

Abstract
Abnormal accumulation of the presynaptic protein α-synuclein has recently been implicated in the pathogenesis of Alzheimer’s and Parkinson’s diseases. Because neurodegeneration in these conditions might be associated with mitochondrial dysfunction and oxidative stress, the effects of α-synuclein were investigated in a hypothalamic neuronal cell line (GT1-7). α-Synuclein overexpression in these cells resulted in formation of α-synuclein-immunopositive inclusion-like structures and mitochondrial alterations accompanied by increased levels of free radicals and decreased secretion of gonadotropin-releasing hormone. These alterations were ameliorated by pretreatment with anti-oxidants such as vitamin E. Taken together these results suggest that abnormal accumulation of α-synuclein could lead to mitochondrial alterations that may result in oxidative stress and, eventually, cell death.
Alpha-synuclein (or the precursor of the non-Aβ component of Alzheimer’s disease amyloid) is a 19-kd presynaptic protein 1-3 from which a 35-amino acid peptide is derived and found in Alzheimer’s disease plaques. 2 α-Synuclein belongs to an expanding family of synuclein proteins and is homologous to rat and torpedo α-synuclein 4 and the zebra finch synelfin. 5 Other members of the synuclein family include β-synuclein 6 and γ-synuclein, or persyn 7,8 and a recently identified protein, synoretin. 9 Recent studies have shown that mutations in the α-synuclein gene lead to rare familial forms of Parkinson’s disease (PD), a disorder characterized by nigral degeneration and formation of inclusions, denominated Lewy bodies (LBs). In these two reports, Polymeropoulos et al 10 identified a mutation (A53T) in an Italian and Greek kindred which conferred susceptibility to early-onset PD. Subsequent analysis by Kruger et al 11 revealed another missense mutation (A30P) found in a German familial case of PD. Furthermore, in the more frequent forms of sporadic PD and other forms of LB disease there is significant α-synuclein aggregation in LBs and neurites. 12-17
The mechanisms through which α-synuclein might lead to neurodegeneration in LB disease are not yet completely understood. Recent studies have shown that overexpression of wild-type and mutant α-synuclein leads to synaptic degeneration 18 and cell death 19,20 in both in vivo and in vitro model systems. The role of abnormal expression of α-synuclein in LB disease is currently being explored. However, in vitro studies have shown that increasing levels, 21 mutations, 22 and oxidative stress 23 conditions lead to α-synuclein aggregation. Furthermore, abnormal accumulation of α-synuclein might trigger mitochondrial dysfunction. Supporting this, there is ample evidence for a central role for mitochondrial dysfunction in the pathogenesis of PD. 24-28 In this context, the main objective of the present study was to determine whether overexpression of α-synuclein might dysregulate cell function via mitochondrial alterations. For this purpose, a murine hypothalamic tumor cell line (GT1-7) 29 was transfected with α-synuclein and analyzed for pathological and physiological effects. We present evidence that overexpression of α-synuclein in this cell system leads to mitochondrial abnormalities and corresponding changes in oxidative stress markers. These studies support the contention that α-synuclein might play a role in the regulation of mitochondrial function and that alterations in this pathway may be involved in the pathogenesis of PD.
Materials and Methods
Cell Culture
The GT1-7 murine hypothalamic tumor cell line (kindly provided by Dr. Pam Mellon, Department of Neurosciences, University of California at San Diego) was selected because of its neuronal origin, expression of many neuronal markers, ability to form synapses in culture, and neurosecretory properties which may represent neurotransmitter-like activity. 29 Cells were maintained at 37°C, 5% CO2 in Dulbecco’s modified Eagle’s medium (high glucose), supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), and 1% v/v penicillin/streptomycin.
cDNA Subcloning
The murine α-synuclein cDNA sequence was obtained as previously described. 30 Briefly, using this plasmid as a template, the coding region was once again amplified except that a simple polymerase chain reaction was used with the annealing temperature of 55°C instead of the touchdown program. The amplified product was run out on a 0.8% agarose gel, purified via the phenol/chloroform extraction technique, and ligated into a pCR3.1 T-vector (Invitrogen, Carlsbad, CA). The clones, which contained either the sense or antisense construct, were selected, verified by sequencing, and amplified via the Qiagen Maxi-Prep kit (Qiagen, Valencia, CA). These clones were referred to as #1 and #9B, respectively.
Transfection and Selection of Stable Transfectants
GT1-7 cells were seeded to ∼50% confluence in 60-mm tissue culture dishes. The Superfect transfection kit (Qiagen) was used to transfect cells with plasmids #1, #9B, and pCR3.1 (vector alone). Briefly, plasmid DNA (7.5 μg) was diluted with 200 μl of serum-free OPTI-MEM (Life Technologies, Inc., Grand Island, NY) and 40 μl of the Superfect reagent was added to this mix. Cells were incubated for 3 hours at 37°C, washed in phosphate-buffered saline (PBS), and cultured for 12 hours in regular cell media.
The optimal concentration of G418 sulfate needed for complete cell death in GT1-7 untransfected cells was obtained after a 3-day treatment with various concentrations. G418 (300 μg/ml) was added to the regular media after transfection and replaced every 3 days. After 3 weeks, cloning rings were used to select colonies and cells were screened for expression via the ribonuclease protection assay (RPA), Western blot, and immunocytochemical analysis. After the initial screening, essentially four clones were selected: 1) sense-transfected (overexpressers), 2) antisense-transfected (underexpressers), 3) vector (VEC)-transfected, and 4) nontransfected wild-type controls.
Analysis of α-Synuclein mRNA Levels by RPA
Briefly, as previously described, riboprobe templates were amplified by polymerase chain reaction. 30 For the murine α-synuclein template, primers (20 mers) started at nucleotide 145 and nucleotide 490 of mouse α-synuclein (GenBank Accession No. AF044672), respectively. An actin riboprobe complementary to nucleotides 480 to 559 of mouse β-actin was used (GenBank Accession No. M18194). RPAs were carried out with [32P]-labeled antisense riboprobes, and signals were quantitated with a PhosphorImager and ImageQuant software, as previously described. 31 Actin signals were used to correct for variations in mRNA content and loading.
Analysis of α-Synuclein Protein Expression by Western Blot
Western blots were analyzed with the affinity-purified rabbit polyclonal antibody directed against C-terminal α-synuclein (amino acids 131 to 140). 32 Briefly, cell pellets were sonicated and separated into cytosolic and particulate fractions 33 and protein levels were determined by the method of Lowry et al. 34 Gels were blotted onto nitrocellulose paper and incubated with antibodies against α-synuclein, (1:1000) followed by 125I-protein A. Blots were also stripped and reprobed with a mouse monoclonal antibody against actin (Chemicon, Temecula, CA) to correct for loading variability. The specific19-kd signals corresponding to murine α-synuclein were quantified using the ImageQuant software.
Analysis of α-Synuclein Immunolocalization by Double-Immunocytochemistry and Laser Scanning Confocal Microscopy (LSCM)
Briefly, as previously described 35 cells were seeded onto poly-l-lysine-coated glass coverslips, grown to 60% confluence, fixed in 4% paraformaldehyde (30 minutes, −4°C), and pretreated with 0.1%Triton X-100 in PBS (20 minutes, room temperature). Cells were incubated overnight at 4°C with antibodies against α-synuclein (rabbit polyclonal) and microtubule-associated protein 2 (mouse monoclonal) (Roche Molecular Biochemicals, Indianapolis, IN). Cells were then incubated with the biotinylated anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA), followed by tyramide signal amplification-Direct Red (Dupont-NEN, Boston, MA) and fluorescein isothiocyanate-conjugated anti-mouse secondary antibody (Vector). Wild-type and VEC-transfected GT1-7 cells were run in parallel as negative controls. Coverslips were air-dried overnight, mounted on slides with antifading media (Vectashield, Vector), and imaged with the confocal microscope (MRC1024; BioRad).
Gonadotropin-Releasing Hormone (GnRH) Radioimmunoassay
To determine whether levels of α-synuclein expression affected the secretory activity of GT1-7 cells, levels of GnRH release were investigated by radioimmunoassay, essentially as previously described. 36 Briefly, tubes were incubated overnight at 4°C with 200 μl of 0.1%Triton-PO4 buffer (0.063 mol/L Na2HPO4, 0.013 mol/L ethylenediaminetetraacetic acid, 0.02% NaN3, 0.1% Triton X-100, pH 7.4), 1% normal rabbit serum, and 100 μl of primary SD-7 antibody, (1:30,000) followed by 100 μl of 125I-GnRH (5000 to 6000 cpm; Dupont-NEN) was added to each tube. On the third day, 100 μl of goat anti-rabbit γ globulin secondary antibody (1:25, Calbiochem, San Diego, CA) in 50 mmol/L ethylenediaminetetraacetic acid buffer (in 0.1% Triton buffer) and 1 ml of 6% polyethylene glycol solution was added per tube and again incubated overnight at 4°C. Tubes were then spun down, inverted, dried overnight, and radioactivity was counted in a γ counter. A duplicate number of wells and triplicate numbers of tubes per well were used. Nonspecific binding, maximum binding, standard curve, and GnRH were added to the assay.
3-(4,5-Dimethythiazolyl)-2.5-Diphenyl-2H-Tetrazolium-Bromide (MTT) Assay
To assess the effects of α-synuclein expression on mitochondrial activity, the MTT colorimetric assay was used to determine the level of NADH production within the four cell clones. For vitamin E treatment, α-tocopherol acetate (stock 1:10 in 100% alcohol; Sigma Chemical Co., St. Louis, MO) was further diluted 1:5 in fetal bovine serum (Hyclone, Logan, UT) and then mixed 1:5 with regular cell culture medium. Briefly, after exposure to treatments, the standard medium was replaced with the MTT dye solution (final concentration, 0.9 mg/ml; Sigma) and cells were incubated for 4 hours at 37°C. Then, 100 μl of solubilization solution (50% dimethylformamide/20% sodium dodecyl sulfate) was added overnight and the blue formazan was detected in a plate reader at 560 nm.
Lactase Dehydrogenase Activity Assay
To assess cell viability, the CytoTox 96 assay (Promega, Madison, WI) was used. A minimum of 12 replicates per clone was used per treatment and each experiment included a plate of control cells. Briefly, after 3 hours of incubation, media containing various treatments was replaced with 100 μl of normal media and 20 μl of CytoTox reagent was added per well. Cells were incubated for an additional 2 to 3 hours and counted on a 96-well plate reader at 492 nm. Additional analysis of cell viability was performed using the Trypan blue staining method.
2′-7′-Dichlorofluorescein (DCF) Analysis
To determine whether free radical production was associated with α-synuclein expression, DCF loading analysis was performed. Briefly, cells were cultured on poly-l-lysine (Sigma)-coated glass coverslips in six-well dishes to 80% confluency. The cell permeable dye, 2′7′-dichlorodihydrofluorescein diacetate (H2DCFDA, 75 μmol/L) (Molecular Probes, Eugene, OR) was added to the media and cells were incubated at 37°C, 5% CO2 for 15 minutes. Loaded cells were then imaged with the LSCM.
Glutathione Assay
Because it has been proposed that glutathione is the primary cellular anti-oxidant response to free radical production, we determined if levels of glutathione were affected in the four clones expressing different levels of α-synuclein. Briefly, as previously described, 37 cells were collected with 1× pancreatin and lysed with cold 10% sulfosalicylic acid. Lysates were incubated on ice for 15 minutes and supernatants were collected by centrifugation. After neutralization with triethanolamine (diluted 1:4), the concentration of total glutathione was determined. In addition, a standard curve of glutathione (0 to 2 nmol/L range; Sigma) was constructed. The A412 was measured at intervals of 0 seconds, 30 seconds, 60 seconds, and 90 seconds, and the total protein cell pellet was dissolved in 50 μl of 0.5 mol/L NaOH. Total protein was measured using the Protein Assay Kit (Pierce, Rockford, IL) and compared to a standard bovine serum albumin curve. Final concentrations of glutathione were obtained via linear regression analysis and expressed as nmol/mg protein.
Electron Microscopy
GT1-7 cells were grown in Lux Permanox culture dishes (Nalgene Nunc International, Naperville, IL) until confluence. Cells were fixed in 1% glutaraldehyde, 2% paraformaldehyde in 0.1 mol/L Na cacodylate buffer (pH 7.4), postfixed in 1% osmium tetroxide, and stained with saturated uranyl acetate in 50% ethanol. The cell layer was dehydrated through a graded series of ethanols to 90% ethanol; 2-hydroxypropyl methacrylate was the intermediate solvent. All infiltrations of 2-hydroxypropyl methacrylate and Scipoxy 812 resin (Energy Beam Sciences, Agawam, MA) were carried out on a shaker at slow speed. After two changes of 100% resin the plates were polymerized in a 65°C oven for 24 hours, the plastic was detached and selected areas were sawed from the plates and glued on dummy blocks. Thin sections (80 nm) were cut on a Reichert Ultracut E Ultramicrotome (Leica, Vienna, Austria), picked up on 200-mesh copper grids, and poststained in ethanolic uranyl acetate followed by bismuth subnitrite. The sections were viewed and photographed with a Zeiss EM10 electron microscope.
Statistical Analysis
To ascertain differences among the various cell lines and treatment groups, one-way analysis of variance (ANOVA) with post hoc Scheffé test was performed using the SuperANOVA program (SAS Institute, Cary, NC).
Results
Establishment of α-Synuclein-Transfected Cell Lines
RPA analysis showed that cells transfected with the sense construct (ST) expressed eightfold to ninefold in- creased levels of murine mRNA α-synuclein compared to VEC-transfected and nontransfected control cells. In contrast, cells transfected with antisense (AST) showed a 50% reduction in α-synuclein mRNA levels (Figure 1 ▶ , A and B). To determine whether the increase in mRNA levels was accompanied by similar changes at the protein level, Western blot analysis was performed. Consistent with previous studies 32 α-synuclein immunoreactivity in GT1-7 cells was identified as an 18-kd band both in cytosolic (Figure 1C) ▶ and particulate fractions (not shown). Semiquantitative Western blot analysis by PhosphorImager of α-synuclein signals showed that ST cells expressed twofold over control, whereas AST cells expressed 50% of control levels (Figure 1D) ▶ .
Figure 1.

Characterization of patterns of murine α-synuclein expression in GT1-7 cells. A: Representative autoradiogram of the RPA showing murine α-synuclein mRNA levels in C (control nontransfected), AST (antisense transfected), VEC (vector-transfected), and ST (sense-transfected) cells. Protected fragment for murine α-synuclein was detected at 385 bp, whereas actin was detected at 85 bp. The left-most lane shows signal of undigested (u/d) radiolabeled riboprobes, whereas the other lanes contain riboprobes of RNase-digested samples. B: PhosphorImager analysis of murine α-synuclein mRNA signals (expressed as percent over control) showing that ST cells expressed ninefold over control, whereas AST cells expressed 50% of control levels. C: Representative autoradiogram of the Western blot showing that the α-synuclein antibody recognized a band at ∼18 kd. The left-most lane shows signal of the recombinant α-synuclein protein. C, control nontransfected; AST, antisense transfected; VEC, vector-transfected; and ST, sense-transfected cells. D: PhosphorImager analysis of α-synuclein signals from semiquantitative Western blots showing, in the cytosolic fractions, that ST cells expressed twofold over control, whereas AST cells expressed 50% of control levels.
Immunocytochemical and Ultrastructural Analyses Reveal Inclusion-Like Structures and Abnormal Mitochondria in α-Synuclein Overexpressing Cells
LSCM of double-immunolabeled control and VEC-transfected GT1-7 cells showed that with the mouse-specific α-synuclein antibody there was a moderate granular cytoplasmic immunoreactivity (Figure 2A) ▶ . In contrast, ST cells showed intense immunoreactivity associated with granular structures (Figure 2 ▶ ; B, C, and E) and dense cytoplasmic aggregates (Figure 2, F–G) ▶ . Control experiments where the primary antibodies were inactivated show no immunoreactivity (Figure 2D) ▶ . AST cells showed microtubule-associated protein 2 immunoreactivity, but mild or no α-synuclein immunostaining (Figure 2H) ▶ . Analysis of the ST cells by transmitted electron microscopy showed that, compared to nontransfected cells (Figure 3, A and B) ▶ , some of the mitochondria were enlarged (Figure 3, C and D) ▶ and displayed abnormal vacuolized cristae (Figure 3, E and F) ▶ . In addition, some of the ST cells presented aggregates of electrodense material localized mainly in the perinuclear region (Figure 4, C and D) ▶ . These inclusion-like structures had a diameter of ∼5 to 7 μm and were usually surrounded by laminated bodies. These alterations were not observed in VEC or control cells (Figure 4, A and B) ▶ . Furthermore, some ST cells displayed numerous secondary lysosomes and laminated bodies in their cytoplasm and a relative lack of neuritic processes (Figure 4, E and F) ▶ .
Figure 2.

Confocal analysis of α-synuclein expression in transfected GT1-7 cells. Cells were double-immunolabeled with antibodies against murine α-synuclein (red) and the neuronal marker microtubule-associated protein 2 (green) and imaged with the laser-scanning confocal microscope. A: Control nontransfected cells showed mild punctuate α-synuclein immunostaining associated with the cytoplasm and some neuritic processes. B: ST cells showed intense immunoreactivity associated with granular cytoplasmic structures. C: Detail at higher magnification of the rectangular area presented in (B). D: Negative control with the primary antibodies inactivated. E, F, and G: ST cells showed intense immunoreactivity associated with granular cytoplasmic structures (*) and dense intracellular inclusions (arrows). H: AST cells showed microtubule-associated protein 2 immunoreactivity but minimal or none α-synuclein immunostaining.
Figure 3.
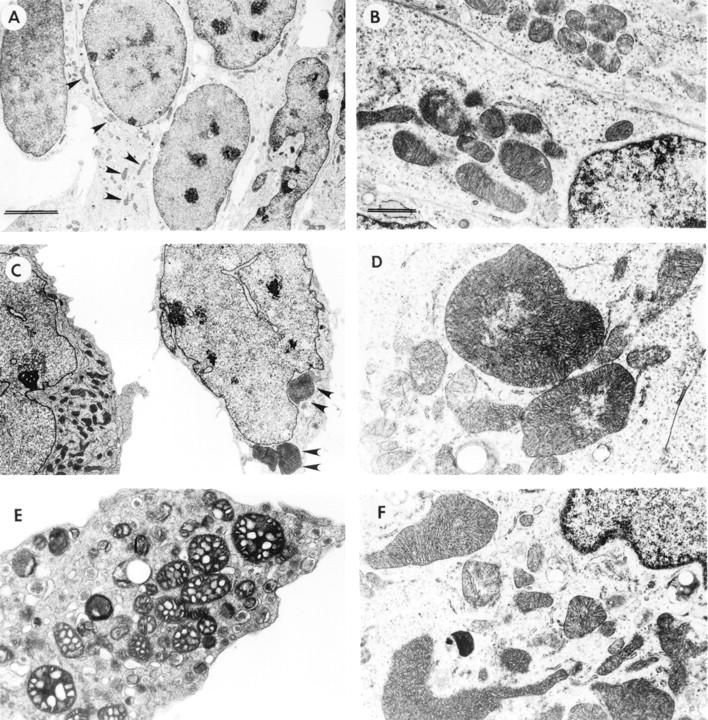
Ultrastructural analysis of mitochondrial alterations in transfected GT1-7 cells. Low power (original magnification, ×5,000; scale bar, 5 μm) (A) and high magnification (original magnification, ×20,000; scale bar, 1 μm) (B) views of vector-transfected cells showing mitochondria of normal characteristics (arrowheads). Low power (original magnification, ×5,000) (C) and high magnification (original magnification, ×20,000) (D) views of ST cells demonstrating abnormally enlarged mitochondria (arrowheads). ST cells also had mitochondria displaying vacuolization of the cristae (original magnification, ×10,000) (E) and distorted morphology (original magnification, ×15,000) (F).
Figure 4.
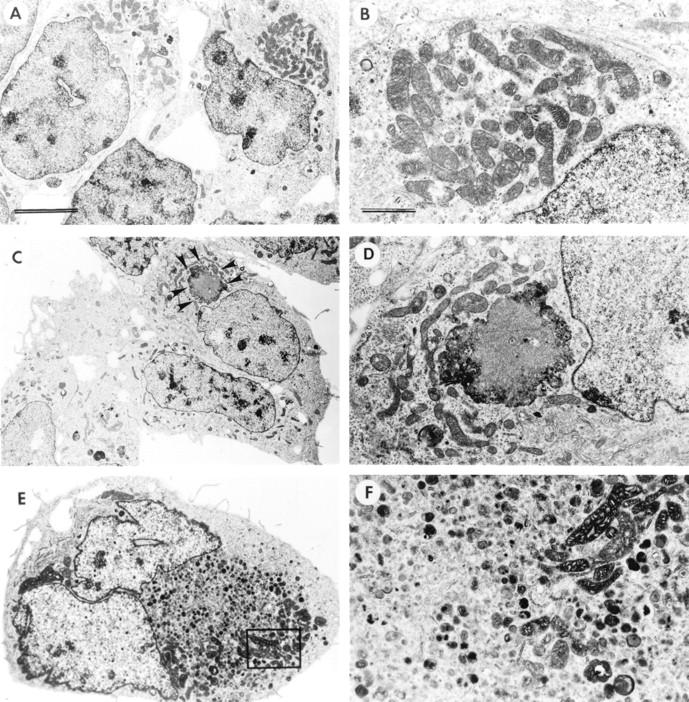
Ultrastructural analysis of other morphological alterations in transfected GT1-7 cells. Low power (original magnification, ×5,000; scale bar, 5 μm) (A) and high magnification (original magnification, ×20,000; scale bar, 1 μm) (B) views of VEC-transfected cells demonstrating normal organelles. Low power (original magnification, ×5,000) (C) and high magnification (original magnification, ×20,000) (D) views of ST cells demonstrating electrodense intracytoplasmic inclusions (arrowheads). E and F: Furthermore ST cells displayed large numbers of secondary lysosomes and laminated bodies in their cytoplasm and complete lack of neuritic processes (original magnification, ×15,000).
Mitochondrial Activity Is Compromised in α-Synuclein Overexpressing Cells
To determine whether structural alterations observed in the mitochondria were associated with functional deficits in the ST cells, the MTT assay (a measure of mitochondrial activity) was performed. In ST cells, this study showed a 20% reduction compared to VEC and control cells (Figure 5) ▶ . Because previous studies have shown that mitochondrial dysfunction might lead to oxidative stress via disruption of the respiratory chain, 27,33,38,39 it is possible that mitochondrial deficiencies might be normalized by treatment with anti-oxidants such as vitamin E. Consistent with this, vitamin E-treated ST cells showed levels similar to VEC and control cells (Figure 5) ▶ . Because it is possible that reduction in mitochondrial activity might be secondary to cell death rather than primarily associated with α-synuclein overexpression, the lactate dehydrogenase assay was performed. No significant differences in cell viability were observed among the clones (data not shown).
Figure 5.

MTT assay in transfected GT1-7 cells. α-Synuclein sense-transfected (ST) cells showed a significant 30% decrease in mitochondrial activity compared to nontransfected control (C). Treatment with the anti-oxidant vitamin E restored normal mitochondrial function in ST cells.
Increased Oxidative Load in α-Synuclein Overexpressing Cells
Because vitamin E pretreatment ameliorated the mitochondrial deficiencies observed in ST cells, we used the membrane permeable dye, DCF to determine whether the cells were under oxidative stress. For this purpose, DCF-loaded GT1-7 cells were imaged with the LSCM. This study showed a mild fluorescent signal in control cells (Figure 6A) ▶ and an increased fluorescent signal in the ST cells (Figure 6C) ▶ . AST cells showed no significant increase in the levels of DCF reactivity (not shown). Experiments where control cells were treated with hydrogen peroxide showed similar increased levels of the fluorescent signal as in the ST cells (Figure 6B) ▶ . To further corroborate that the increased DCF signal observed in the ST cells was associated with generation of oxidative stress, cells were pretreated with vitamin E. This anti-oxidant treatment resulted in a reduction in the signal in ST cells (Figure 6D) ▶ to levels similar to those found in nontransfected control and VEC cells (Figure 6A) ▶ .
Figure 6.
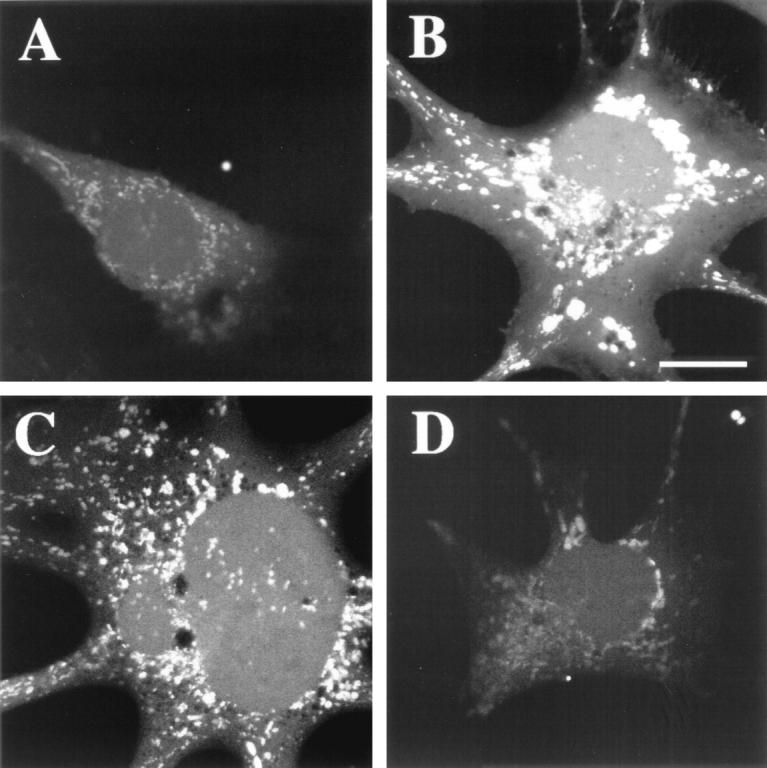
DCF staining in GT1-7-transfected cells. All images were obtained by LSCM. A: Control cells showed mild fluorescence. B: Positive-control experiment where GT1-7 cells were exposed to 1 μmol/L hydrogen peroxide for 15 minutes showed a strong fluorescent signal (scale bar, 10 μm). C: α-Synuclein sense-transfected (ST) cells showed a strong fluorescent signal associated with the vesicular structures, indicative of oxidative stress. D: Treatment with the anti-oxidant vitamin E restored normal mitochondrial function in ST cells.
Because the ST cells responded to vitamin E treatment and the DCF imaging experiment indicated that these cells seemed to be in a heightened state of oxidative stress, but were still equally viable compared to the VEC-transfected and wild-type counterparts, we hypothesized that the selected ST cell clone might be able to compensate for the oxidative stress by producing endogenous anti-oxidants. To investigate this possibility we examined the levels of glutathione expression. The rationale for performing this assay is based on previous studies showing that cells respond to oxidative stress by changing the levels of this anti-oxidative factor. 37 The present study showed that ST cells displayed a 75% increase in levels of glutathione compared to VEC and control cells (Figure 7A) ▶ . AST cells showed no significant alterations in the levels of glutathione expression (Figure 7A) ▶ .
Figure 7.

Glutathione and GnRH levels in transfected GT1-7 cells. A: α-Synuclein sense-transfected (ST) cells showed a significant 85% increase in glutathione levels compared to nontransfected controls (C), vector-transfected cells (VEC), and antisense-transfected (AST) cells. B: α-Synuclein sense-transfected (ST) cells showed a significant 60% decrease in GnRH levels compared to nontransfected control (C). Treatment with the anti-oxidant vitamin E restored normal levels of GnRH secretion in ST cells.
GnRH Secretion Is Decreased in α-Synuclein Overexpressing Cells
To further confirm that the mitochondrial alterations and oxidative stress conditions affected the function of ST cells, levels of GnRH release were determined by radioimmunoassay. This study showed that in ST cells there was a 60% reduction in the levels of GnRH secretion compared to VEC and control cells and treatment of ST cells with the anti-oxidant vitamin E resulted in normalization of GnRH secretion (Figure 7B) ▶ . GnRH release was within normal limits in AST cells (not shown).
Discussion
The present study showed that overexpression of α-synuclein in GT1-7 cells leads to mitochondrial dysfunction, as reflected by the presence of giant mitochondria, formation of laminated bodies and autophagozomes, and decreased MTT levels. Consistent with this possibility, a recent study has shown that α-synuclein exhibits homology with 14-3-3 proteins and is capable of interacting with BAD, a Bcl-2 homologue that controls mitochondrial function and promotes cell death. 40 In this regard, BAD interacts with Bcl-x1 to form a heterodimer which displaces Bcl-2 leading to release of cytochrome c from mitochondria, which is thought to be one of the earliest steps involved in apoptotic cell death. 41 Because recent studies have also shown that cytochrome c interacts with α-synuclein in LBs, 21,42 then release of this mitochondrial molecule might not only trigger cell death, but also promote α-synuclein aggregation.
As to other potentially important consequences of the α-synuclein-mediated mitochondrial dysfunction, the present study showed that overexpression of α-synuclein in GT1-7 cells resulted in decreased GnRH release and cellular changes consistent with oxidative stress. Regarding the reduced vesicular secretion of GnRH in ST cells, it has been demonstrated that cellular energy stores affect functioning of vesicular transport proteins such as N-ethylmaleimide-sensitive fusion protein. 43 Vesicle activity correlates well with synaptic function or neural plasticity and levels of α-synuclein seem to follow those of other proteins important for synaptogenesis and development. 30 In addition, it has been shown that in the zebra finch model, levels of α-synuclein homologue, synelfin, are developmentally regulated in the brain during critical periods of song learning. 5 Furthermore, α-synuclein has been shown to be capable of binding to synthetic membranes of acidic phospholipids and preferentially to vesicles of smaller diameter suggestive of selective regulation at the nerve terminal. 44 α-Synuclein has also been shown to inhibit the activity of phospholipase D2, 45 an enzyme that has been implicated in the vesicular activity pathway. 46 Taken together these studies suggest that decreased neurosecretory activity in transfected GT1-7 cells may be indirectly associated with mitochondrial dysfunction. Alternatively, the neurosecretory alterations might also be associated with other toxic changes such as generation of radical oxygen species. In this regard, consistent with previous reports showing that mitochondrial dysfunction leads to oxidative stress, 27,47 the present study indicated that α-synuclein overexpression results in mitochondrial alterations, accompanied by increased DCF fluorescence and altered glutathione levels. Furthermore, these alterations were reversible by treatment with the anti-oxidant, vitamin E. It should be noted that whereas we observe an increase in cellular glutathione levels, this is in contrast to the observed decrease in glutathione levels in the brains of patients with PD. 48 It is possible that this difference can be accounted for by the selection process of viable cell clones and corroborates the hypothesis that the clones have altered their cellular anti-oxidant defenses to counter the increased oxidative load.
These results are of significant importance for the understanding of PD pathogenesis, because in this disorder mitochondrial function is significantly compromised as reflected by alterations, among others, in mitochondrial complex I levels 23,25,49-54 and oxidative stress has been postulated to play a central role in the neurodegenerative process in PD. 27 In this context, it is possible that overexpression of α-synuclein leads to mitochondrial alterations which, in turn, lead to generation of oxygen radicals. This is notable in view of recent studies showing that oxidative stress leads to α-synuclein aggregation 23 and that cytochrome c, which is centrally involved in the mitochondrial-associated apoptotic pathway, is closely associated with α-synuclein in LBs and triggers α-synuclein aggregation. 42 These results suggest that oxidative stress might further accelerate α-synuclein aggregation which, in turn, may result in LB formation.
In summary, the present study supports the contention that α-synuclein might be involved in regulating mitochondrial activity and alterations in α-synuclein expression may lead to mitochondrial dysfunction and generation of radical oxygen species, that eventually results in neurodegeneration in conditions associated with LBs such as PD.
Footnotes
Address reprint requests to Dr. E. Masliah, Department of Neurosciences, University of California, San Diego, La Jolla, CA 92093-0624. E-mail: emasliah@ucsd.edu.
Supported by National Institutes of Health grants AG5131 and AG10869.
References
- 1.Maroteaux L, Campanelli JT, Scheller RH: Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 1988, 8:2804-2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Otero D, Kondo J, Ihara Y, Saitoh T: Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1993, 90:11282-11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jakes R, Spillantini MG, Goedert M: Identification of two distinct synucleins from human brain. FEBS Lett 1994, 345:27-32 [DOI] [PubMed] [Google Scholar]
- 4.Maroteaux L, Scheller RH: The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res 1991, 11:335-343 [DOI] [PubMed] [Google Scholar]
- 5.George JM, Jin H, Woods WS, Clayton DF: Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15:361-372 [DOI] [PubMed] [Google Scholar]
- 6.Nakajo S, Tsukada K, Omata K, Nakamura Y, Nakaya K: A new brain-specific 14-kDa protein is a phosphoprotein. Its complete amino acid sequence and evidence for phosphorylation. Eur J Biochem 1993, 217:1057-1063 [DOI] [PubMed] [Google Scholar]
- 7.Ninkina NN, Alimova-Kost MV, Paterson JW, Delaney L, Cohen BB, Imreh S, Gnuchev NV, Davies AM, Buchman VL: Organization, expression and polymorphism of the human persyn gene. Hum Mol Genet 1998, 7:1417-1424 [DOI] [PubMed] [Google Scholar]
- 8.Alimova-Kost MV, Ninkina NN, Imreh S, Gnuchev NV, Adu J, Davies AM, Buchman VL: Genomic structure and chromosomal localization of the mouse persyn gene. Genomics 1999, 56:224-227 [DOI] [PubMed] [Google Scholar]
- 9.Surguchov A, Surgucheva I, Solessio E, Baehr W: Synoretin-A new protein belonging to the synuclein family. Mol Cell Neurosci 1999, 13:95-103 [DOI] [PubMed] [Google Scholar]
- 10.Polymeropoulos MH, Lavedant C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Diorio G, Golbe LI, Nussbaum RL: Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276:2045-2047 [DOI] [PubMed] [Google Scholar]
- 11.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen J, Schols L, Riess O: Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998, 18:106-108 [DOI] [PubMed] [Google Scholar]
- 12.Wakabayashi K, Matsumoto K, Takayama K, Yoshimoto M, Takahashi H: NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett 1997, 239:45-48 [DOI] [PubMed] [Google Scholar]
- 13.Goedert M, Spillantini MG: Lewy body diseases and multiple system atrophy as alpha-synucleinopathies. Mol Psychol 1998, 3:462-465 [DOI] [PubMed] [Google Scholar]
- 14.Mezey E, Dehejia AM, Harta G, Suchy SF, Nussbaum RL, Brownstein MJ, Polymeropoulos MH: Alpha synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Mol Psychol 1998, 3:493-499 [DOI] [PubMed] [Google Scholar]
- 15.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M: Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 1998, 95:6469-6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeda A, Mallory M, Sundsmo M, Honer W, Hansen L, Masliah E: Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol 1998, 152:367-372 [PMC free article] [PubMed] [Google Scholar]
- 17.Trojanowski JQ, Lee VM: Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch Neurol 1998, 55:151-152 [DOI] [PubMed] [Google Scholar]
- 18.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L: Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 2000, 287:1265-1269 [DOI] [PubMed] [Google Scholar]
- 19.El-Agnaf OM, Jakes R, Curran MD, Middleton D, Ingenito R, Bianchi E, Pessi A, Neill D, Wallace A: Aggregates from mutant and wild-type alpha-synuclein proteins and NAC peptide induce apoptotic cell death in human neuroblastoma cells by formation of beta-sheet and amyloid-like filaments. FEBS Lett 1998, 440:71-75 [DOI] [PubMed] [Google Scholar]
- 20.Feany MB, Bender WW: A Drosophila model of Parkinson’s disease. Nature 2000, 404:394-398 [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto M, Hsu LJ, Sisk A, Xia Y, Takeda A, Sundsmo M, Masliah E: Human recombinant NACP/α-synuclein is aggregated and fibrillated in vitro: Relevance for Lewy body disease. Brain Res 1998, 799:301-306 [DOI] [PubMed] [Google Scholar]
- 22.Narhi L, Wood SJ, Stevenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitnev K, Denis P, Louis JC, Wypych J, Biere AL, Citron M: Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J Biol Chem 1999, 274:9843-9846 [DOI] [PubMed] [Google Scholar]
- 23.Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sundsmo M, Masliah E: Oxidative stress induces amyloid-like aggregate formation of NACP/α-synuclein in vitro. NeuroReport 1999, 10:717-721 [DOI] [PubMed] [Google Scholar]
- 24.Shin-ichiro I, Tanaka M, Ozawa T: Point mutations of mitochondrial genome in Parkinson’s disease. Mol Brain Res 1995, 28:281-295 [DOI] [PubMed] [Google Scholar]
- 25.Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Jr, Davis RE, Parker WD, Jr: Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol 1996, 40:663-671 [DOI] [PubMed] [Google Scholar]
- 26.Blandini F, Nappi G, Greenamyre T: Quantitative study of mitochondrial complex I in platelets of parkinsonian patients. Mov Disord 1998, 13:11-15 [DOI] [PubMed] [Google Scholar]
- 27.Mizuno Y, Yoshino H, Shin-ichiro I, Hattori N, Kobayashi T, Shimoda-Matsubayashi S, Matsumine H, Kondo T: Mitochondrial dysfunction in Parkinson’s disease. Ann Neurol 1998, 44:S99-S109 [DOI] [PubMed] [Google Scholar]
- 28.Schapira AHV, Gu M, Taanman J-W, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM: Mitochondria in the etiology and pathogenesis of Parkinson’s disease. Ann Neurol 1998, 44:S89-S98 [DOI] [PubMed] [Google Scholar]
- 29.Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI: Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 1990, 5:1-10 [DOI] [PubMed] [Google Scholar]
- 30.Hsu LJ, Mallory M, Xia Y, Veinbergs I, Hashimoto M, Yoshimoto M, Thal LJ, Saitoh T, Masliah E: Expression pattern of synucleins (non-Abeta component of Alzheimer’s disease amyloid precursor protein/alpha-synuclein) during murine brain development. J Neurochem 1998, 71:338-344 [DOI] [PubMed] [Google Scholar]
- 31.Rockenstein EM, McConlogue L, Tan H, Power M, Masliah E, Mucke L: Levels and alternative splicing of amyloid β-protein precursor (APP) transcripts in brains of APP transgenic mice and humans with Alzheimer’s disease. J Biol Chem 1995, 270:28257-28267 [DOI] [PubMed] [Google Scholar]
- 32.Iwai A, Masliah E, Yoshimoto M, de Silva R, Ge N, Kittel A, Saitoh T: The precursor protein of non-Aβ component of Alzheimer’s disease amyloid (NACP) is a presynaptic protein of the central nervous system. Neuron 1994, 14:467-475 [DOI] [PubMed] [Google Scholar]
- 33.Schapira AHV: Mitochondrial involvement in Parkinson’s disease, Huntington’s disease, hereditary spastic paraplegia and Friedreich’s ataxia. Biochim Biophys Acta 1999, 1410:159-170 [DOI] [PubMed] [Google Scholar]
- 34.Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ: Protein measurement with Folin phenol reagent. J Biol Chem 1951, 193:265-272 [PubMed] [Google Scholar]
- 35.Van Uden E, Carlson G, Mallory M, Rockenstein E, Orlando R, Masliah E: Aberrant Presenilin-1 expression downregulates LDL receptor-related protein (LRP): is LRP central to Alzheimer’s disease pathogenesis? Mol Cell Neurosci 1999, 14:129-140 [DOI] [PubMed] [Google Scholar]
- 36.Lopez FL, Moretto M, Merchenthaler I, Negro-Vilar A: Nitric oxide is involved in the genesis of pulsatile LHRH secretion from immortalized LHRH neurons. J Neuroendocrinol 1997, 9:647-654 [DOI] [PubMed] [Google Scholar]
- 37.Sagara Y: Induction of reactive oxygen species in neurons by haloperidol. J Neurochem 1998, 71:1002-1012 [DOI] [PubMed] [Google Scholar]
- 38.Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC: Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. J Biol Chem 1997, 272:11369-11377 [DOI] [PubMed] [Google Scholar]
- 39.Tatton WG, Olanow CW: Apoptosis in neurodegeneration diseases: the role of mitochondria. Biochim Biophys Acta 1999, 1410:195-213 [DOI] [PubMed] [Google Scholar]
- 40.Osterova N, Petrucelli L, Farref M, Mahta N, Choi P, Hardy J, Wolozin B: α-Synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci 1999, 19:5782-5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X, Kim CN, Yang J, Jemmerson R, Wang X: Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996, 86:147-157 [DOI] [PubMed] [Google Scholar]
- 42.Hashimoto M, Takeda A, Hsu LJ, Takenouchi T, Masliah E: Role of cytochrome c as a stimulator of α-synuclein aggregation in Lewy body disease. J Biol Chem 1999, 274:28849-28852 [DOI] [PubMed] [Google Scholar]
- 43.Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE: N-ethylmaleimide-sensitive fusion protein: a trimeric ATPase whose hydrolysis of ATP is required for membrane fusion. J Cell Biol 1994, 126:945-954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davidson WS, Jonas A, Clayton DF, George JM: Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998, 273:9443-9449 [DOI] [PubMed] [Google Scholar]
- 45.Jenco JM, Rawlingson A, Daniels B, Morris AJ: Regulation of phospholipase D2: selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 1998, 37:4901-4909 [DOI] [PubMed] [Google Scholar]
- 46.Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, Frohman MA: Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol 1991, 7:191-201 [DOI] [PubMed] [Google Scholar]
- 47.Merad-Boudia M, Nicole A, Santiard-Baron D, Saille C, Ceballos-Picot I: Mitochondrial impairment as an early event in the process of apoptosis induced by glutathione depletion in neuronal cells: relevance to Parkinson’s disease. Biochem Pharmacol 1998, 56:645-655 [DOI] [PubMed] [Google Scholar]
- 48.Jenner P, Olanow CW: Understanding cell death in Parkinson’s disease. Ann Neurol 1998, 44:S72-S84 [DOI] [PubMed] [Google Scholar]
- 49.Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y: Deficiencies of the respiratory chain in Parkinson’s disease. Biochem Biophys Res Commun 1989, 163:1450-1455 [DOI] [PubMed] [Google Scholar]
- 50.Parker WD Jr, Boyson SJ, Parks JK: Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol 1989, 26:719–723 [DOI] [PubMed]
- 51.Schapira AHV, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD: Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1:1269. [DOI] [PubMed] [Google Scholar]
- 52.Bindoff LA, Birch-Machin M, Cartilidge NE, Parker Jr WD, Turnbull DM: Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J Neurol Sci 1991, 104:203–208 [DOI] [PubMed]
- 53.Mann VM, Cooper JM, Kriige D, Daniel SE, Schapira AHV, Marsden D: Brain, skeletal muscle, and platelet homogenate mitochondrial functions in Parkinson’s disease. Brain 1992, 115:333-342 [DOI] [PubMed] [Google Scholar]
- 54.Singer TP, Ramsay RR: Mechanisms of the neurotoxicity of MPTP. FEBS Lett 1990, 274:1-8 [DOI] [PubMed] [Google Scholar]