

Abstract
Microsatellite instability (MSI) is characteristic of hereditary nonpolyposis colorectal cancer, and occurs in a subset (10 to 15%) of unselected colorectal cancer cases. In hereditary nonpolyposis colorectal cancer, MSI is caused by defects in five mismatch repair genes, and in sporadic cases the main cause seems to be somatic MLH1 promoter methylation. Most likely additional hereditary nonpolyposis colorectal cancer genes remain to be discovered. Genes with simple repeats in their coding region are often targets for deletions in MSI-positive tumors. Several genes (TGFβ RII, IGFIIR, MSH3, MSH6, BAX, MBD4) with significance in tumorigenesis harbor repeats in their coding regions and are often somatically inactivated because of deletions causing frameshifts. Recently, a novel human mismatch repair gene, MLH3, was cloned and shown to be involved in mammalian mismatch repair. To evaluate the possible role of MLH3 in hereditary cancer, we performed germline single-strand conformation polymorphism-analysis for 52 patients displaying features of inherited colorectal cancer. Forty-six of these had been diagnosed with MSI-positive tumors. No germline mutations were found. Similar to MSH3 and MSH6, MLH3 harbors mononucleotide repeats, ie, (A6)-(A9), in its coding region, which makes it a putative target for somatic mutations in MSI-positive tumors. To evaluate its somatic inactivation we performed a deletion search focusing on eight exonic MLH3 mononucleotide repeats in a series of 93 MSI-positive tumors. Somatic deletions were found in 8.6% of the samples, a frequency similar to one detected in neutral noncoding mononucleotide repeats. No evidence of involvement of MLH3 in MSI tumorigenesis was obtained.
Microsatellite instability (MSI) is a characteristic of hereditary nonpolyposis colorectal cancer and is seen in a subset (10 to 15%) of sporadic colorectal cancers as well. 1-3 It is typically caused by a defect in DNA mismatch repair. To date germline mutations in five mismatch repair genes (MSH2, 4 MLH1, 5,6 PMS1 and PMS2, 7 and MSH6ref 8 ) have been associated with hereditary nonpolyposis colorectal cancer. Because the causative mutation is found only in a subset of kindreds with MSI-positive colorectal tumors, it is possible that more genes underlying MSI-positive familial colorectal cancer remain to be discovered. In sporadic cases a major cause of MSI seems to be MLH1 promoter methylation. 9
Mismatch repair complexes in humans are multiple. Mismatches and insertion/deletion loops are recognized by MSH2/MSH6 heterodimers. 10 In the absence of MSH6 the recognition of insertion/deletion loops can be mediated by MSH2/MSH3 heterodimers, indicating redundant functions of MSH6 and MSH3. 10,11 The actual mismatch repair is performed by the MLH1/PMS2 heterodimer 12 with the help of exonucleases and polymerases.
Recently, a novel mismatch repair gene, MLH3, was cloned and shown to be associated with mammalian MSI. 13 The amino terminus of MLH3 contains a predicted ATPase motif and the carboxyl terminus encodes a predicted MLH1 interaction domain. 13 Thus, it is possible that MLH3 replaces PMS2 in mismatch repair complex, and they might have similarly redundant functions as MSH6 and MSH3. The putative MSH2/MSH3-MLH1/MLH3 complex is predicted to address a subset of insertion/deletion loops. 10 Because MLH3 has a greater similarity to yeast Mlh3p than to PMS2, it might have a specific role in human mismatch repair distinct from that of PMS2. 13 MLH3 is ubiquitously expressed. In the postnatal mouse the highest expression has been detected in epithelial tissues, including colon, skin, small bowel, and ventricle. 13 Therefore its function may have importance in maintaining genomic stability in the epithelium of the large bowel.
MSI-positive tumors acquire somatic deletions and insertions in simple repeated sequences. Several genes involved in tumorigenesis contain mononucleotide repeats in their coding regions. Somatic deletions have been seen in MSH3, MSH6, 14 BAX, 15 IGFIIR, 16 MBD4, 17,18 and TGF-β RII. 19-21 . Frameshift mutations of secondary mutators are presumably induced by primary mutators such as MLH1 and MSH2. 22 Accumulation of somatic mutations in mismatch repair genes such as MSH3 and MSH6 is believed to further increase genetic instability. MSH3 harbors an (A)8 repeat, whereas MSH6 has a (C)8 repeat. Both genes have confirmed roles in human mismatch repair, and germline MSH6 mutations are associated with a minor subset of hereditary nonpolyposis colorectal cancer cases. Although these genes do not have prominent roles in hereditary susceptibility to MSI-positive tumors, they seem to be targets for secondary hits. The newly identified mismatch repair gene, MLH3, harbors as many as eight mononucleotide repeats, (A)6-(A)9, in its coding region, which makes it an attractive target for somatic deletions in mismatch repair deficient cells.
To test whether germline mutations of MLH3 predispose to colorectal cancer, we analyzed 52 colorectal cancer patients displaying features of hereditary cancer for all coding exons and exon-intron boundaries by SSCP. To study whether MLH3 is, similar to MSH3 and MSH6, a target gene for MSI, we performed mononucleotide repeat length analysis on 93 MSI-positive tumor samples for eight polyA repeats, ie, (A6)-(A9), residing in the MLH3 coding region.
Materials and Methods
We analyzed 46 MSI-positive colorectal cancer patients with features of hereditary cancer by genomic SSCP analysis on normal tissue DNA. The MSI status of these samples has been previously determined using BAT26 mononucleotide marker, and MLH1 and MSH2 mutations have been excluded by genomic sequencing. 23,24 Of the 46 patients, 43 (93%) had at least one additional first or second degree relative with cancer (in 18 cases colorectal cancer, in six cases endometrial, in eight cases stomach, and in 11 cases some nonhereditary nonpolyposis colorectal cancer) and three (7%) patients were selected based on young age (mean, 40 years).
In addition, we analyzed six MSI-negative patients whose tumors had shown 14q deletions in comparative genomic hybridization and a possible common haplotype in chromosome 14q in loss of heterozygosity-studies (unpublished data). These six MSI-negative patients all had one to four additional first-degree relatives with colorectal cancer.
MLH3 is indicated to have 12 coding exons, one of which is alternatively spliced. 13 The GenBank sequence (AF195657) only includes 11 coding exons. We used the Genscan1.0 exon prediction program available on the web (http://bioweb.pasteur.fr/seqanal/interfaces/genscan.html) and found a 96.9% probability for an exon located between coding exons 5 and 6 (which is the location indicated by Lipkin and colleagues). 13 We performed reverse transcriptase-polymerase chain reaction (RT-PCR) to confirm that our interpretation was correct (data not shown). In this study we call this alternatively spliced exon “6A.” This alternative form lacks the MLH1 interaction domain, and its role, if any, in DNA mismatch repair is unclear. All of the exons were amplified separately except exons 8 and 9, because they only have 102 bp of intron sequence in between. Exon 1 was divided into 14 overlapping fragments. Because of the limitations of SSCP analysis, the length of the fragments was always <330 bp (between 158 and 329 bp; mean, 273 bp) (Table 1 ▶ .).
Table 1.
Primer Sequences, the Length of the PCR Fragments, Annealing Temperatures, and SSCP Running Times for each Fragment
| Primer sequence | Length, bp | Annealing temperature, °C | SSCP, hours |
|---|---|---|---|
| MLH3 F1-1: TCC AGT CAG AGA AGG AAA CCA | 294 | 58 | 22 |
| MLH3 R1-1: CGA GTG GCA TTT ACT GGT GA | |||
| MLH3 F1-2: TGT AGA GAA AGT GGG AAA TCG TT | 276 | 60 | 22 |
| MLH3 R1-2: ACA GGA AGC TGG TAA AAT AGG TT | |||
| MLH3 F1-3: CTG ATG TGA CTA GAG CAA GCG | 309 | 60 | 22 |
| MLH3 R1-3: AGC CAC TAA GCT CAA ACT CTT TAT | |||
| MLH3 F1-4: GGG AAA GTC CCA AAA GCT AA | 305 | 60 | 22 |
| MLH3 R1-4: CAT CAT ACT CAC AGA ATT GGC AC | |||
| MLH3 F1-5: ATG TGC AGT GCC AAT TCT GT | 245 | 60 | 22 |
| MLH3 R1-5: GAA ATT GCT CCT CTC ATC GG | |||
| MLH3 F1-6: TCA GGT GAG GAT ATT AAG GAA TTT | 312 | 61 | 34 |
| MLH3 R1-6: TTT GTT TTG TAA AGA TGG CTC TG | |||
| MLH3 F1-7: CCA GGC CAT AGC AAA ATG AC | 275 | 60 | 30 |
| MLH3 R1-7: GCA GCC ATG CCA TTA ACA G | |||
| MLH3 F1-8: TGG GCA GGA TCT AGA AAT ATG G | 316 | 60 | 22 |
| MLH3 R1-8: TGA ATG TTC TGT TTC AGT TGA TTT | |||
| MLH3 F1-9: GGG CGA GTT AAA TTA TGT TCC A | 309 | 60 | 22 |
| MLH3 R1-9: GCT ACC TTC CTG AAA AGC AGA A | |||
| MLH3 F1-10: GGC TAG AGA ATG AAC CTA CAG CA | 312 | 60 | 22 |
| MLH3 R1-10: CTT GAA GAC TGA GAT TGG TAG TGA | |||
| MLH3 F1-11: TGG GAA GGT TGA AAA TCC TC | 318 | 60 | 22 |
| MLH3 R1-11: TCA AGG TCC AAA GGT TTT CTA TT | |||
| MLH3 F1-12: TCC TAT GAC CCT GAA GGA GTT | 293 | 60 | 22 |
| MLH3 R1-12: AGG AAT TAT CCT GTG TGG CAG | |||
| MLH3 F1-13: CAG AGA ATG GTG TCA TCC CAA | 265 | 60 | 30 |
| MLH3 R1-13: CCA TTT TGG TCA CCT GTG G | |||
| MLH3 F1-14: ATC CGG TAG AAG ATG CCA CA | 320 | 60 | Does not separate |
| MLH3 R1-14: CCT TGT CCA GCA TTC CCA T | |||
| MLH3 F2: TGT CTT GAC TCA GTT TGT GCA G | 247 | 62 | 22 |
| MLH3 R2: ACG ATG TGT ACT GTG TGC CC | |||
| MLH3 F3: TGG TTC TGG ATG CCA ACT TT | 229 | 59 | 26 |
| MLH3 R3: ATT TCA GTC TGG GCA ACA GG | |||
| MLH3 F4: CAA TTA TAT TTT GCT GAG TC | 158 | 52 | 14 |
| MLH3 R4: ATG AGA TTT TGA AGT TAA TC | |||
| MLH3 F5: CCC AGT CTC AAA GAA AGG AGT G | 239 | 57 | 20 |
| MLH3 R5: AGC TGG TTA GTC ATT CAG GC | |||
| MLH3 F6: TTC CCT TCC TAC TCT TAA CCC A | 265 | 59 | 20 |
| MLH3 R6: TGT AAC CTC TCT TGG TCT CAT CTG | |||
| MLH3 F7: TTT GGA ACC AGT AGT GAA GTG C | 267 | 62 | 20 |
| MLH3 R7: CAG CAA TTT CCT TAA CAT CTG C | |||
| MLH3 F8/9: CGT AGA TTA AAG CCG ATT TTC | 329 | 59 | 22 |
| MLH3 R8/9: TGT ACC CTC TGC CTC TTT CG | |||
| MLH3 F10: GTC AGC ATT GGT TTC CCA CT | 251 | 59 | 20 |
| MLH3 R10: AAA CTT TGC TCC CTC CTG CT | |||
| MLH3 F11: GCC CAG CCT GTA TGC TAC CT | 226 | 59 | 20 |
| MLH3 R11: CAG TGA CAC TCC CTT TGT TCC | |||
| MLH3 6AF: CAT GAT GGT TGT CGT CTT GC | 185 | 60 | 14 |
| MLH3 6AR: GGT GTA CTG ATT CTG CTG GGA |
PCR-reactions were carried out in 20 μl of reaction volume containing 100 ng genomic DNA, 1× PCR buffer (Perkin Elmer Applied Biosystems Division, Foster City, CA), 500 μmol/L of each dNTP (Finnzymes, Espoo, Finland), 1 μmol/L of each primer, and 1 U of AmpliTaqGOLD polymerase (Perkin Elmer). The MgCl2 concentration was 2.75 mmol/L. The following PCR cycles were used for amplification: 95°C for 10 minutes, 40 cycles at 95°C for 45 seconds, corresponding annealing temperature for 45 seconds, and 72°C for 45 seconds (30 seconds for exons 4 and 6A). Final extension was 72°C for 10 minutes. The length of the PCR fragments, primer sequences, and the corresponding annealing temperatures for each fragment are listed in Table 1 ▶ .
SSCP analysis was performed using mutation detection enhancement gel solution (FMC BioProducts, Rockland, Maine). PCR products were run on 0.6× mutation detection enhancement gels, 4 W for 14 to 34 hours. The running buffer was 0.6× Tris borate-ethylenediaminetetraacetic acid. SSCP gels were silver stained according to standard procedure. The SSCP running times for each fragment are indicated in Table 1 ▶ . The complementary strands were not separated in exon 1 fragment 1 to 14 even after a 40-hour run, and this fragment was analyzed using genomic sequencing. Direct sequencing was performed using the ABI PRISM Dye Terminator or ABI PRISM dRhodamine cycle sequencing ready reaction kit (Perkin Elmer). Cycle sequencing products were electrophoresed on 6% Long Ranger gels (FMC BioProducts) and analyzed on an Applied Biosystems model 373A or 377 automated DNA sequencer (Perkin Elmer).
Tumor DNA of 93 MSI-positive colorectal cancer patients was analyzed for deletions and insertions in eight mononucleotide repeats, ie, (A6)-(A9) in the coding region of MLH3. New sets of primers for shorter PCR fragments (100 bp or less) were designed to maximize resolution in polyacrylamide gel electrophoresis analysis. For primer sequences see Table 2 ▶ . The following repeats were analyzed: (A)6 starting at nucleotide 1410 in mRNA, 1576 (A)6, 1861 (A)9, 1961(A)6, 2128 (A)8, 2218 (A)6, 2897 (A)6, and 3796 (A)6. Out of the 93 cases chosen 18 (19%) had an MLH1 or MSH2 germline mutation. Thirty-three (35%) out of 93 patients (none with a known germline mutation) were also included in the germline SSCP analysis; 30 because of family history of cancer, and three because they were diagnosed before the age of 50.
Table 2.
Primer Sequences, the Length of the PCR Fragments, and Annealing Temperature for PAGE Analysis
| Primer sequence | Length, bp | Annealing temperature |
|---|---|---|
| MLH3 F1-6s: CTG CAG AAA ACG TAA ACA CAC A | 100 | 54°C, 1 minute |
| MLH3 R1-6s: CCT GGA CCA CCT GAT TCA TAA | ||
| MLH3 F1-7s: AAT TGT AGC ATC AGA AGC TGG AG | 90 | 54°C, 1 minute |
| MLH3 R1-7s: AAA CTG GTT CCA CAC GGA TT | ||
| MLH3 F1-8s: TGC AAC AAC ATT ATG GGG AGT | 100 | 60°C, 45 seconds |
| MLH3 R1-8s: TCG CCC ATA ACT AAA AAC ATT TC | ||
| MLH3 F1-9as: TGT TCC ACT GGC TTT ATA ACT CA | 97 | 54°C, 1 minute |
| MLH3 R1-9as: TGG GAC CAG GTC TAA CAT AAT TT | ||
| MLH3 F1-9bs: CAT CAA AGA TTT AGC CAG CAC T | 99 | 54°C, 45 seconds |
| MLH3 R1-9bs: GGT TCA TTC TCT AGC CCA TAA CTT | ||
| MLH3 F1-10s: CAG CAA CTT ATA CAA TGT TTT CTG C | 90 | 54°C, 45 seconds |
| MLH3 R1-10s: GGA AAG AGG GGG ATG TAT CA | ||
| MLH3 F1-12s: CAG CAA GTT GTG CAG TGT GTT | 99 | 55°C, 45 seconds |
| MLH3 R1-12s: GCA GAA TCT GAT GTT GGG ATG | ||
| MLH3 F6s: TTC CTA CGA GAA GCA ACA GG | 94 | 54°C, 50 seconds |
| MLH3 R6s: TTC CTC TGT CAC TGT TAT CTC TAG C |
PCR reactions were carried out in 10 μl of reaction volume containing 50 ng genomic DNA, 1× PCR buffer (Perkin Elmer), 1 μmol/L of each primer (2 μmol/L of primers 1–6s, 1–7s, and 1–9as), dCTP at 20 μmol/L, 200 μmol/L each of dATP, dGTP, dTTP (Amersham Pharmacia Biotech, Piscataway, NJ), 0.7 μCi [α-32P]dCTP (3,000 Ci/mmol; Amersham Pharmacia Biotech UK Ltd., Buckinghamshire, UK), and 0.5 units of AmpliTaqGOLD polymerase (Perkin Elmer). The MgCl2 concentration was 3.75 mmol/L (1.25 mmol/L for exon 1–8s). The following PCR cycles were used for amplification: 95°C for 10 minutes, 37 cycles of 95°C for 45 seconds, for annealing conditions see Table 2 ▶ , and 72°C for 1 minute (45 seconds for exon 1–8). Final extension was 72°C for 10 minutes. Ten μl of PCR-product was mixed with 7 μl of loading buffer and run on 6.7% polyacrylamide gels, with 0.6× TBE as a running buffer, at a constant power of 80 W. All aberrant bands were sequenced in both directions as previously described. In all aberrant cases the corresponding normal DNA was analyzed to confirm the somatic nature of the changes.
To determine the polyA background mutation rate in MSI-positive colorectal tumors, we analyzed an (A)8 and an (A)9 repeat in the intron of OBR-gene, coding for leptin receptor (GenBank accession codes U62509 and U62513, respectively) by radioactive polyacrylamide gel electrophoresis in the 93 tumors. All aberrant bands as well as the corresponding normal DNA were sequenced in both directions. Primer sequences and PCR conditions are available on request.
Results
No germline mutations were detected in any of the 52 normal tissue samples analyzed. Multiple aberrant SSCP bands were discovered, and sequencing revealed several polymorphisms. Some of the aberrant SSCP bands are shown in Figure 1 ▶ , and the corresponding sequences are shown in Figure 2 ▶ . In exon 1 fragment 1 to 6, three samples showed aberrant bands. Genomic sequencing revealed a missense type of change 1258G→A (Val→Ile) (allele frequency, 2.9%). The same change was seen in three out of 56 cancer-free controls (allele frequency, 2.7%) confirming its benign nature. In exon 11 a silent change 4377G→A (Gln→Gln) was detected in 28 patients in heterozygous form and in 10 patients in homozygous form, giving an allele frequency of 46%. The same change was seen in 30 out of 57 and in 13 out of 57 cancer-free controls in heterozygous and homozygous forms, respectively (allele frequency, 49%). In the intron between exons 9 and 10 (exon 9 + 55) a heterozygous G→C change was found in three samples (allele frequency, 2.9%). When sequencing the PCR product of these three samples showing aberrant bands in SSCP, also a A→G change was discovered in the intron between exons 8 and 9 (exon 8 + 66). This change was not visible on SSCP, because other samples not showing aberrant bands were found to have it. Out of seven sequenced samples, five were heterozygous for this variant, and one was homozygous. In analysis of exons 3 and 7 some aberrant SSCP bands were seen (five in exon 3 and one in exon 7). Sequencing did not reveal any changes in the coding region or in the splice sites.
Figure 1.

Some of the aberrant SSCP bands are shown here. A: MLH3 exon 11, 4377G→A (Gln→Gln), arrow points at the mutant band. B: MLH3 exon 1, fragment 1 to 6, 1258G→A (Val→Ile). C: MLH3 exon 8/9, a G→C in the intron between exons 8 and 9.
Figure 2.

Direct sequencing results of the polymorphisms presented in Figure 1 ▶ . A: MLH3 exon 11, 4377G→A (Gln→Gln), arrow points at a heterozygous change. B: MLH3 exon 1 fragment 1 to 6, 1258G→A (Val→Ile). C: MLH3 exon 8/9, a G→C in the intron between exons 8 and 9.
Somatic deletions were detected in eight out of 93 (8.6%) MSI-positive tumor samples. Seven patients showed a 1-bp deletion in the (A)9 tract in exon 1 (fragment 1–8s, starting at nucleotide 1861). One patient showed a deletion of 1-bp in the (A)8 tract in exon 1 (fragment 1–9bs, starting at nucleotide 2128). Deletions of 1 bp in these polyA tracts cause frameshifts and predicted premature stop at codons 609 and 679, respectively. None of these mutations were present in the corresponding normal DNA. One of these eight patients also has a germline MLH1 mutation. An example sequence of a deletion in the (A)9 tract is shown in Figure 3 ▶ . During the sequencing of aberrant bands a 1234A→G (Lys→Glu) change was seen in one out of 13 samples in exon 1 (within fragment 1–6) (allele frequency 3.8%). The change was present both in the tumor as well as the normal DNA of the patient. The same change was seen in two out of 92 (allele frequency, 1.1%) cancer-free controls. Also the fact that the patient showing this change has a germline MLH1 mutation favors the benign nature of this change.
Figure 3.
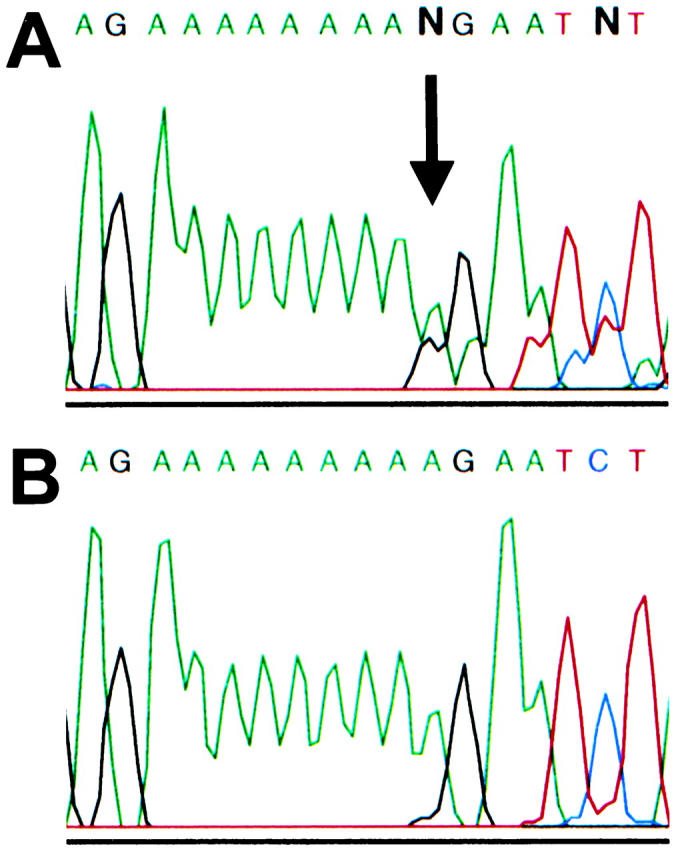
Direct sequencing results of a somatic deletion in the (A)9 repeat in MLH3 exon 1 (starting at nucleotide 1861). A: Tumor DNA displays a 1-bp deletion causing frameshift. The arrow depicts the deletion point. B: The corresponding normal DNA shows wild-type sequence.
Somatic deletions in the intronic OBR (A)8 and (A)9 repeats were seen in three out of 93 (3.2%), and in three out of 93 (3.2%) tumors, respectively. None of these mutations were present in the corresponding normal DNA.
Discussion
Recently, a novel mutL homologue, MLH3, was cloned. 13 An association between MLH3 and human DNA mismatch repair was proposed based on the following evidence: first, MLH3 is predicted to have an MLH1 interaction domain. Second, overexpressed dominant-negative MLH3 induces MSI in mammalian cell culture. 13 Third, MLH3 is predicted to be involved in mismatch repair in yeast. 13
An interesting question is whether MLH3 has a role in MSI tumorigenesis in vivo. The predicted MLH1 interaction domain indicates that MLH3 might replace PMS2 in the mismatch repair complex. Very few germline mutations have been found in PMS2, and Pms2−/− mice do not display colon cancer susceptibility. 25 However, MLH3 does not show great similarity to PMS2, so it may have a different role in mismatch repair.
To study whether MLH3 is involved in MSI-positive colorectal tumorigenesis, we performed germline mutation analysis by SSCP on 46 samples from MSI-positive colorectal cancer patients with features of hereditary cancer. No deleterious mutations were found in any of the samples. Six MSI-negative familial colon cancer patients whose tumors had shown 14q deletions (data not shown) did not show germline mutations either.
MLH3 harbors eight polyadenine repeats in its coding sequence, ie, six discrete (A)6 repeats, one (A)8-repeat, and one (A)9-repeat. These repeats provide a tool for evaluating the possible role of MLH3 in somatic tumorigenesis. Analysis of 93 MSI-positive colorectal carcinomas revealed somatic deletions in eight (8.6%) patients in the mononucleotide repeats in the coding region of MLH3. For comparison, 90 out of these 93 MSI-positive samples had previously been analyzed for somatic TGFβ RII deletions and insertions, and 87% had shown deletions in the (A)10 tract (data not shown).
Analysis of the intronic (A)8 and (A)9 repeats in the OBR-gene revealed somatic deletions in three out of 93 (3.2%), and in three out of 93 (3.2%) tumors, respectively. These intronic mutations most likely have no impact on tumor development, and are thus not selected for. Therefore they serve as an ideal tool for evaluating the background mutation rate in MSI tumors. The frequency of MLH3 deletions (8.6%) is similar to the background mutation frequency in equal-sized repeats in the intron of the OBR gene (six out of 93, 6.5%). Our data thus suggest that the deletions observed in MLH3 are merely a consequence of the MSI phenotype rather than selected events driving tumor progression.
It seems that MLH3 is not frequently involved in MSI-positive colorectal cancer as a hereditary or somatic component. Redundant function of MLH3 and PMS2 may explain the rarity of mutations, like in the case of MSH6 and MSH3. 10 The absence of frequent somatic MLH3 changes even though the sequence provides abundant targets for MSI emphasizes the significance of the previous finding of somatic MSH3 deletions in 39 to 57% 14,26 and MSH6 deletions in 30% 14 of MSI-positive colorectal cancers. Also another mismatch repair gene, MBD4, has been shown to acquire somatic deletions in its (A)10 tract in 25 to 40% of MSI-positive tumors. 17,18 In light of the MLH3 results MSH3, MSH6, and MBD4 indeed appear as important target genes of MSI. Presumably somatic deletions occur also in MLH3, but unlike MSH3, MSH6, and MBD4 deletions, these are not selected for. This data supports the hypothesis that the effect of the putative MLH1/MLH3 complex to insertion/deletion loop repair is not significant. 10
Acknowledgments
We thank Kirsi Laukkanen and Annika Lahti for excellent technical assistance and Paula Kristo and Elvi Karila for helping with the sequencing.
Footnotes
Address reprint requests to Dr. Lauri A. Aaltonen at the Department of Medical Genetics, Haartman Institute, P.O. Box 21, FIN-00014 University of Helsinki, Finland. E-mail: lauri.aaltonen@helsinki.fi.
Supported by grants from the Finnish Cancer Society, the Academy of Finland, Sigrid Juselius Foundation, Duodecim, Ida Montin Foundation, Jalmari and Rauha Ahokas Foundation, Emil Aaltonen Foundation, Finnish-Norwegian Medical Foundation, Nordisk Cancer Union, and Helsinki University Central Hospital.
A. L. is a Ph.D. student of the Helsinki Biomedical Graduate School.
References
- 1.Aaltonen LA, Peltomäki P, Leach F, Sistonen P, Pylkkänen L, Mecklin J-P, Järvinen H, Powell S, Jen J, Hamilton SR, Petersen GM, Kinzler KW, Vogelstein B, de la Chapelle A: Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260:812-816 [DOI] [PubMed] [Google Scholar]
- 2.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M: Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363:558-561 [DOI] [PubMed] [Google Scholar]
- 3.Thibodeau SN, Bren G, Schaid D: Microsatellite instability in cancer of the proximal colon. Science 1993, 260:816-819 [DOI] [PubMed] [Google Scholar]
- 4.Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomäki P, Sistonen P, Aaltonen LA, Nyström-Lahti M, Guan X-Y, Zhang J, Meltzer PS, Yu J-W, Kao F-T, Chen DJ, Cerosaletti KM, Fournier REK, Todd S, Lewis T, Leach RJ, Naylor SL, Weissenbach J, Mecklin J-P, Järvinen H, Petersen GM, Hamilton SR, Green J, Jass J, Watson P, Lynch HT, Trent JM, de la Chapelle A, Kinzler KW, Vogelstein B: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75:1215-1225 [DOI] [PubMed] [Google Scholar]
- 5.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, Tannergård P, Bollag RJ, Godwin AR, Ward DC, Nordenskjöld M, Fishel R, Kolodner R, Liskay RM: Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368:258-261 [DOI] [PubMed] [Google Scholar]
- 6.Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Hamilton SR, Petersen GM, Watson P, Lynch HT, Peltomäki P, Mecklin J-P, de la Chapelle A, Kinzler KW, Vogelstein B: Mutation of a mutL homolog in hereditary colon cancer. Science 1994, 263:1625-1629 [DOI] [PubMed] [Google Scholar]
- 7.Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Dunlop MG, Hamilton SR, Petersen GM, de la Chapelle A, Vogelstein B, Kinzler KW: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994, 371:75-80 [DOI] [PubMed] [Google Scholar]
- 8.Miyaki M, Konishi M, Tanaka K, Kikuchi-Yanoshita R, Muraoka M, Yasuno M, Igari T, Koike M, Chiba M, Mori T: Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 1997, 17:271-272 [DOI] [PubMed] [Google Scholar]
- 9.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R: Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997, 57:808-811 [PubMed] [Google Scholar]
- 10.Jiricny J: Mediating mismatch repair. Nat Genet 2000, 24:6-8 [DOI] [PubMed] [Google Scholar]
- 11.Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA: Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics 1998, 148:1637-1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li GM, Modrich P: Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc Natl Acad Sci USA 1995, 92:1950-1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lipkin SM, Wang V, Jacoby R, Banerjee-Basu S, Baxevanis AD, Lynch HT, Elliot RM, Collins FS: MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet 2000, 24:27-35 [DOI] [PubMed] [Google Scholar]
- 14.Malkhosyan S, Rampino N, Yamamoto H, Perucho M: Frameshift mutator mutations. Nature 1996, 382:499-500 [DOI] [PubMed] [Google Scholar]
- 15.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M: Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275:967-969 [DOI] [PubMed] [Google Scholar]
- 16.Souza RF, Appel R, Yin J, Wang S, Smolinski KN, Abraham JM, Zou TT, Shi YQ, Lei J, Cottrell J, Cymes K, Biden K, Simms L, Leggett B, Lynch PM, Frazier M, Powell SM, Harpaz N, Sugimura H, Young J, Melzer SJ: Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet 1996, 14:255-257 [DOI] [PubMed] [Google Scholar]
- 17.Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, Masciullo V, Genuardi M, Paravatou-Petsotas M, Bassi DE, Ruggeri BA, Klein-Szanto AJ, Testa JR, Neri G, Bellacosa A: The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat Genet 1999, 23:266-268 [DOI] [PubMed] [Google Scholar]
- 18.Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper M, Wyllie A: Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene 1999, 18:8044-8047 [DOI] [PubMed] [Google Scholar]
- 19.Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B: Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res 1995, 55:5548-5550 [PubMed] [Google Scholar]
- 20.Lu SL, Akiyama Y, Nagasaki H, Saitoh K, Yuasa Y: Mutations of the transforming growth factor-beta type II receptor gene and genomic instability in hereditary nonpolyposis colorectal cancer. Biochem Biophys Res Commun 1995, 216:452-457 [DOI] [PubMed] [Google Scholar]
- 21.Akiyama Y, Iwanaga R, Ishikawa T, Sakamoto K, Nishi N, Nihei Z, Iwama T, Saitoh K, Yuasa Y: Mutations of the transforming growth factor-beta type II receptor gene are strongly related to sporadic proximal colon carcinomas with microsatellite instability. Cancer 1996, 78:2478-2484 [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto H, Sawai H, Perucho M: Frameshift somatic mutations in gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res 1997, 57:4420-4426 [PubMed] [Google Scholar]
- 23.Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomäki P, Chadwick RB, Percesepe A, Kääriäinen H, Ahtola H, Eskelinen M, Härkönen N, Julkunen R, Kangas E, Ojala S, Tulikoura J, Valkamo E, Järvinen H, Mecklin J-P, de la Chapelle A: Incidence of hereditary nonpolyposis colorectal cancer, and molecular screening for the disease. N Engl J Med 1998, 338:1481-1487 [DOI] [PubMed] [Google Scholar]
- 24.Salovaara R, Loukola A, Kristo P, Kääriäinen H, Ahtola H, Eskelinen M, Härkönen N, Julkunen R, Kangas E, Ojala S, Tulikoura J, Valkamo E, Järvinen H, Mecklin J-P, Aaltonen LA, de la Chapelle A: Population-wide molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000, 18:2193-2200 [DOI] [PubMed] [Google Scholar]
- 25.Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM: Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet 1998, 18:276-279 [DOI] [PubMed] [Google Scholar]
- 26.Akiyama Y, Tsubouchi N, Yuasa Y: Frequent somatic mutations of hMSH3 with reference to microsatellite instability in hereditary nonpolyposis colorectal cancer. Biochem Biophys Res Commun 1997, 236:248-252 [DOI] [PubMed] [Google Scholar]