

Abstract
Inflammatory myofibroblastic tumors (IMTs) are neoplastic mesenchymal proliferations featuring an inflammatory infiltrate composed primarily of lymphocytes and plasma cells. The myofibroblastic cells in some IMTs contain chromosomal rearrangements involving the ALK receptor tyrosine-kinase locus region (chromosome band 2p23). ALK—which is normally restricted in its expression to neural tissues—is expressed strikingly in the IMT cells with 2p23 rearrangements. We now report a recurrent oncogenic mechanism, in IMTs, in which tropomyosin (TPM) N-terminal coiled-coil domains are fused to the ALK C-terminal kinase domain. We have cloned two ALK fusion genes, TPM4-ALK and TPM3-ALK, which encode ∼95-kd fusion oncoproteins characterized by constitutive kinase activity and tyrosylphosphorylation. Immunohistochemical and molecular correlations, in other IMTs, implicate non-TPM ALK oncoproteins that are predominantly cytoplasmic or pre- dominantly nuclear, presumably depending on the subcellular localization of the ALK fusion partner. Notably, a TPM3-ALK oncogene was reported recently in anaplastic lymphoma, and TPM3-ALK is thereby the first known fusion oncogene that transforms, in vivo, both mesenchymal and lymphoid human cell lineages.
The inflammatory myofibroblastic tumor (IMT) 1-4 is a clinicopathologically distinctive but biologically controversial entity that was defined originally as a nonneoplastic lesion. IMTs arise usually in the soft tissues, most often in the abdomen of children and adolescents, and they are composed of myofibroblastic spindle cells admixed with a prominent inflammatory infiltrate of lymphocytes, plasma cells, and, less often, acute inflammatory cells. Notably, many patients with IMTs present with constitutional symptoms of fever and weight loss, and these same patients are often anemic and thrombocythemic. However, it is unclear whether the constitutional symptoms and inflammatory infiltrate are induced by tumor-cell factors or, alternately, whether the constitutional symptoms are secondary to the inflammatory process.
The uncertain pathogenesis of IMTs, and the ongoing question of its neoplastic versus reactive nature, is reflected in the large number of names which have been bestowed on this disorder. IMTs arising in the lung, particularly those that are well circumscribed, are generally cured by surgical excision. Until recently, these were referred to as “plasma cell granulomas,” “inflammatory pseudotumors,” or “pseudosarcomatous myofibroblastic proliferations.” Intra-abdominal IMTs may pursue a somewhat more aggressive clinical course with multiple recurrences and with potential for metastatic spread in rare cases. Such tumors have often been referred to as “inflammatory myofibroblastic tumor” or “inflammatory fibrosarcoma.” 1,2 However, most IMTs have overlapping histological characteristics and it is difficult to distinguish those with neoplastic potential from the potentially reactive subset that belong under the umbrella category of “inflammatory pseudotumor.” Another complicating feature, with respect to classification, is that differences in clinical behavior, between different IMT subcategories, do not exclude a common pathogenesis.
Cytogenetic banding studies were the first assays to demonstrate unequivocal clonal mutations—indicative of a neoplastic pathogenesis—in IMTs. 5-10 Approximately 50% of soft-tissue IMT karyotypes contain clonal rearrangements of the chromosome 2 short arm, 5-10 and each of two cytogenetically characterized IMTs arising in bone contained rearrangements of the HMGIC region on chromosome band 12q15. 8 Recently, Griffin et al 10 showed that IMT 2p rearrangements fall within an ∼100-kb region containing the ALK receptor tyrosine-kinase locus, on chromosome band 2p23. These rearrangements were associated with striking ALK expression in the IMT myofibroblastic spindle cells. 10 ALK is a receptor tyrosine kinase that was originally characterized as a component of the anaplastic large-cell lymphoma NPM-ALK fusion oncoprotein. 11 ALK expression is normally restricted to the central nervous system, 12-14 and expression of constitutively activated ALK, in anaplastic large-cell lymphoma, seems to be a pivotal transforming event. High-level ALK expression, in IMTs, is notable because ALK is expressed at low or undetectable levels in nonneoplastic fibroblasts. These findings suggest that IMTs might be neoplasms in which the myofibroblastic component is transformed, in some cases, by chromosomal mechanisms targeting ALK.
In the present study we show that chromosome 2p rearrangements in IMTs create ALK fusion genes. We characterize two such genes, tropomyosin 4 (TPM4)-ALK and tropomyosin 3 (TPM3)-ALK, and provide evidence for at least two additional fusion mechanisms that remain to be characterized. These studies also identify a diagnostically useful molecular marker in IMTs.
Materials and Methods
Eleven IMTs were obtained as either frozen or paraffin-embedded specimens. The cytogenetic features of cases 1, 2, 6, and 11 have been described previously. 6,8,9 Cytogenetic banding analyses had not been performed in the remaining seven cases.
Immunostaining
All cases were re-evaluated histologically by three of the authors (APA, BPR, and CDMF). ALK immunostaining was performed using monoclonal antibody ALK-1 (DAKO, Carpinteria, CA) at a dilution of 1:50 after antigen retrieval, by pressure steaming, in an ethylenediaminetetraacetic acid solution (pH 8.0). Antibody detection was accomplished using a labeled streptavidin-biotin LSAB+ (DAKO) method.
5′-Rapid Amplification of cDNA Ends (RACE) and DNA Sequencing
Total RNA was extracted from frozen tissue using Trizol (Life Technologies, Inc., Gaithersburg, MD), according to the manufacturer’s protocol. One μg of RNA was then used for RACE using the Marathon Kit (Clontech, Palo Alto, CA) according to the manufacturer’s protocol. First round polymerase chain reaction (PCR) was with the Marathon kit AP1 adapter primer and ALK reverse primer ALK/4436/R (5′-CTGTTGAGAGACCAGGAGAGGAGGAA) at 94°C for 30 seconds and 68°C for 8 minutes for 25 cycles. Second round PCR was with the Marathon kit AP2 adapter primer and ALK reverse primer ALK/4161/R, (5′-ACTTCCTGGTTGCTTTTGCTGGGGTAT) at 94°C for 30 seconds and 68°C for 8 minutes for 30 cycles. A single RACE product band was observed and was gel purified using the Qiaquick gel extraction kit (Qiagen, Valencia, CA), and the RACE fragment was then cycle sequenced in forward and reverse directions using the AP2 primer and ALK reverse primer ALK/3638/R (5′-AATCCAGTTCGTCCTGTTCAGAGCACA), respectively. Cycle sequencing was performed using ABI BigDye terminators, and the sequences were analyzed using an ABI Prism 310 automated sequencer (Applied Biosystems, Foster City, CA).
Fluorescence in Situ Hybridization
Interphase cells from IMT case 1 were spread on glass slides and denatured according to standard protocols. Hybridization and washing steps were performed as described. 15 ALK and TPM4 region rearrangements were evaluated using a dual-color ALK split-apart probe (Vysis, Downers Grove, IL) and a yeast artificial chromosome clone, 766_E_7, mapping to chromosome band 19p13.1, which was labeled 50:50 biotin:digoxigenin by random octamer priming. 15 Detection was with streptavidin-fluorescein isothiocyanate (Zymed Laboratories, South San Francisco, CA) and rhodamine anti-digoxigenin (Zymed), and nuclei were counterstained with 0.4 mg/ml 4,6-diamidino-2-phenylindole-dihydrochloride. Images were captured using a charge-coupled device camera (Photometrics, Tucson, AZ).
RT-PCR and DNA Sequencing
Total RNAs were extracted from frozen tissue specimens using Trizol (Life Technologies, Inc.) and from paraffin-embedded tissues after a 5-day sodium dodecyl sulfate-proteinase K digestion. 16 One μg of the RNAs were reverse transcribed (GeneAmp Kit; Perkin Elmer, Norwalk, CT) using the ALK/3638/R primer. TPM3-ALK and TPM4-ALK fusion genes were evaluated using a nested PCR method that was optimized for detection of either TPM3 or TPM4 fusions in the extensively degraded RNAs isolated from paraffin sections. The TPM primers correspond to regions of near sequence identity in TPM3 and TPM4 and, hence, anneal effectively to both genes. First-round PCR was with TPM3/249/F (5′-ACTGATAAACCCAAGGAGGCAGAGA) and ALK/3455/R (5′- AGGTCTTGCCAGCAAAGCAGTAGTT) at 94°C for 30 seconds, 67 to 62°C (touchdown) for 30 seconds, and 72°C for 30 seconds for 10 cycles, and then 94°C for 30 seconds, 62°C for 30 seconds, and 72°C for 30 seconds for 25 cycles. Second-round PCR was with TPM3/269/F (5′- AGAGACCCGTGCTGAGTTTGCTGA) and ALK/3399/R (5′- CGGAGCTTGCTCAGCTTGTACTC) at 94°C for 30 seconds, and 68°C for 30 seconds for 30 cycles. Five microliters of the second-round PCR product were gel purified and cycle sequenced using the ALK/3399/R primer to determine whether the tropomyosin-derived sequence was contributed by TPM3 or TPM4. TPM-ALK fusions were also subtyped using first-round TPM-ALK reverse transcriptase (RT)-PCR template (as described above) in a second-round PCR with a TPM3-specific forward primer, TPM3/284/F (5′-GTTTGCTGAGCGATCGGTAGCCAAGC) and ALK/3399R at 94°C for 30 seconds, and 68°C for 30 seconds for 30 cycles. Nested RT-PCR was required for consistent detection of the TPM-ALK fusion transcripts, irrespective of whether RNAs were isolated from frozen or paraffin specimens.
Integrity of both frozen tumor and paraffin-derived RNAs was evaluated by nested RT-PCR for the ubiquitously expressed glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. Reverse transcription was performed using random primers (GeneAmp Kit, Perkin Elmer), and first-round PCR was performed using GAPDH/177/F (5′-CCTCAACTACATGGTTTACATGTTC) and GAPDH/488/R (5′-CTGTTGTCATACTTCTCATGGTTCA) at 94°C for 30 seconds and 70 to 60°C (touchdown) for 1 minute for 10 cycles, and then 94°C for 30 seconds, and 60°C for 1 minute for 25 cycles. Second-round PCR was with GAPDH/250/F (5′-AACGGGAAGCTTGTCATCAAT) and GAPDH/427/R (5′-CAGAGATGATGACCCT- TTTGG) at 94°C for 30 seconds, and 62°C for 30 seconds for 30 cycles.
In Vitro Kinase Assay and Western Blotting
Protein lysates were isolated from frozen tumor tissue in IMT cases 1 and 2 and from a non-IMT spindle-cell sarcoma. Three-mm 3 tumor pieces were minced in ice-cold lysis buffer (1% Nonidet P-40, 50 mmol/L Tris, pH 8.0, 100 mmol/L sodium fluoride, 30 mmol/L sodium pyrophosphate, 2 mmol/L sodium molybdate, 5 mmol/L ethylenediaminetetraacetic acid, 2 mmol/L sodium vanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 100 μg/ml phenylmethylsulfonyl fluoride), homogenized further by 10 to 20 strokes of a Dounce homogenizer, then rocked for 45 minutes at 4°C. Residual cell debris was removed by centrifugation, and supernatant protein concentrations were determined using the BioRad MMT assay. Cell lysates (1.5 mg) were precleared with 20 μl of Protein G Sepharose (Zymed Laboratories) for 1 hour at 4°C, followed by sequential additions of 50 μl of anti-ALK hybridoma supernatant (ALK1; DAKO) and 20 μl of Protein G Sepharose with end-to-end rotation for 2 hours at 4°C after each addition. The immunoprecipitates were then washed three times in lysis buffer and twice in kinase buffer (20 mmol/L HEPES, pH 7.4, 10 mmol/L sodium fluoride, 10 mmol/L magnesium chloride, 10 mmol/L manganese chloride, 1 mmol/L sodium vanadate) before incubation in 20 μl of fresh kinase buffer containing 5 μCi 32P-(γ)-ATP for 15 minutes at 25°C. Kinase reactions were stopped by addition of sodium dodecyl sulfate-polyacrylamide gel electrophoresis loading buffer, and 15 μl of each reaction were resolved on 4 to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gradient gels followed by electrophoretic transfer to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and autoradiography. Tyrosylphosphorylation was evaluated by staining the membranes with PY99 anti-phosphotyrosine monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were then stripped and restained with a rabbit anti-ALK polyclonal antiserum (Sanbio, Uden, The Netherlands). Both PY99 and anti-ALK were detected by chemiluminescence (ECL; Pierce, Rockford, IL).
Results
Histology and ALK Expression
The 11 cases showed typical histomorphological features of IMTs, being composed of a variably cellular proliferation of plump, spindle-shaped fibroblasts/myofibroblasts set in a collagenous or myxoid stroma containing numerous lymphocytes and plasma cells. The IMT fibroblasts/myofibroblasts showed mild to moderate variation in nuclear size but there was no significant nuclear hyperchromasia or pleomorphism. Seven of 11 IMTs were ALK immunopositive, all having strong staining of 50 to 90% of the lesional spindle cells (Table 1 ▶ , Figure 1A ▶ ). The ALK immunostaining was cytoplasmic in six cases and nuclear in one, the latter being concentrated in the nuclear membrane with no nucleolar accentuation. ALK immunostaining was not detected in the IMT stromal inflammatory cells (Figure 1A) ▶ .
Table 1.
Clinicopathological Data, ALK Expression, and ALK Fusion Events in 11 IMTs
| Case no. | Age/sex | Tumor location | ALK expression | ALK fusion* |
|---|---|---|---|---|
| 1 | 1 /M | Abdomen | Cytoplasmic | TPM4-ALK |
| 2 | 30 /F | Lung | Cytoplasmic | TPM3-ALK |
| 3 | 23 /F | Abdomen | Cytoplasmic | TPM3-ALK |
| 4 | 8 /M | Abdomen | Cytoplasmic | – |
| 5 | 1 /F | Abdomen | Cytoplasmic | – |
| 6 | 27 /M | Femur | Cytoplasmic | – |
| 7 | 4 /M | Abdomen | Nuclear | – |
| 8 | 40 /M | Paratesticular | – | – |
| 9 | 50 /M | Abdomen | – | – |
| 10 | 50 /F | Lung | – | – |
| 11 | 40 /M | Femur | – | – |
IMT, Inflammatory myofibroblastic tumor; –, not detected.
*By RT-PCR screening for TPM4-ALK and TPM3-ALK.
Figure 1.
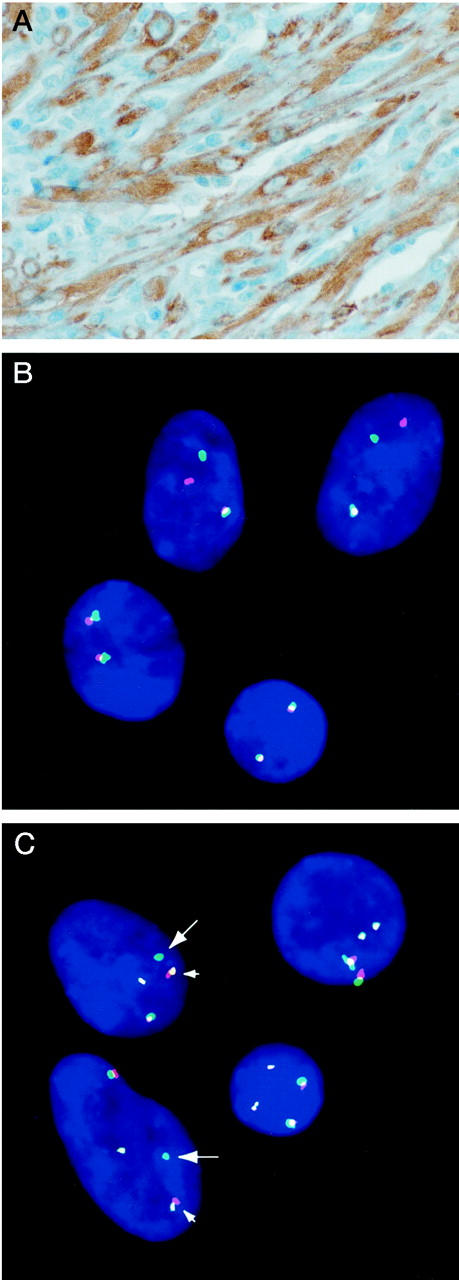
ALK subcellular localization (A) and FISH evaluations (B–C) in IMT case 1: Strong cytoplasmic ALK staining is evident in the spindle cells, whereas ALK is undetectable in the intervening inflammatory cells (A). FISH probes centromeric and telomeric in relationship to ALK are detected with Spectrum Green and Spectrum Orange, respectively (B-C). B: Centromeric and telomeric ALK probes are translocated apart in the top two cells. Bottom two cells, presumably inflammatory, have normal FISH signals. C: A chromosome band 19p13 yeast artificial chromosome clone, 766_E_7, centromeric to the TPM4 gene, is detected with 50:50 Texas Red: fluorescein isothiocyanate. Small arrows indicate colocalized yeast artificial chromosome 766_E_7 (yellow) and telomeric/red ALK probe, whereas large arrows indicate corresponding centromeric/green ALK probe. The two cells at right have normal FISH signals.
Identification of a TPM4-ALK Fusion Gene
Cytogenetic analysis of IMT case 1 demonstrated a translocation involving the chromosome band 2p23 ALK locus region and an unidentified partner chromosome. FISH analysis demonstrated that the 2p23 rearrangement was within 200 kb of the ALK locus (Figure 1B) ▶ . 5′-RACE was then performed to evaluate the possibility of a translocation-related ALK fusion transcript. A single RACE product of ∼1,650 bp was obtained using ALK-specific primers corresponding to the ALK kinase domain. Sequence analysis demonstrated that the 1,650-bp RACE product was an in-frame fusion of the TPM4 and ALK genes. The predicted fusion protein contains all but the 27 most C-terminal amino acids of TPM4, joined (TIDDLE::VYRRKH) to the ALK juxtamembrane and kinase domains. The ALK fusion breakpoint is identical to those in NPM-ALK and TPM3-ALK fusions in anaplastic large-cell lymphoma. 11,17 The TPM4 locus maps to chromosome band 19p13.1, and FISH confirmation of TPM4-ALK fusion was obtained by co-hybridization of a dual-color ALK FISH probe and a yeast artificial chromosome clone, 766_E_7, mapping centromeric to the chromosome band 19p13.1 TPM4 locus (Figure 1C) ▶ .
The first 186 bp of the TPM4-ALK 5′-RACE sequence differs from the TPM4 sequence published previously. Both the variant and previously published TPM4 transcripts contain three TPM signature sequences, and virtually the entire protein sequence, in each case, is expected to form a coiled-coil structure. However, the variant TPM4 transcript lacks the first two in-frame ATG codons found in the published TPM4, and hence has a smaller open reading frame. Alternate TPM4-ALK fusion transcripts containing the variant (designated type 1 hereafter) and published (designated type 2 hereafter) 5′ ends were evaluated by RT-PCR using type 1- (5′-CGAGGCTCCCCCGCCTCGTC-3′) or type 2- (5′-GCCATGGCCGGCCTCAACTCC-3′) specific forward primers and a common reverse primer, ALK/3369/R (5′-TGCAGCTCCATCTGCATGGCTTG-3′). TPM4-ALK type 1 (609 bp) and TPM4-ALK type 2 (716 bp) RT-PCR products were detected reproducibly on each of three separate PCR runs (Figure 2) ▶ and confirmed by sequencing (TPM4-ALK type 1 = GenBank AF186110, and TPM4-ALK type 2 = GenBank AF186109).
Figure 2.

Expression of alternate TPM4-ALK transcripts in IMT case 1. Type 1 TPM4-ALK was identified by RACE, whereas the type 2 transcript was predicted based on published TPM4 sequence. Both type 1 (609 bp) and type 2 (716 bp) transcripts were identified by RT-PCR (A). Primer locations are as schematized (B).
ALK Is Fused to Another TPM Family Member in IMT
The coding sequences for TPM4 and TPM3 are highly homologous, and the TPM3 locus maps to chromosome bands 1q22-23. Notably, IMT case 2 contained a balanced translocation, t(1;2)(q21;p23), suggestive of a TPM3-ALK fusion event. We directed primers to homologous and nonhomologous regions of TPM3 and TPM4, to establish RT-PCR screening approaches detecting both TPM3-ALK and TPM4-ALK fusions (method 1) versus TPM3-ALK fusion only (method 2). Evaluation of 11 IMTs confirmed detection of a TPM-ALK fusion in case 1 (TPM4-ALK) by RT-PCR method 1 but not by method 2 (Figure 3) ▶ . By contrast, fusion sequences, consistent with TPM3-ALK, were identified by methods 1 and 2 in IMT cases 2 and 3 (Figure 3) ▶ . Sequence analysis confirmed TPM3-ALK fusions in the latter two cases. The TPM3-ALK fusion breakpoints were identical to those reported recently in anaplastic large-cell lymphoma, 17 and the TPM3 and ALK breakpoints were homologous and identical, respectively, to the TPM4 and ALK breakpoints in IMT case 1 (Figure 4) ▶ .
Figure 3.
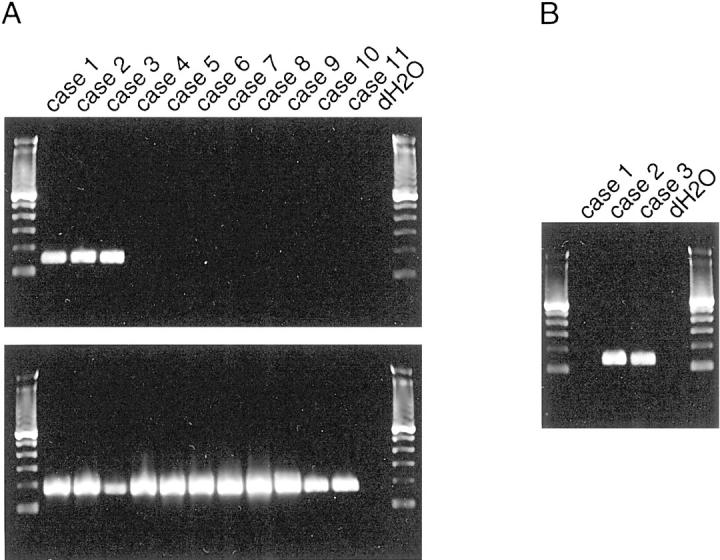
A: Expression of TPM4-ALK and TPM3-ALK in 11 IMT. RT-PCR with universal TPM4/TPM3 primers identifies 147-bp transcripts in IMT cases 1, 2, and 3 (top), whereas positive control (177 bp) GAPDH transcripts are amplified from all cases (bottom). B: RT-PCR with primers specific for TPM3-ALK reveals fusion transcripts (130 bp) in cases 2 and 3. The identity of all transcripts was confirmed by sequencing (data not shown).
Figure 4.

Homology between TPM4-ALK and TPM3-ALK cDNA fusion regions. Sequence mismatches, used to distinguish the TPM4-ALK and TPM3-ALK RT-PCR products, are set in boxes. ALK sequence is shown in a gray box.
TPM4-ALK and TPM3-ALK Are Constitutively Activated
Detection of putative ALK fusion proteins, and evaluation of tyrosine kinase activities, was accomplished by anti-ALK immunoprecipitation in protein lysates from IMT case 1 (TPM4-ALK) and case 2 (TPM3-ALK). In vitro kinase assays revealed similar patterns of putative ALK fusion proteins in the IMT but not in a non-IMT spindle cell sarcoma (Figure 5A) ▶ . Both IMTs contained three kinase-active proteins ranging in size from 80 to 95 kd. Confirmation of tyrosylphosphorylated ALK forms was then obtained by sequential restaining with anti-phosphotyrosine and a polyclonal anti-ALK antibody (Figure 5, B–C) ▶ . Notably, anti-ALK staining also revealed a 200-kd protein, consistent with native ALK, which was expressed strongly and weakly in IMT cases 1 and 2, respectively, and which lacked in vitro kinase activity and tyrosylphosphorylation.
Figure 5.

In vitro kinase (A), phosphotyrosine (B), and ALK (C) evaluations in IMT case 1 (TPM4-ALK), IMT case 2 (TPM3-ALK), and a non-IMT spindle-cell sarcoma. Three kinase-active, tyrosylphosphorylated ALK forms (80 to 95 kd) are seen in the IMT. Native ALK (200 kd), seen in IMT case 1 and also demonstrable in case 2 at longer exposures, lacked constitutive kinase activity and tyrosylphosphorylation.
Discussion
IMT are clinicopathologically distinctive tumors of disputed pathogenesis. 1-4 The studies described herein show that ALK activation is a recurrent oncogenic event in IMTs. ALK activation, in some cases, is accomplished by chromosomal fusion with the TPM4 or TPM3 loci. In other cases, ALK is likely activated by fusion with other genes.
Receptor tyrosine-kinase oncogenes have been described in many tumors, and the usual mechanisms of activation are point mutations or gene fusions that result in constitutive, ligand-independent, receptor oligomerization. Fusion receptor tyrosine-kinase oncoproteins are typically composed of an N-terminal sequence, encoded by the non-RTK member of the fusion pair, containing one or more oligomerization domains and replacing the RTK transmembrane and/or extracellular domains. Our studies indicate that either TPM3 or TPM4 can contribute oligomerization domains to ALK fusion oncoproteins in IMTs. Notably, the TPM3 sequences contained in IMT ALK oncoproteins are identical to those found in TPM3-NTRK1 oncoproteins in papillary thyroid carcinoma. 18 TPM3 contributes a coiled-coil self-association domain to the TPM3-NTRK1 oncoprotein and consequently enables constitutive oligomerization and NTRK1 kinase activation. Moreover, while our studies were in progress, TPM3-ALK oncogenic fusions, containing the same TPM3 sequence, were reported in a subset of anaplastic large-cell lymphomas. 17 The TPM3-ALK and TPM4-ALK IMT fusion genes are highly homologous in that the TPM gene sequences in each case encode a related series of coiled-coil domains. In addition, the fusion breakpoints are found in the corresponding location for both genes (Figure 4) ▶ . TPM3-ALK and TPM4-ALK are constitutively active oncoproteins, as evidenced by high-level, ligand-independent, kinase activity and resultant tyrosylphosphorylation (Figure 5) ▶ .
TPM3 and TPM4 are highly expressed in mesenchymal cell lineages: hence, both genes are expected to contribute active promoter regions to the IMT ALK fusion genes. 19-21 Physiological ALK expression, by contrast, is primarily restricted to the central nervous system and is low, specifically, in nonneoplastic mesenchymal cells. 12 Nonetheless, our studies reveal 200-kd ALK, consistent with expression of a native, nonfusion, ALK protein and encoded presumably by the nontranslocated allele, in neoplastic IMT myofibroblasts (Figure 5C) ▶ . Therefore, ALK signaling pathways might be important biologically in IMT nonneoplastic progenitor cells. These pathways, when activated constitutively, might contribute to neoplastic transformation. It will be intriguing to determine whether other myofibroblastic neoplasms, particularly those arising in children and young adults, result from ALK activation. A speculative, but intriguing, aspect of the TPM-ALK oncogenic mechanism is the possible interference, through heterodimerization, with normal TPM cellular functions. Although there is no direct evidence for a dominant-negative TPM-ALK mechanism, several observations indicate a tumor suppressor role for certain TPM isoforms. It is known that TPM expression can suppress the transformed phenotype in both src- and ras-transformed fibroblasts. 22 Furthermore, TPMs are down-modulated, in part through decreased synthesis, in various retrovirus-induced neoplasms. 23,24 These findings suggest that interference with normal TPM function, which would potentially result from heterodimerization with TPM-ALK oncoproteins, might contribute to neoplastic transformation of IMT progenitor cells.
Both TPM4-ALK and TPM3-ALK fusions were identified in this series of IMTs (Table 1) ▶ . Myofibroblasts with TPM4-ALK and TPM3-ALK fusions featured intense, predominantly cytoplasmic, ALK staining patterns (Figure 1A) ▶ , in keeping with that reported for the TPM3-ALK oncoprotein in anaplastic large-cell lymphoma. 17 Four other IMTs expressed ALK at high levels but did not seem to contain TPM-ALK (cases 4 to 7; Table 1 ▶ and Figure 3 ▶ ) or NPM-ALK (data not shown) fusions. These cases included three in which ALK localization was cytoplasmic (cases 4, 5, and 6) and one in which ALK localization was seen mainly in the nuclear membrane (case 7). None of the cases demonstrated the nucleolar localization typical of NPM-ALK fusion oncoproteins. Previous studies, in anaplastic large-cell lymphoma, have shown that cellular sublocalization patterns, for the various ALK fusion oncoproteins, are consistent and distinctive. 25-27 In some cases, eg, NPM-ALK, a characteristic localization results from shuttling of fusion oncoprotein heterodimers to those cellular structures with which the ALK fusion partner is normally associated. 28 The subcellular localization patterns establish morphological signatures for the different ALK fusion proteins, notwithstanding the fact that the major site of oncogenic action—at least with respect to activation of ALK signaling pathways—is undoubtedly cytoplasmic. 26-28 Therefore, our data imply the existence of at least two alternative ALK fusion mechanisms in IMTs, and these putative fusions likely involve proteins that are predominantly cytoplasmic (cases 4, 5, and 6) and predominantly nuclear (case 7) in their native localization.
The studies described herein are of clinical relevance inasmuch as the clinical behavior of IMT is often difficult to predict. IMTs can be confused with various benign and malignant spindle-cell neoplasms or, alternatively, with various inflammatory nonneoplastic processes. Consequently, evaluation of ALK rearrangement and ALK expression will likely be useful in distinguishing IMTs from other myofibroblastic proliferations. Our preliminary studies show that ALK is not expressed in a variety of non-IMT lesions, including nodular fasciitis, inflammatory liposarcoma, inflammatory leiomyosarcoma, and angiomatoid malignant fibrous histiocytoma, which—akin to IMT—are characterized by neoplastic spindle cells admixed with an inflammatory component (CDM Fletcher, unpublished data). Based on our initial data, it seems that a substantial subset of IMTs will contain TPM-ALK fusions, whereas other cases will likely contain ALK fusions involving genes other than TPM3 and TPM4. Still other IMTs, as evidenced by the absence of immunohistochemical ALK staining in a subset of cases (Table 1 ▶ , cases 8 to 11), probably lack ALK fusion genes. This pathogenetic heterogeneity is reminiscent of that in anaplastic large-cell lymphomas, where tumors are increasingly divided into ALK-positive and ALK-negative categories. Indeed, there is a growing sense that ALK status (rearranged and expressed versus wild-type and nonexpressed) discriminates between two fundamentally different types of anaplastic large-cell lymphomas. 29-31 Notably, the four ALK-negative IMTs in this series (Table 1) ▶ were diagnosed in 40- to 50-year-old patients, whereas the seven ALK-positive IMTs were diagnosed in patients younger than 30 years old. These data suggest that IMTs in children and young adults might depend generally on ALK activation, whereas IMTs in older adults might represent a different disease and/or have different transforming mechanisms. However, additional studies are required to determine whether ALK status correlates with histopathological features and clinical behavior in IMT.
It is intriguing that ALK fusion genes, including an identical TPM3-ALK fusion, are found in both anaplastic large-cell lymphoma and IMTs. 11,17,32-34 However, the preferred ALK fusion partners differ in anaplastic large-cell lymphoma and IMT. NPM-ALK fusions, found in 80% of anaplastic large-cell lymphomas, have not yet been identified in IMTs, and TPM3-ALK fusions are rare in anaplastic large-cell lymphoma. Anaplastic large-cell lymphoma and IMT are associated classically with systemic signs, particularly fever, and one wonders whether this is a direct consequence of unregulated and/or unscheduled ALK signaling. Recent studies show that specific RTK activating mechanisms can bring about transformation of different human cell lineages. For example, activating KIT point mutations are found in human mast cell neoplasms, myeloid leukemia, germ cell tumors, and gastrointestinal stromal tumors, 35-38 and ETV6-NTRK3 fusion oncogenes are found in both myeloid leukemia and congenital fibrosarcoma. 39,40 Irrespective, our studies reveal the first example of an identical RTK fusion oncogene in lymphoid and mesenchymal neoplasia. This finding is in keeping with the observation that activated RTKs are generally transforming in both lymphoid (Ba/F3 cell) and mesenchymal (3T3 cell) models, in vitro. In summary, we have demonstrated a recurring oncogenic mechanism, involving ALK activation, in IMTs. These findings highlight the substantial relevance of RTK mechanisms in human mesenchymal neoplasia, and they underscore that such mechanisms are not lineage-specific.
Footnotes
Address reprint requests to Jonathan A. Fletcher, M.D., Department of Pathology, Brigham and Women’s Hospital, 75 Francis St., Boston, MA 02115. E-mail: jfletcher@rics.bwh.harvard.edu.
References
- 1.Meis JM, Enzinger FM: Inflammatory fibrosarcoma of the mesentery and retroperitoneum. A tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol 1991, 15:1146-1156 [DOI] [PubMed] [Google Scholar]
- 2.Coffin CM, Watterson J, Priest JR, Dehner LP: Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol 1995, 19:859-872 [DOI] [PubMed] [Google Scholar]
- 3.Coffin CM, Dehner LP, Meis-Kindblom JM: Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma, and related lesions: an historical review with differential diagnostic considerations. Semin Diagn Pathol 1998, 15:102-110 [PubMed] [Google Scholar]
- 4.Hasegawa SL, Schofield DE, Fletcher CDM: Inflammatory myofibroblastic tumor or inflammatory fibrosarcoma? Pathol Case Review 1998, 3:128-134 [Google Scholar]
- 5.Treissman SP, Gillis DA, Lee CL, Giacomantonio M, Resch L: Omental-mesenteric inflammatory pseudotumor. Cytogenetic demonstration of genetic changes and monoclonality in one tumor. Cancer 1994, 73:1433-1437 [DOI] [PubMed] [Google Scholar]
- 6.Snyder CS, Dell’Aquila M, Haghighi P, Baergen RN, Suh YK, Yi ES: Clonal changes in inflammatory pseudotumor of the lung: a case report. Cancer 1995, 76:1545-1549 [DOI] [PubMed] [Google Scholar]
- 7.Hojo H, Newton WA, Jr, Hamoudi AB, Qualman SJ, Wakasa H, Suzuki S, Jaynes F: Pseudosarcomatous myofibroblastic tumor of the urinary bladder in children: a study of 11 cases with review of the literature. An Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol 1995, 19:1224–1236 [DOI] [PubMed]
- 8.Sciot R, Dal Cin P, Fletcher CD, Hernandez JM, Garcia JL, Samson I, Ramos L, Brys P, Van Damme B, Van den BH: Inflammatory myofibroblastic tumor of bone: report of two cases with evidence of clonal chromosomal changes. Am J Surg Pathol 1997, 21:1166–1172 [DOI] [PubMed]
- 9.Su LD, Atayde-Perez A, Sheldon S, Fletcher JA, Weiss SW: Inflammatory myofibroblastic tumor: cytogenetic evidence supporting clonal origin. Mod Pathol 1998, 11:364-368 [PubMed] [Google Scholar]
- 10.Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ: Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res 1999, 59:2776-2780 [PubMed] [Google Scholar]
- 11.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT: Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263:1281-1284 [DOI] [PubMed] [Google Scholar]
- 12.Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, Delsol G, Mason DY: Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997, 89:1394-1404 [PubMed] [Google Scholar]
- 13.Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP: ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin’s lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997, 14:2175-2188 [DOI] [PubMed] [Google Scholar]
- 14.Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T: Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997, 14:439-449 [DOI] [PubMed] [Google Scholar]
- 15.Xiao S, Renshaw AA, Cibas ES, Hudson TJ, Fletcher JA: Novel fluorescence in situ hybridization approaches in solid tumors: characterization of frozen specimens, touch preparations, and cytologic preparations. Am J Pathol 1995, 147:896-904 [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson DP, Hayden JD, Quirke P: PCR, A Practical Approach. 1994:pp 29-50 Oxford University Press, New York
- 17.Lamant L, Dastugue N, Pulford K, Delsol G, Mariame B: A new fusion gene TPM3-ALK in anaplastic large cell lymphoma created by a (1;2)(q25;p23) translocation. Blood 1999, 93:3088-3095 [PubMed] [Google Scholar]
- 18.Butti MG, Bongarzone I, Ferraresi G, Mondellini P, Borrello MG, Pierotti MA: A sequence analysis of the genomic regions involved in the rearrangements between TPM3 and NTRK1 genes producing TRK oncogenes in papillary thyroid carcinomas. Genomics 1995, 28:15-24 [DOI] [PubMed] [Google Scholar]
- 19.Prasad GL, Fuldner RA, Braverman R, McDuffie E, Cooper HL: Expression, cytoskeletal utilization and dimer formation of tropomyosin derived from retroviral-mediated cDNA transfer. Metabolism of tropomyosin from transduced cDNA. Eur J Biochem 1994, 224:1-10 [DOI] [PubMed] [Google Scholar]
- 20.Novy RE, Lin JL, Lin CS, Lin JJ: Human fibroblast tropomyosin isoforms: characterization of cDNA clones and analysis of tropomyosin isoform expression in human tissues and in normal and transformed cells. Cell Motil Cytoskeleton 1993, 25:267-281 [DOI] [PubMed] [Google Scholar]
- 21.MacLeod AR, Houlker C, Reinach FC, Smillie LB, Talbot K, Modi G, Walsh FS: A muscle-type tropomyosin in human fibroblasts: evidence for expression by an alternative RNA splicing mechanism. Proc Natl Acad Sci USA 1985, 82:7835-7839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prasad GL, Masuelli L, Raj MH, Harindranath N: Suppression of src-induced transformed phenotype by expression of tropomyosin-1. Oncogene 1999, 18:2027-2031 [DOI] [PubMed] [Google Scholar]
- 23.Janssen RA, Veenstra KG, Jonasch P, Jonasch E, Mier JW: Ras- and Raf-induced down-modulation of non-muscle tropomyosin are MEK-independent. J Biol Chem 1998, 273:32182-32186 [DOI] [PubMed] [Google Scholar]
- 24.Cooper HL, Feuerstein N, Noda M, Bassin RH: Suppression of tropomyosin synthesis, a common biochemical feature of oncogenesis by structurally diverse retroviral oncogenes. Mol Cell Biol 1985, 5:972-983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pittaluga S, Wiodarska I, Pulford K, Campo E, Morris SW, Van den BH, Wolf-Peeters C: The monoclonal antibody ALK1 identifies a distinct morphological subtype of anaplastic large cell lymphoma associated with 2p23/ALK rearrangements. Am J Pathol 1997, 151:343–351 [PMC free article] [PubMed]
- 26.Falini B, Pulford K, Pucciarini A, Carbone A, Wolf-Peeters C, Cordell J, Fizzotti M, Santucci A, Pelicci PG, Pileri S, Campo E, Ott G, Delsol G, Mason DY: Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood 1999, 94:3509-3515 [PubMed] [Google Scholar]
- 27.Pulford K, Falini B, Cordell J, Rosenwald A, Ott G, Muller-Hermelink HK, MacLennan KA, Lamant L, Carbone A, Campo E, Mason DY: Biochemical detection of novel anaplastic lymphoma kinase proteins in tissue sections of anaplastic large cell lymphoma. Am J Pathol 1999, 154:1657-1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bischof D, Pulford K, Mason DY, Morris SW: Role of the nucleophosmin (NPM) portion of the non-Hodgkin’s lymphoma-associated NPM-anaplastic lymphoma kinase fusion protein in oncogenesis. Mol Cell Biol 1997, 17:2312-2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiota M, Nakamura S, Ichinohasama R, Abe M, Akagi T, Takeshita M, Mori N, Fujimoto J, Miyauchi J, Mikata A: Anaplastic large cell lymphomas expressing the novel chimeric protein p80NPM/ALK: a distinct clinicopathologic entity. Blood 1995, 86:1954-1960 [PubMed] [Google Scholar]
- 30.Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, Verhoef G, Menestrina F, Todeschini G, Paulli M, Lazzarino M, Giardini R, Aiello A, Foss HD, Araujo I, Fizzotti M, Pelicci PG, Flenghi L, Martelli MF, Santucci A: ALK+ lymphoma: clinico-pathological findings and outcome. Blood 1999, 93:2697-2706 [PubMed] [Google Scholar]
- 31.Gascoyne RD, Aoun P, Wu D, Chhanabhai M, Skinnider BF, Greiner TC, Morris SW, Connors JM, Vose JM, Viswanatha DS, Coldman A, Weisenburger DD: Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood 1999, 93:3913-3921 [PubMed] [Google Scholar]
- 32.Pileri SA, Pulford K, Mori S, Mason DY, Sabattini E, Roncador G, Piccioli M, Ceccarelli C, Piccaluga PP, Santini D, Leone O, Stein H, Falini B: Frequent expression of the NPM-ALK chimeric fusion protein in anaplastic large-cell lymphoma, lympho-histiocytic type. Am J Pathol 1997, 150:1207-1211 [PMC free article] [PubMed] [Google Scholar]
- 33.Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, Haralambieva E, Pulford K, Pileri S, Morris SW, Mason DY, Delsol G: ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 1998, 91:2076-2084 [PubMed] [Google Scholar]
- 34.Rosenwald A, Ott G, Pulford K, Katzenberger T, Kuhl J, Kalla J, Ott MM, Mason DY, Muller-Hermelink HK: t(1;2)(q21;p23) and t(2;3)(p23;q21): two novel variant translocations of the t(2;5)(p23;q35) in anaplastic large cell lymphoma. Blood 1999, 94:362-364 [PubMed] [Google Scholar]
- 35.Nakahara M, Isozaki K, Hirota S, Miyagawa J, Hase-Sawada N, Taniguchi M, Nishida T, Kanayama S, Kitamura Y, Shinomura Y, Matsuzawa Y: A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology 1998, 115:1090-1095 [DOI] [PubMed] [Google Scholar]
- 36.Kanakura Y, Furitsu T, Tsujimura T, Butterfield JH, Ashman LK, Ikeda H, Kitayama H, Kanayama Y, Matsuzawa Y, Kitamura Y: Activating mutations of the c-kit proto-oncogene in a human mast cell leukemia cell line. Leukemia 1994, 8:S18-S22 [PubMed] [Google Scholar]
- 37.Tian Q, Frierson HF, Jr, Krystal GW, Moskaluk CA: Activating c-kit gene mutations in human germ cell tumors. Am J Pathol 1999, 154:1643-1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakata Y, Kimura A, Katoh O, Kawaishi K, Hyodo H, Abe K, Kuramoto A, Satow Y: c-kit point mutation of extracellular domain in patients with myeloproliferative disorders. Br J Haematol 1995, 91:661-663 [DOI] [PubMed] [Google Scholar]
- 39.Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH: A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998, 18:184-187 [DOI] [PubMed] [Google Scholar]
- 40.Eguchi M, Eguchi-Ishimae M, Tojo A, Morishita K, Suzuki K, Sato Y, Kudoh S, Tanaka K, Setoyama M, Nagamura F, Asano S, Kamada N: Fusion of ETV6 to neurotrophin-3 receptor TRKC in acute myeloid leukemia with t(12;15)(p13;q25). Blood 1999, 93:1355-1363 [PubMed] [Google Scholar]