Zou Y, Hu Y, Metzler B, Xu Q: Signal transduction in arteriosclerosis: mechanical stress-activated MAP kinases in vascular smooth muscle cells. Int J Mol Med 1998, 1:827-834 [DOI] [PubMed] [Google Scholar]
- 33.Xu Q, Liu Y, Gorospe M, Udelsman R, Holbrook NJ: Acute hypertension activates mitogen-activated protein kinases in arterial wall. J Clin Invest 1996, 97:508-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Y, Cheng L, Hochleitner BW, Xu Q: Activation of mitogen-activated protein kinases (ERK/JNK) and AP-1 transcription factor in rat carotid arteries after balloon injury. Arterioscler Thromb Vasc Biol 1997, 17:2808-2816 [DOI] [PubMed] [Google Scholar]
- 35.Hu Y, Böck G, Wick G, Xu Q: Activation of PDGF receptor α in vascular smooth muscle cells by mechanical stress. FASEB J 1998, 12:1135-1142 [DOI] [PubMed] [Google Scholar]
- 36.Li C, Hu Y, Mayr M, Xu Q: Cyclic strain stress-induced mitogen-activated protein kinase (MAPK) phosphatase-1 expression in vascular smooth muscle cells is regulated by ras/rac-MAPK pathways. J Biol Chem 1999, 274:25273-25280 [DOI] [PubMed] [Google Scholar]
- 37.Bennett MR, Littlewood TD, Schwartz SM, Weissberg PL: Increased sensitivity of human vascular smooth muscle cells from atherosclerotic plaques to p53-mediated apoptosis. Circ Res 1997, 81:591-599 [DOI] [PubMed] [Google Scholar]
- 38.Bennett MR, Macdonald K, Chan SW, Boyle JJ, Weissberg PL: Cooperative interactions between RB and p53 regulate cell proliferation, cell senescence, and apoptosis in human vascular smooth muscle cells from atherosclerotic plaques. Circ Res 1998, 82:704-712 [DOI] [PubMed] [Google Scholar]
- 39.Granville DJ, Carthy CM, Hunt DWC, McManus BM: Apoptosis: molecular aspects of cell death and disease. Lab Invest 1998, 78:893-913 [PubMed] [Google Scholar]
- 40.Bennett MR, Boyle JJ: Apoptosis of vascular smooth muscle cells in atherosclerosis. Atherosclerosis 1998, 138:3-9 [DOI] [PubMed] [Google Scholar]
- 41.Horino K, Nishiura H, Ohsako T, Shibuya Y, Hiraoka T, Kitamura N, Yamamoto T: A monocyte chemotactic factor, S19 ribosomal protein dimer, in phagocytic clearance of apoptotic cells. Lab Invest 1998, 78:603-617 [PubMed] [Google Scholar]
- 42.Nishiura H, Shibuya Y, Yamamoto T: S19 ribosomal protein cross-linked dimer causes monocyte-predominant infiltration by means of molecular mimicry to complement C5a. Lab Invest 1998, 78:1615-1623 [PubMed] [Google Scholar]