

Abstract
Pemphigus vulgaris (PV) is a potentially fatal autoimmune mucocutaneous blistering disease. It was assumed that PV is caused by anti-desmoglein (Dsg) 3 autoimmunity because absorption of PV sera with a chimeric baculoprotein containing the Dsg 3 and IgG1 portions, rDsg3-Ig-His, eliminated disease-causing antibodies. In this study we demonstrate that rDsg3-Ig-His adsorbs out autoantibodies to different keratinocyte antigens, including a non-Dsg 3 130-kd polypeptide. Because the pool of disease-causing PV IgGs contains antibodies against the keratinocyte acetylcholine receptor (AChR), we sought to identify the targeted receptor(s). Preincubation of monkey esophagus with PV antibodies blocked specific staining of the keratinocyte cell membrane with rabbit monoepitopic antibody to α9 AChR, indicating that this first of its kind AChR with dual, muscarinic and nicotinic pharmacology is targeted by PV autoimmunity. Anti-α9 antibody stained keratinocytes in a fishnet-like intercellular pattern, and visualized a single band at ∼50 kd in Western blots of keratinocyte membrane proteins. Using step-by-step reverse transcription polymerase chain reactions with primers based on known α9 sequence regions, we identified the complete reading frame of human α9. Its amino acid sequence showed 85% similarity with rat α9. Treatment of keratinocyte monolayers with anti-α9 antibody induced pemphigus-like acantholysis, which could be reversed either spontaneously or by using the cholinergic agonist carbachol. We conclude that α9 is coupled to physiological regulation of keratinocyte adhesion, and its interaction with PV IgG may lead to blister development.
Pemphigus vulgaris (PV) is a life-threatening autoimmune disease of mucocutaneous adhesion. PV autoantibodies bind to the cell membrane of oral and skin epithelial cells, keratinocytes, and cause these cells to separate from one another and round up (acantholysis). 1 The occurrence of acantholysis in PV is currently attributed to the autoantibody action. 2 The self-antigens targeted by pemphigus antibodies include ∼20 different polypeptides. 3 The specificities of pemphigus antibodies identified to date include both keratinocyte adhesion molecules and the molecules that can regulate the keratinocyte adhesive function. Some pemphigus antigens represent adhesion molecules such as desmoglein (Dsg) 1, Dsg 3, desmocollins, plakoglobin, and collagen XVII/BP180. 4-10 Other pemphigus antigens identified to date include keratinocyte acetylcholine receptor(s) (AChR) targeted by autoantibodies in pemphigus patients with or without coexistent myasthenia gravis, 3 annexins, 11 and the α chain of the high-affinity IgE receptor, FcεRIα. 12 The remaining self-antigens await to be identified and named.
A number of reports have produced circumstantial evidence for the unique association of acantholysis in PV with an antibody to a 130-kd polypeptide. 2 Because an antibody eluted from the 130-kd band identified an adhesion molecule, Dsg 3, 13 it has become a common wisdom in the field of pemphigus research to believe that the 130-kd band recognized by PV IgGs is represented only by Dsg 3 and that Dsg 3 is the only pathophysiologically important target of PV autoimmunity, whereas other self-antigens may not be important. 14-18 However, the intuitive notion that a disease of skin adhesion, PV, is caused by an antibody to an adhesion molecule, Dsg 3, needed experimental validation. Several recombinant Dsg 3 proteins were produced and used to show that adsorbed antibodies can elicit skin blistering in neonatal mice on passive transfer. 19-23 The preabsorption with a chimeric baculoprotein that included both the extracellular portion of Dsg 3 and an Fc portion of human IgG1, 20,23 but not with the extracellular portion of Dsg 3 alone, 19 could eliminate all disease-causing antibodies from pemphigus sera. Although both recombinant proteins could absorb PV autoantibodies to Dsg 3, only the eluant from the chimeric baculoprotein could induce gross skin blisters in neonatal mice. Surprisingly, an antigenic profile of the supposedly monospecific antibody adsorbed by the chimeric baculoprotein has never been shown.
We have recently demonstrated that the PV antibodies that do not include anti-Dsg 1 IgG can induce intra-epidermal acantholysis and gross skin blisters characteristic of PV on passive transfer to the neonatal Dsg3null mice. 3 In that study, anti-AChR antibody was detected in 85% of pemphigus patients by a radioimmunoprecipitation assay using as antigen the AChRs solubilized from the membrane of human epidermal keratinocytes and covalently labeled with the muscarinic radioligand [3H]propylbenzilylcholine mustard ([3H]PrBCM). 3 Earlier, we had demonstrated that PV IgGs compete directly with the reversible muscarinic radioligand [3H]atropine for binding to intact keratinocytes. 24 The AChRs expressed by human keratinocytes are coupled to regulation of transmembrane Ca2+ flux and intracellular metabolism and have been implicated in mediating physiological control of keratinocyte adhesion and motility. 25 Pharmacological blockade of AChRs with either muscarinic or nicotinic antagonists, atropine and mecamylamine, respectively, in both cases results in pemphigus-like acantholysis in the monolayers of human oral and epidermal keratinocytes. 24,26,27 Likewise, electron microscopic examination of human skin preincubated with 1 μmol/L of atropine or the nicotinic antagonist tubocurarine in both cases reveals a widening of the intercellular space and loosening of desmosomes. 28 This phenomenon is explained by antagonist-mediated interruption of the constant activation of keratinocyte AChRs with the endogenously supplied cytotransmitter acetylcholine (ACh), which is synthesized and released in an autocrine and paracrine manner by keratinocytes. 25 The stimulatory effect of ACh on the adhesive function of keratinocytes can be reproduced by both muscarinic agonists and nicotinic agonists. Both classes of cholinergic agonists can also protect keratinocyte monolayers from the acantholytic effects of PV antibody, and reverse acantholysis produced by PV IgGs, by the serine proteinase trypsin and by the calcium chelator ethylenediaminetetraacetic acid. 24 Taken together, these observations suggest that PV IgGs act as antagonists at the AChR of keratinocytes, interrupting stimulation of these receptors with ACh, and thus altering normal control of keratinocyte adhesion and motility via the cholinergic signaling pathway, which may lead to acantholysis.
In this study, we attempted to identify the type of keratinocyte AChRs targeted by PV IgG and examine acantholytic potential of antibody against this AChR. To reconcile our findings that non-Dsg molecules, such as keratinocyte AChRs, can be targeted by disease-causing antibodies with the notion that anti-Dsg 3 antibody is the sole cause of acantholysis in various forms of pemphigus, 29-31 we also had to characterize reactivities of PV IgGs absorbed by the Dsg3-Ig-His chimera. The results of the study show that Dsg3-Ig-His absorbs a mixture of anti-keratinocyte antibodies which may explain the ability of this chimeric protein to remove all disease-causing antibodies from PV sera. We obtained the full sequence of human α9, and visualized this novel human ionotropic and metabotropic receptor/Ca2+ channel with dual muscarinic and nicotinic pharmacology on the cell membrane of epidermal and oral keratinocytes, predominantly at the sites of cell-to-cell contacts. Antibody-mediated blockade of keratinocyte α9 AChR resulted in reversible acantholysis in keratinocyte monolayers, which could be abolished by the cholinergic agonist carbachol. We therefore conclude that a pool of disease-causing antibodies in PV includes anti-α9 autoantibody, and that the cholinergic agonists that can compete with this pathogenic PV IgG at keratinocyte α9 AChR may be potentially useful in the treatment of pemphigus.
Materials and Methods
Immunoabsorption Experiments with rDsg3-Ig-His
The rDsg3-Ig-His baculovirus was provided by Dr. Masayuki Amagai (Department of Dermatology, Keio University, Tokyo, Japan). The authenticity of the rDsg3-Ig-His was established by polymerase chain reaction (PCR) using two primers: the forward primer 5′-caggcacggtatatgttagag-3′ specific for the Dsg 3 portion of the chimera and the reverse primer 5′-gcacggtgggcatgtgtg-3′ specific for the Fc IgG1 portion. The production of baculoprotein and absorption of PV sera with this construct were performed following the protocol described by Amagai et al. 20 Briefly, the baculovirus rDsg3-Ig-His was amplified in the Sf9 cells (Life technologies, Inc., Rockville, MD) and used to infect the cultures of High Five cells (Invitrogen, San Diego, CA) from which the baculoprotein was isolated 4 days later. The baculoprotein rDsg3-Ig-His was then immobilized on the Talon affinity metal resin (Clontech, Palo Alto, CA), and incubated with PV sera at 4°C overnight. After washing 10 times with Tris-buffered saline containing 300 mmol/L NaCl and 20 mmol/L CaCl2, the bound PV IgGs were eluted with the Immunopure Gentle Elution buffer (Pierce Chemical Co., Rockford, IL), and desalted on the D-Salt Exellulose Plastic Desalting column (Pierce). The immunoreactivity profile of these affinity-purified PV autoantibodies was characterized by immunoblotting (below).
Development and Immunoaffinity Purification of Rabbit Antibodies to Keratinocyte AChRs
Although human keratinocytes express functional AChRs of both muscarinic and nicotinic classes, 25 the facts that all muscarinic AChR subtypes can bind atropine and [3H]PrBCM 32 and that novel α9 AChR, which we found in human epidermal keratinocytes, 33 can exhibit dual, muscarinic and nicotinic ligand-binding properties 34 identified these keratinocyte AChR types as most likely candidates for being a target for PV autoantibody. The anti-AChR rabbit antibodies were generated and characterized using the procedures detailed by us elsewhere. 35 Briefly, each female New Zealand White rabbit (Myrtle’s Rabbitry Inc., Thompson Station, TN) was immunized with the mixture of peptides representing a highly homologous sequence present in the second extracellular loop of either odd-numbered, m1, m3, and m5 (ertvlagqcyiqflsqpiitfgtamc, qyfvgkrtvppgecfiqflseptitf, and krtvpldecqiqflseptitfgtaic; residues 170–194, 208–233, and 175–199, respectively), or even numbered, m2 and m4 (vrtvedgecyiqffsnaavtfgtaic and qfvvgkrtvpdnhcfiqflsnpavtf; residues 168–192 and 172–197, respectively), muscarinic AChRs, 36-38 or the amino terminus of α9 AChR 33,34 (cwhdayltwdrdqydrld and cnkaddessepvntn; residues 65–81 and 99–112, respectively) synthesized at Genemed Synthesis, Inc. (San Francisco, CA). To immunoaffinity purify rabbit anti-AChR antibodies, the peptides were covalently conjugated to thiopropyl Sepharose 6B (Amersham Pharmacia Biotechnology, Piscataway, IL), following a protocol provided by the manufacturer, washed five times with phosphate-buffered saline (PBS), and incubated overnight at 4°C with rabbit immune sera diluted 1:10 in PBS. After incubation, the beads with adsorbed IgGs were thoroughly washed and the antibody was eluted and desalted as described above.
Immunoblotting Experiments
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting were performed in accordance with the procedure described by us elsewhere. 3 Briefly, the monolayers of second passage oral human keratinocytes or epidermal sheets separated from the skin of Dsg 3−/− (ie, Dsg3null) mice by incubation with 200 mmol/L ethylenediaminetetraacetic acid for 90 minutes at 37°C and 5% CO2 39 (substrate for PV IgG), both were lysed with a solution containing 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, and 2 mmol/L phenylmethylsulfonyl fluoride in 0.5 mol/L Tris-HCl (pH 6.8) (all from Sigma Chemical Co., St. Louis, MO). The lysates were centrifuged at 40,000 × g to remove cell debris. Twenty μg of the obtained partially purified keratinocyte membrane protein fraction were loaded into each lane and the proteins were separated on a 7.5% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and electroblotted onto a nitrocellulose membrane (Millipore Corp., Bedford, MA). The membranes were blocked with 3% nonfat milk or 2% bovine serum albumin (Sigma) for 1 hour at room temperature and cut into 4-mm wide strips, each of which was exposed to a primary antibody diluted 1:10 for 3 hours at room temperature, and then washed thoroughly. The anti-AChR rabbit antiserum was diluted in PBS supplemented to contain 1% bovine serum albumin and 0.05% Tween-20 (Sigma), and PV IgG fraction eluted from rDsg3-Ig-His fusion protein was diluted in Tris-buffered saline supplemented to contain 1% nonfat milk, 10 mmol/L CaCl2, and 0.05% Tween-20. Binding of rabbit anti-AChR antibody was visualized using a secondary horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (BioRad, Hercules, CA) and 4-chloro-1-naphthol substrate (Opti-4CN; BioRad) for development, whereas binding of pemphigus IgG was visualized using a biotinylated goat anti-human IgG antibody (Pierce, Rockford, IL) with the biotin/avidin system (Vectastain ABC system; Vector Laboratories, Burlingame, CA) for development. In negative control experiments, the primary antibody was omitted. The specificity of binding of the affinity-purified anti-α9 antibody to the substrate was tested in blocking experiments using the immunogen peptides.
Immunofluorescence Experiments
Immunofluorescence experiments were performed as detailed previously. 3 To visualize AChR expressed on the cell membrane of keratinocytes, the tissue and culture substrates were fixed for 3 minutes in 3% freshly depolymerized paraformaldehyde that contained 7% sucrose, thus avoiding permeabilization. The fixed specimens were incubated overnight at 4°C with the primary anti-AChR rabbit antibody and its binding was detected by staining with the secondary fluorescein-isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG antibody (Pierce) at room temperature for 1 hour. The specificity of binding of primary antibody was determined in control experiments in which staining was eliminated because of either omitting primary antibody or replacing it with pre-immune serum.
To allow direct blocking experiments with PV antibodies, a portion of rabbit anti-AChR antibodies were labeled with FITC as follows: 10 mg of total IgG, isolated from the immune sera by 40% ammonium sulfate precipitation, were dissolved in 100 mmol/L carbonate-bicarbonate buffer (pH 9.4), incubated for 2 hours at room temperature with 10× concentration of the FITC-isomer I (Sigma) diluted in the same buffer. The excess of FITC was removed by desalting on Sephadex-50 (Sigma) and then by dialysis against PBS for 24 hours at 4°C. The FITC-labeled anti-AChR antibodies were stabilized with 1% bovine serum albumin and affinity purified on peptide-conjugated Sepharose 6B, as detailed above. In blocking experiments, before staining with FITC-conjugated anti-AChR antibodies, the specimens were preincubated overnight at 4°C with PV (experiment) or normal human (control) serum samples, diluted 1:10 in Tris-buffered saline supplemented to contain 1% nonfat milk, 10 mmol/L CaCl2, and 0.05% Tween-20, washed thoroughly with this buffer, and fixed with a 3% freshly depolymerized paraformaldehyde solution containing 7% sucrose.
PCR Experiments and Sequence Analysis
The total RNA was extracted using the guanidinium thiocyanate-phenol-chloroform extraction procedure 40 from confluent monolayers of second passage oral keratinocytes, as detailed by us elsewhere. 27 The DNase-treated RNA samples (5 μg) underwent reverse transcription (RT) in 1× Superscript II buffer, 5 mmol/L dithiothreitol, 60 U RNase block, 10 μmol/L random decamer primers (dN)10 (Life Technologies, Inc.), 600 U RNase H-free reverse transcriptase (Superscript; Life Technologies, Inc.) and 1 mmol/L each of dATP, dCTP, dGTP, and dTTP in a final volume of 60 μl. The synthesis of single-stranded cDNA was performed for 60 minutes at 42°C. The PCRs were performed using primers described in the Results section in a final volume of 50 μl, containing the RT product, 1× PCR buffer (Promega, Madison, WI), 0.2 mmol/L each of dATP, dCTP, dGTP, dTTP, 2 U Taq DNA polymerase (Promega), and 1 μmol/L each of the forward and reverse primers. The reaction mixture was first heated at 95°C for 5 minutes and hot-started with 2 U of DNA Taq polymerase (Life Technologies, Inc.) followed by 35 cycles of denaturing at 95°C for 60 seconds, annealing at a gradient of temperature of 52, 54, 56, 58, and 60°C for 60 seconds, and extension at 72°C for 120 seconds. In the final cycle, the extension was increased to 8 minutes. The PCR products were purified from the gel using the silica membrane spin-column technology (QIAquick Spin; Qiagen, Santa Clarita, CA) and sequenced using an automated DNA sequencing system (ABI Prism 377; Perkin-Elmer-Cetus, Emeryville, CA).
In Vitro Antibody Transfer Experiments
The acantholytic activity of anti-α9 antibody was tested in keratinocyte monolayers. The cultures of oral keratinocytes were established from cuts of normal human attached gingiva left after periodontal surgical procedures, and maintained in a humidified 5% CO2 incubator at 37°C exactly as described by us elsewhere. 27 The purity of the cultures used in each experiment was demonstrated by staining for the cytokeratins that are known to be expressed by gingival keratinocytes in vitro. 41 Antibody-induced changes in keratinocyte morphology were observed in the monolayers grown in 6-well tissue-culture plates, which were placed onto the heated (37°C) stage of a computer-linked, video-monitored inverted microscope, and exposed to the immunoaffinity-purified rabbit anti-α9 antibody dissolved in growth medium. Control monolayers were treated with the IgG fraction of pre-immune serum from the same rabbit.
In Vivo Antibody Transfer Experiments
Following a standard procedure for passive transfer of antibodies to neonatal mice, 3,42 the 10- to 12-hours-old neonatal mice were injected intraperitoneally through a 30-gauge needle with increasing doses (from 1 to 20 mg/g body weight) of IgG fractions of pre-immune (control) or immune (experiment) rabbit sera which were isolated by precipitation with 40% ammonium sulfate followed by dialysis in PBS, and concentrated by lyophilization. In a separate set of experiments, the neonates were injected with PV IgG either precipitated from the whole sera or eluted from rDsg3-Ig-His.
Results
rDsg3-Ig-His Absorbs Pathogenic Non-Dsg 3 PV Antibodies
The correct sequence of the rDsg3-Ig-His baculovirus and the authenticity and purity of the rDsg3-Ig-His baculoprotein used in our experiments were confirmed by PCR (Figure 1A) ▶ and Western blotting with PV IgG (Figure 1B) ▶ , respectively. Passive transfer to neonatal BALB/c mice of up to 10 mg/g body weight per day of IgG from PV serum preabsorbed with rDsg3-Ig-His induced no gross or microscopic lesions on the skin or oral mucosa of experimental mice, whereas IgG from nonpreabsorbed PV sera (10 mg/g body weight) as well as IgG eluted from rDsg3-Ig-His (1 mg/g body weight) caused both gross and microscopic lesions characteristic of PV (data not shown). These observations confirmed the results reported by Amagai et al 20 and suggested that concentrated anti-Dsg 3 antibody alone can produce PV-like skin blisters. If correct, this conclusion would eliminate the need to characterize other self-antigens targeted by antibodies produced by PV patients, because it would imply that anti-Dsg 3 antibody is the sole cause of PV. In marked disagreement with such conclusion were the results of our previously published study, 3 in which we found that neither antibody to Dsg 3 nor to Dsg 1 were required to induce gross skin blisters in neonatal mice. Therefore, we hypothesized that in addition to anti-Dsg 3 antibody, the rDsg3-Ig-His chimeric baculoprotein absorbed PV antibodies of other specificities. To test this hypothesis, we injected neonatal Dsg3nullmice with the similar doses (ie, up to 20 mg/g) of IgGs from either preabsorbed PV sera or rDsg3-Ig-His eluant. The lack of Dsg 3 in neonatal mice used in experiments was confirmed by genotyping with two sets of primers, the Neo primers for the knockout Dsg3 gene and the Dsg3 primers for the intact Dsg3 gene (Figure 1C) ▶ , as we described previously. 3 Preabsorption with rDsg3-Ig-His eliminated the ability of PV IgG to cause lesions in Dsg3nullmice, whereas eluted antibodies caused PV-like skin blisters which were similar to those observed in Dsg 3+/+ mice (data not shown). Because keratinocytes in Dsg3nullmice do not express Dsg 3, 3,43 which was confirmed by genotyping (Figure 1C) ▶ , the anti-Dsg 3 antibody was not supposed to cause skin blisters in these animals. These results therefore indicated that: 1) rDsg3-Ig-His absorbs out non-Dsg 3 antibodies from PV sera; and 2) these non-Dsg 3 PV IgG(s) is/are pathogenic. These findings prompted further characterization of the antigenic profile of the adsorbed antibodies.
Figure 1.

Characterization of disease-causing PV antibodies affinity purified on rDsg3-Ig-His. A: RT-PCR amplification of the sequence encompassing the junction region in the rDsg3-Ig-His baculovirus with a forward primer matching the Dsg 3 sequence and a reverse primer matching the human Fc IgG1 sequence. A PCR product appeared at the expected size when genomic DNA was isolated from infected Sf9 cells (lane 2), but not from noninfected insect cells (lane 3). The PCR product size was estimated using the 100 bp DNA ladder run in parallel (lane 1). B: Western blotting of the affinity-purified rDsg3-Ig-His baculoprotein used in absorption experiments. The chimeric protein migrated with an apparent molecular weight of 105 kd where it was recognized by a PV serum (lane 1). No protein band can be seen in the control lane in which primary antibody was omitted (lane 2). C: Genotyping of a Dsg 3−/− (Dsg3null) mouse versus a Dsg 3+/− mouse. The Dsg3null mice were used both as a recipient in passive transfer experiments and as a source of epidermal proteins in immunoblotting experiments. Note: both the Neo band (220 bp) and the Dsg3 band (500 bp) were amplified from Dsg 3+/− mouse, whereas amplification of the Dsg 3 knockout mouse DNA gave only the Neo band. D: Immunoblotting characterization of PV IgGs eluted from rDsg3-Ig-His after absorption of the PV serum-coded PRC-45, using epidermal proteins from a Dsg3null mouse as a substrate. The eluted PV IgGs specifically recognized polypeptides with apparent molecular weights of 38, 55, 60, 80, 85, 110, 120, 130, 140, 170, and 190 kd (lane 1). A 115-kd protein was excluded because it was nonspecifically visualized in a negative control experiment in which the primary antibody was omitted (lane 2). Positions of the molecular weight markers in kd are shown on the right.
The Antigenic Reactivities of Pathogenic PV IgGs Adsorbed on rDsg3-Ig-His
The ability of PV IgG to recognize the Dsg 3-Fc IgG1 chimeric baculoprotein was demonstrated in the past, but the immunoreactivity of PV IgG eluted from this chimera was not. 20,23 Because PV IgG affinity purified with a recombinant protein representing only the extracellular epitope of Dsg 3, rDsg3, reacted selectively with the wild-type or recombinant Dsg 3, 19 it was assumed that rDsg3-Ig-His, too, was monospecific for anti-Dsg 3 antibody. 20,23 However, the PV serum preabsorbed with rDsg3 alone did not lose its ability to cause skin blisters in neonatal mice, 19 whereas the serum preabsorbed with rDsg3-Ig-His did. 20,23 Therefore, addition of the Fc IgG1 portion to the Dsg 3 extracellular epitope might have created new secondary and tertiary epitopes that were recognized by disease-causing non-Dsg 3 PV IgGs. To test this hypothesis, we performed immunoblotting experiments with PV IgG immunoaffinity purified on the rDsg3-Ig-His chimeric baculoprotein using the epidermal proteins from Dsg 3−/− mice as a substrate. The rationale for using Dsg3−/− epidermal protein extract was to avoid a possibility that multiple bands could result from reactivities of anti-Dsg 3 antibody with products of the proteolytic degradation of Dsg 3. Repeated experiments, using sera from different PV patients, demonstrated that chimera absorbs a mixture of PV IgGs which recognize several different keratinocyte protein bands, depending on the PV serum tested. For example, as seen in Figure 1D ▶ , from the PV serum coded “PRC-45,” rDsg3-Ig-His adsorbed out autoantibodies directed against the 38, 55, 60, 80, 85, 110, 120, 130, 140, 170, and 190 kd antigens. The 130-kd band represented a keratinocyte protein other than Dsg 3 because there was no Dsg 3 in the epidermis of the Dsg3null mouse used in this immunoblotting assay. These findings convincingly demonstrated the pemphigus immunopathology is not limited to anti-Dsg antibodies and urged identification of novel self-antigens targeted by PV autoimmunity.
PV IgG Recognizes Novel Human α9 AChR
Our previous works indicated that the pool of disease-causing PV antibodies includes an antibody to a putative keratinocyte AChR binding the muscarinic drugs [3H]atropine 24 and [3H]PrBCM. 3 In addition to classical m1, m3, m4, and m5 muscarinic AChRs, 35 we previously found in human keratinocytes a putative homologue of the rat α9 AChR 33 that can bind muscarinic as well as nicotinic drugs. 34 Therefore in our search for keratinocyte AChR(s) targeted by PV autoantibodies we included both classical muscarinic AChR subtypes and α9 AChR. The blocking experiments were performed to examine if staining produced by anti-AChR antibody could be blocked by preincubating the specimens with PV antibodies. The serum samples from PV patients’ coded 230, 245, and 246, which showed the highest titers of anti-AChR antibodies in the radioimmunoprecipitation experiments reported elsewhere, 3 were included in a series of immunofluorescence blocking experiments.
We tested a hypothesis that pretreatment of the stratified epithelium with the PV sera/IgG fractions that contain anti-AChR antibody should prevent subsequent tissue staining with FITC-conjugated, immunoaffinity-purified rabbit antibody to this receptor. Three of three test PV sera had antibodies that completely blocked staining produced by FITC-labeled anti-α9 nAChR antibody (Figure 2, A and B) ▶ . Preincubation with normal human sera did not alter binding of anti-α9 antibody, indicating that the blockade with PV antibodies was disease-specific. None of test PV patients had antibodies that could block or diminish the specific staining of keratinocytes produced by antibodies to muscarinic AChRs subtypes (not shown). These results indicated that human α9 AChR homologue is a novel and specific target for PV autoimmunity.
Figure 2.

Visualization of keratinocyte α9 AChR—a novel PV antigen. A and B: Specific blocking of the immunostaining of the cell membranes of keratinocytes by FITC-conjugated rabbit anti-α9 monoepitopic antibody because of preincubation with the PV serum PRC-45. The strong intercellular staining of the cryostat section of monkey esophagus resulting from anti-α9 antibody binding to the cell membrane (A) was completely eliminated in the specimen preincubated with PV (B) but not normal human serum (not shown). Scale bars, 50 μm. C: Characterization of the affinity-purified monoepitopic anti-α9 AChR rabbit antibody. The sodium dodecyl sulfate-polyacrylamide gel electrophoresis-resolved membrane protein extract of cultured human oral keratinocytes was used as the substrate for immunoblotting. The anti-α9 antibody recognized a protein that migrated with an apparent molecular weight of 50 kd (lane 1). The specificity of this single-band antibody binding was illustrated by the ability to abolish the staining in control experiments, in which a strip of the immunoblotting membrane was exposed to the anti-α9 antibody that had been preincubated overnight with 150 nmol of the peptide immunogens (lane 2). In another set of control experiments, no staining was detected when the primary antibody was omitted (lane 3). The molecular weight markers in kd are shown on the right. D–F: Visualization of α9 AChR on the keratinocyte cell membrane. In a series of indirect immunofluorescence experiments, nonpermeabilized specimens of normal human attached gingiva (D) and a second passage preconfluent (E) or confluent (F) monolayers of normal human gingival keratinocytes were incubated with immunoaffinity-purified rabbit anti-α9 AChR antibody, and its binding was visualized by using secondary, FITC-conjugated donkey anti-rabbit IgG antibody, as detailed in the Material and Methods. The antibody visualized α9 AChR on the cell membrane of keratinocytes, producing a characteristic intercellular, or pemphigus-like fishnet staining of the epithelium (D). In keratinocyte monolayers, the antibody decorated the cell surface of preconfluent cultured keratinocytes (E), and was found clustered at the sites of cell-to-cell contacts in confluent cultures (F), indicating that α9 AChRs are localized predominantly to the areas of keratinocyte cell membrane associated with cell-to-cell contacts. Scale bars, 50 μm.
We further hypothesized that anti-α9 PV IgGs might recognize the α9 epitope that was used to raise anti-α9 rabbit antibody. To test this hypothesis, we preabsorbed PV sera with the α9 peptides used for immunization and examined reactivities of the preabsorbed PV sera with the monkey esophageal epithelium by indirect immunofluorescence. The differences in staining intensity produced by preabsorbed versus nonpreabsorbed PV sera, however, were not sufficient to make any definitive conclusion. It can be postulated therefore that the α9 epitope(s) other than those represented by the peptides used for immunization is/are targeted by anti-α9 autoantibodies present in serum of test PV patients. Consequently, the ability of PV IgGs to block binding of rabbit anti-α9 antibody to keratinocytes is most likely explained by stereotypic hindrance of the epitope recognized by rabbit antibody by the PV antibody attached to a different epitope on the same α9 molecule.
Visualization of Human α9 AChR in Tissue and Keratinocyte Monolayers
The monospecificity of rabbit anti-α9 antibody used in blocking experiments was demonstrated in immunoblotting experiments in which 20 μg of human oral keratinocyte protein were separated by a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The affinity-purified anti-α9 antibody recognized a single major protein band that migrated with an apparent molecular weight of 50 kd (Figure 2C) ▶ . By indirect immunofluorescence, this antibody produced a classical pemphigus-like staining of the stratified epithelium (Figure 2D) ▶ and also specifically stained the cell membranes of cultured human oral (Figure 2, E and F) ▶ and epidermal (not shown) keratinocytes. The specificity of antibody binding was confirmed in control experiments showing that staining of the cell and tissue substrates as well as immunoblotting membranes could be eliminated by: 1) omitting primary antibody; and 2) preincubating the primary antibody solution with the α9 peptides used for immunization, but not with an irrelevant peptide.
Antibody Against α9-AChR Causes Acantholysis in Keratinocyte Monolayers
To determine whether antibody binding to α9 AChR may be relevant to the clinical and pathological features of PV, we investigated the acantholytic activity of rabbit anti-α9 antibody in monolayers of human oral keratinocytes. The experimental monolayers grown in standard 6-well plates, which were installed onto a heated (37°C) stage of an inverted microscope, received 10 μg/ml of rabbit monoepitopic anti-α9 antibody dissolved in the culture medium, and the control monolayers received an equivalent amount of IgGs precipitated from the pre-immune serum of the same rabbit. The typical sequence of the morphological changes that could be observed during the time-course study of the experimental, but not control, cultures is shown in Figure 3 ▶ . The intact keratinocytes had polygonal morphology and were tightly attached to each other (Figure 3A) ▶ . Within 30 to 45 minutes after the addition of anti-α9 antibody, the exposed cells retracted their cytoplasmic aprons, shrunk, and rounded-up, revealing large gaps in the monolayer (Figure 3, B–F) ▶ . These changes were very similar to acantholysis observed in keratinocyte monolayers treated with the IgG fraction of PV sera. 24 The acantholysis produced by rabbit anti-α9 antibody could reverse spontaneously if the antibody was washed out after exposing the cells for no longer than 60 minutes. The reverse could be hastened by adding to the medium 50 μmol/L of the cholinergic agonist carbachol (data not shown). During the reverse, the round-shaped keratinocytes flattened and expanded their cytoplasmic aprons. The cell borders exhibited peripheral ruffling which smoothened after the cytoplasmic protrusions had reached the borders of neighboring cells and formed new intercellular contacts. Thus, blocking α9 AChR on the membrane of cultured keratinocytes with rabbit anti-receptor antibody resulted in pemphigus-like acantholysis.
Figure 3.
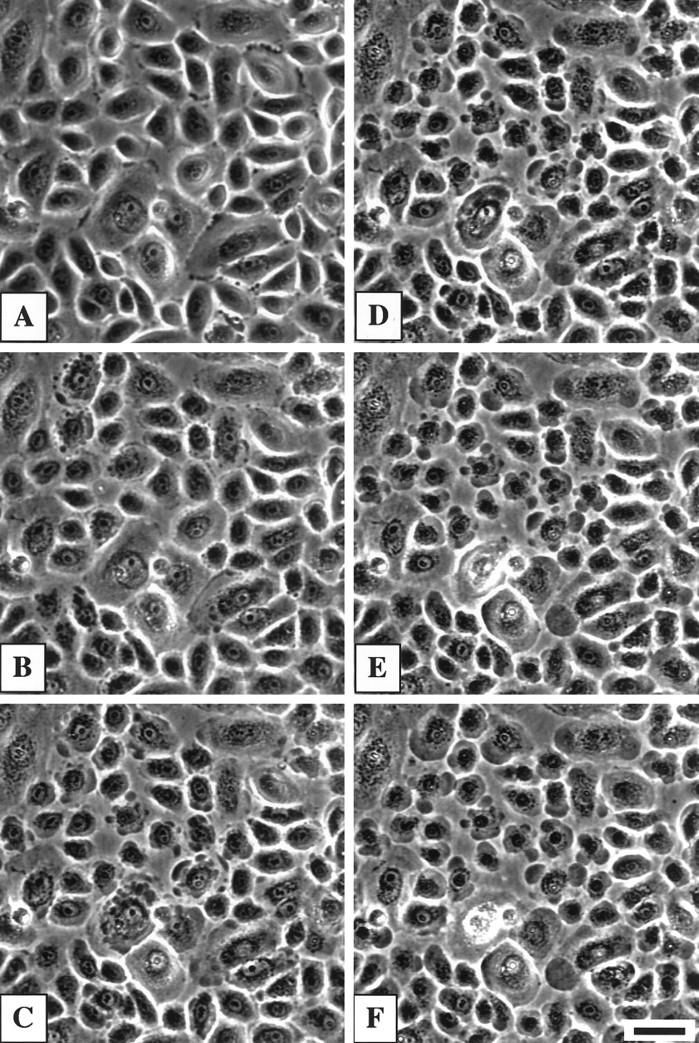
Pemphigus-like acantholysis in a monolayer of second passage human oral keratinocytes treated with immunoaffinity-purified rabbit anti-α9 AChR monoepitopic antibody. The phase-contrast images were obtained in monolayers, grown to confluence in serum-free keratinocyte growth medium in a standard 6-well cell culture plate, before (A), and 15 (B), 30 (C), 45 (D), 60 (E), and 75 (F) minutes after the addition of 10 μg/ml of anti-α9 AChR antibody. Note: separation of individual keratinocytes from neighboring cells is followed by peripheral cytoplasmic blebbing and cell rounding-up. The pictures were taken from the same microscopic field before and after addition of antibody. No changes in cell morphology were observed in the monolayer treated with the equal amount of pre-immune serum from the same rabbit (not shown). Scale bar, 50 μm.
We further asked if anti-α9 antibody could induce acantholysis in vivo. The passive transfer experiments were performed using both neonatal BALB/c and Dsg3null mice. The pups received from 1 to 20 mg IgG of rabbit IgG per g of body weight per day during at least 3 days. Approximately 48 hours after last intraperitoneal injection, the mice were sacrificed and their skin and oral mucosa were examined by light microscopy and direct immunofluorescence. All animals survived at least three injections, after which no significant macroscopic or microscopic alterations consistent with the PV phenotype could be found. The antibody, however, did bind to murine keratinocytes in the skin and oral mucosa, as judged from positive direct immunofluorescence staining of these tissue samples with FITC-conjugated anti-rabbit IgG antibody (data not shown). These results indicated that either the conformational epitope of the α9 AChR expressed by keratinocytes residing in tissue is different from that expressed by cultured keratinocytes, or that anti-α9 antibody alone is not sufficient at inducing the PV phenotype in vivo.
Identification of Complete Reading Frame of Human α9 AChR
To allow further characterization of antigenic epitopes of α9 in the future, in this study we sought to obtain the whole sequence of the human α9 AChR protein. In the past, we used RT-PCR with degenerate primers to amplify from human epidermal keratinocyte mRNA a portion of human α9 AChR, which seemed to be highly homologous to rat α9. 33 In this study, we used a step-by-step RT-PCR approach to obtain the complete human α9 sequence using RNA isolated from cultured human oral keratinocytes. We first amplified the middle portion of the translated region of the human α9 gene using a pair of primers, 5′-tgggaggtccatggcatgcc-3′ (forward) and 5′-tccttctttctggaaaggtctttgtt-3′ (reverse), which matched within the sequences present in rat α9. 34 This PCR product, termed “product 1,” showed the size of 577 bp (Figure 4) ▶ , as predicted from the rat α9 AChR sequence. 34 Subsequent sequencing of product 1 revealed its 85% similarity with the homologous sequence region of rat α9. A second RT-PCR experiment was aimed to identify a region upstream to the 5′ end of product 1 (Figure 5A) ▶ . The forward primer 5′-ggcacgatgcgtacctcacg-3′ was designed to match the rat sequence of this α9 region, and the reverse primer 5′-gggatgaggaggttgacgat-3′ was based on the authentic human α9 sequence revealed by sequencing product 1. This RT-PCR amplified a 481-bp product 2 (Figures 4 and 5A) ▶ ▶ . Following the same strategy, a third RT-PCR experiment was conducted using the primer pair 5′-agatgaaccggccccattcc-3′ and 5′-gcctccacacgaggtcactg-3′ which amplified a 345-bp product 3 representing the remaining portion of the 5′ end (Figures 4 and 5A) ▶ ▶ . Finally, the 3′ end region of human α9, a 329-bp product 4 (Figures 4 and 5A) ▶ ▶ , was amplified using the forward primer 5′-tagcaaactcccagagtctaacctg-3′, matching the 3′ end region of human α9 product 1, and the reverse primer 5′-ctctttcctgctaatctgctcttgc-3′, matching the 3′ end of rat α9. The overlapping sequences of the four PCR products rendered the complete nucleic acid sequence coding for human α9 AChR (Figure 5B) ▶ . Computer analysis of sequence homology was performed using the Gene Jockey III software (Biosoft, Cambridge, UK). The nucleotide and amino acid sequences of rat and human α9 AChRs were found to be ∼85% and 92% similar, respectively (Figure 5C) ▶ .
Figure 4.

Agarose gel electrophoresis of overlapping portions of the human α9 AChR mRNA amplified in the course of step-by step RT-PCR experiments. Total RNA was extracted from second passage human gingival keratinocytes and used as a template in RT-PCR experiments detailed in the Material and Methods section. The PCR products amplified from DNase-treated RNA samples (lanes 2) appeared at expected sizes, which were deduced from rat α9 sequence. The DNase nontreated, nonreverse-transcribed RNA samples (lanes 1), which contained some residual genomic DNA, were used to test the efficacy of DNase I treatment in the negative control experiments (lanes 3) in which the RT step was omitted. The absence of the bands with the expected sized in lanes 1 of the “products 1–3” indicates that the primers amplified genomic sequences containing introns, whereas the presence of the band with the expected size in lane 1 of the “product 4” indicates that the primers amplified an intronless genomic sequence. An additional negative control experiment included the use of water instead of the RNA template (lanes 4) to control for any possible contamination with the genetic material throughout the entire procedure. No PCR products were obtained in either negative control experiment, ie, no bands in lanes 3 or 4.
Figure 5.

The complete reading frame of human α9 AChR. A: Schematic representation of the overlapping sequences of human α9 AChR cDNAs amplified in the course of step-by-step RT-PCR experiments, showing positions of the nucleotide sequences of the amplified products. B: The nucleotide sequence and the predicted amino acid sequence of human α9 AChR. The putative signal sequence and the membrane-spanning regions (USR) are indicated. Positions of the peptides used to generate monoepitopic anti-α9 antibody are underlined. Double-underlined are the cysteines in the positions 133, 147, 197, and 198, which can provide reducible double-disulfide bridges, and the negatively charged Asp 136 and 143, Thr 138, Gln 145 and 146, which also can be involved in formation of the ACh binding pocket. C: Amino acid sequence alignment of rat and human α9 AChR homologues. The dots indicate identical residues of the two protein sequences exhibiting 92% similarity.
In all RT-PCR experiments, the adequacy of DNase treatment of RNA was verified in two sets of negative control experiments. In a first set, the omission of the RT step abolished amplification, whereas in a second set no PCR product was generated when a tube containing H2O instead of the RNA extract received the same treatments as the experimental sample (Figure 4) ▶ .
Discussion
The results of this study demonstrate that disease-causing PV autoantibodies are not limited to anti-Dsg antibodies because we documented that the rDsg3-Ig-His chimera, which, in contrast to the rDsg3 alone, can eliminate all acantholytic IgGs from PV sera 19,23 actually absorbs out a mixture of anti-keratinocyte antibodies. A pool of disease-causing PV IgGs includes an autoantibody that targets novel human α9 AChR expressed in oral and epidermal keratinocytes, which extends our previously published observations that 85% of pemphigus patients develop antibodies to keratinocyte AChRs. 3,24 In this study, we identified the complete reading frame of human α9 AChR—the first known representative of a novel class of cholinergic receptors with dual, muscarinic and nicotinic pharmacology and mixed metabotropic and ionotropic functions coupled to regulation of intracellular Ca2+ metabolism. 34,44 Rabbit monoepitopic antibody visualized α9-made AChRs at the sites of keratinocyte cell-to-cell attachments both in the stratified epithelium and in cell cultures. In keratinocyte monolayers, anti-α9 antibody produced an antagonist-like effect, causing pemphigus-like acantholysis, which can be abolished by the cholinergic agonist carbachol.
α9 is a distant member of the nicotinic AChR gene family which may form Ca2+-permeable ACh-gated ion channels. 34 It is the only AChR with dual, nicotinic and muscarinic pharmacology identified to date. Electrophysiological studies demonstrated that the response of α9-injected Xenopus laevis oocytes to ACh can be blocked by the muscarinic antagonist atropine as well as the nicotinic antagonist tubocurarine. 34 Although α9 shares ∼40% sequence homology with all other known nicotinic AChR subunits, 34 it forms homo-oligomeric channels that undergo conformational changes upon binding of both nicotinic and muscarinic drugs, as well as professional ligands of the type A γ-aminobutyric acid, glycine, and type 3 serotonin receptors 34,45
Demonstration in frog keratinocytes of a low-affinity ACh-responsive receptor modulating Ca2+ metabolism with mixed, muscarinic and nicotinic pharmacological profile 46,47 prompted our search for α9 in human keratinocytes. We used the degenerate primer RT-PCR approach to amplify portions of human α9 from mRNA isolated from epidermal keratinocytes, which were highly homologous with respective rat α9 regions. 33 Indeed, mammalian α9 sequences are highly homologous. 48,49 Therefore, in this study, the PCR primers were designed based on the reported rat α9 sequence. 34 Comparison of the complete human and rat α9 sequences revealed 85% and 92% similarity of the nucleic and amino acid, respectively. In addition to human keratinocytes, α9 sequences have so far been cloned from mammalian nasal epithelium, cochlear and vestibular hair cells, and pars tuberalis of the hypophysis. 34,48,50 In situ hybridization studies also showed that in the rat the α9 gene is expressed in the skeletal muscle of the tongue. 34 Surprisingly, α9 has not been found in neurons. 51 Substantial nonneuronal expression of α9 mRNA suggested that this receptor has functions separate from its role in neurotransmission. 52 The results of this study indicate that one of the biological functions of α9 AChRs in the stratified epithelium is control of the adhesive function of keratinocytes.
A possibility that keratinocyte shape and adhesion is controlled by a putative AChR with dual, muscarinic and nicotinic pharmacology was predicted by our observations that the muscarinic and nicotinic effects on keratinocyte adhesion are similar. 25 For instance, human keratinocytes in cell culture respond with reversible acantholysis to the reversible muscarinic antagonist atropine in the exact same way as they respond to the reversible nicotinic antagonist mecamylamine. This is in marked contrast with the opposite effects produced by muscarinic and nicotinic drugs on other cellular functions, such as proliferation, migration, and differentiation. 25,53 Thus, the presence in human keratinocytes of an AChR with mixed pharmacological characteristics, such as α9, in addition to classical nicotinic and muscarinic types of AChRs, can help explain why certain nicotinic and muscarinic drugs produce similar effects on keratinocyte adhesion, while at the same time, exhibit opposite effects on other keratinocyte functions which are probably controlled through classical AChR types.
The peculiar pharmacology of α9 AChR, which includes binding of the pan-muscarinic ligand atropine, 34 also helps explain how PV IgG that presumably should be directed against a structurally nicotinic type of AChR (based on the relationship between the myasthenia gravis and pemphigus 54,55 ) in effect compete with [3H]atropine for binding to keratinocytes 24 and precipitate a keratinocyte AChR covalently labeled with the predominantly muscarinic radioligand [3H]PrBCM. 3 The fact that keratinocyte α9 is targeted by pemphigus antibody is also in keeping with the notion that the biochemical events elicited because of PV IgG binding to the keratinocyte cell surface include both the nicotinic-like effects (ie, Ca2+ influx) and the muscarinic-like effects (ie, activation of phospholipase C, production of inositol 1,4,5-trisphosphate, rapid transient increase of intracellular Ca2+, changes in the intracellular cAMP/cGMP ratios, and activation and translocation of protein kinase C from the cytosol to the particulate/cytoskeletal fractions). 56-60 Similar ionotropic and metabotropic effects altering the concentrations of intracellular free Ca2+ can be induced by acting on α9 AChR expressed in cochlear outer hair cells. 44
Altering of the α9-coupled signaling pathway may lead to PV acantholysis. It has been recently shown that the whole PV IgG fraction can induce phosphorylation of Dsg 3 and its dissociation from plakoglobin, 61 resulting in a separation of desmosomal cadherins from the cytoskeleton proteins, which may provide a mechanism for pemphigus acantholysis. Interestingly, stimulation of α9 AChR also induces phosphorylation of yet unidentified cell membrane proteins with molecular weights of 120 and 220 kd. 62 These may represent the epithelial adhesion molecules, such as the 120-kd E-cadherin 63 and the 220-kd desmoplakin II. 64 Furthermore, the cholinergic agonist carbachol, which can reverse acantholysis produced by both PV IgG 24 and anti-α9 antibody has been shown to activate the cell adhesive function via stimulation of E-cadherin activity/function. 65 Altogether, these findings suggest that pemphigus acantholysis results, at least in part, from antagonism of anti-receptor IgG and the cytotransmitter ACh at keratinocyte α9 AChR, and that cholinergic control of cell adhesion mediated through this receptor-coupled pathways can be restored in the excess of a pharmacological agonist which protects the receptor from its ligation by the antibody that acts as a high-affinity ligand.
In contrast to distinct in vitro acantholytic activity of anti-α9 antibody, intraperitoneal administration of anti-α9 antibody to neonatal mice did not induce PV-like mucocutaneous changes. The lack of symptoms in injected animals was not surprising. We believe that the integrity of the epidermal barrier in higher species relies on more than a single molecule. It is important to emphasize that although all mice carrying a null mutation for either the α9 AChR 66 or the Dsg 3 43,67 gene survive their genetic defect, the majority of nontreated PV patients die because of their of disease. 68 Indeed, the keratinocyte self-antigens targeted by PV autoimmunity are not limited to adhesion molecules and so-called “professional” AChRs. For instance, using an immunoaffinity-purified PV antibody we have screened human keratinocyte cDNA expression library and found another novel PV antigen, which we termed “pemphaxin.” 69 Pemphaxin is a low-affinity AChR upregulated by methylprednisolone. 70 Pemphaxin is similar to annexin II, and, therefore, may play an important role in the physiological control of actin polymerization, 71 which is a major driving force for keratinocyte cell-to-cell adhesion. 72 Therefore, the pathophysiology of PV cannot be explained by autoimmunity to a single molecule or to a single group of molecules. Additionally, the importance of cell-mediated immune reactions in pemphigus acantholysis, which was demonstrated a decade ago, 73-75 has become fully appreciated only recently. For instance, interleukin 10 was found to exhibit an anti-acantholytic activity. 76 Involvement of cellular immune reactions in PV pathogenesis helps explain why some PV patients with a high titer of circulatory anti-keratinocytes antibodies do not develop skin lesions, whereas some other patients with a low antibody titer have extensive skin lesions. 77 Thus, physiological protection of keratinocytes by endogenous anti-acantholytic cytokine(s), on the one hand, and a lack of pro-acantholytic cellular immune reactions, on the other, may explain inability to disrupt the integrity of the epidermis in neonatal mice by passive transfer of anti-α9 antibody. We therefore propose that a constellation of autoantibodies to different groups of keratinocyte self-antigens, including both cell-to-cell adhesion molecules and AChRs, and activation of effectors of cellular immunity, and involvement of mediators of inflammation on the favorable genetic background all are required to break up the integrity of keratinocyte adhesion in vivo and produce clinical symptoms of PV.
In summary, the pathophysiological significance of autoimmunity against keratinocyte α9 AChR in PV is supported by the following sets of data: 1) the vast majority of PV patients develop autoantibodies that: i) precipitate [3H]PrBCM-labeled keratinocyte membrane proteins; 3 ii) displace the cholinergic radioligand [3H]atropine from its specific binding site on the cell membrane of intact human keratinocytes; 24 and iii) block staining of human keratinocytes with anti-α9 antibody in reactions of indirect immunofluorescence (Figure 2, A and B) ▶ . 2) Treatment of cultured keratinocytes with a cholinergic antagonist of either muscarinic or nicotinic class 24,26 or with an anti-α9 antibody (Figure 3) ▶ in all cases results in the acantholysis. 3) Cholinergic agonists can stimulate the keratinocyte adhesive function, 25 and prevent, attenuate, or reverse acantholysis, depending on the timing of addition of cholinomimetic drugs or PV IgG, 24 or anti-α9 IgG to keratinocyte monolayers.
We conclude that whereas the structure and function of keratinocyte self-antigens recognized by PV IgG await their further identification and characterization, the established role of the cholinergic pathway in regulating keratinocyte adhesion and the growing body of evidence that this pathway is altered in PV strongly suggest that cholinomimetic drugs may be a useful adjunct to treatment of PV patients and may even facilitate the development of a nonhormonal treatment of pemphigus patients. The recently reported case of PV that had improved by cigarette smoking 78 and reports of successful use of nicotinamide as a steroid-sparing agent in pemphigus 79 hint that such expectation is realistic, since cigarette smoke contains the cholinomimetic nicotine and nicotinamide exhibits a cholinomimetic effect 80 because of both stimulation of ACh release 81 and inhibition of acetylcholinesterase. 82 Further studies should be also directed toward the development of recombinant full-length α9 to be used in PV antibody absorption experiments, to obtain more direct evidence of the pathophysiological significance of anti-α9 PV IgG, and define the pathogenic epitope(s) of the α9 antigen.
GenBank Accession Number
The accession number of the human α9 sequence reported in this paper is AF227732.
Acknowledgments
The colony of mice with targeted disruption of the Dsg3 gene was established from animals generously donated to The Induced Mutant Resource of The Jackson Laboratory by Dr. John R. Stanley (University of Pennsylvania). We thank Dr. Leonard D. Shultz (The Jackson Laboratory) for providing Dsg3null mice and Dr. Peter Koch (Baylor College of Medicine) for providing the primer sequences used for typing Dsg3null mice, and Dr. Masayuki Amagai (Keio University, Tokyo, Japan) for providing the Dsg3-Ig-His baculovirus used to produce the respective baculoprotein for immunoadsorption experiments.
Footnotes
Address reprint requests to Sergei A. Grando, M.D., Ph.D., D.Sc., Department of Dermatology, University of California, Davis, UC Davis Medical Center, 4860 Y Street, #3400, Sacramento, CA 95817. E-mail: sagrando@ucdavis.edu.
Supported in part by the International Pemphigus Research Fund and by The Smokeless Tobacco Research Council, Inc. (research grant #0713).
References
- 1.Cohen LM, Skopicki DK, Harrist TJ, Clark WH, Jr: Noninfectious vesicobullous and vesicopustular diseases. ed 8 Elder D Elenitas R Jaworsky C Johnson BJ eds. Lever’s Histopathology of the Skin, 1997, :pp 209-252 Lippincott-Raven, Philadelphia [Google Scholar]
- 2.Stanley JR: Pemphigus and pemphigoid as paradigms of organ-specific, autoantibody-mediated diseases. J Clin Invest 1989, 83:1443-1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen VT, Lee TX, Ndoye A, Shultz LD, Pittelkow MR, Dahl MV, Lynch PJ, Grando SA: The pathophysiological significance of non-desmoglein targets of pemphigus autoimmunity. Pemphigus vulgaris and foliaceus patients develop antibodies against keratinocyte cholinergic receptors. Arch Dermatol 1998, 134:971-980 [DOI] [PubMed] [Google Scholar]
- 4.Olague-Alcala M, Giudice GJ, Diaz LA: The 80 KD pemphigus foliaceus antigen extracted from the epidermal envelope fraction is a fragment of desmoglein 1. J Invest Dermatol 1994, 102:608(abstr.) [DOI] [PubMed] [Google Scholar]
- 5.Ding X, Aoki V, Mascaro JM, Jr, Lopez-Swiderski A, Diaz LA, Fairley JA: Mucosal and mucocutaneous (generalized) pemphigus vulgaris show distinct autoantibody profiles. J Invest Dermatol 1997, 109:592-596 [DOI] [PubMed] [Google Scholar]
- 6.Dmochowski M, Hashimoto T, Garrod DR, Nishikawa T: Desmocollins I and II are recognized by certain sera from patients with various types of pemphigus particularly Brazilian pemphigus foliaceus. J Invest Dermatol 1993, 100:380-384 [DOI] [PubMed] [Google Scholar]
- 7.Korman NJ, Eyre RW, Klaus-Kovtun V, Stanley JR: Demonstration of an adhering-junction molecule (plakoglobin) in the autoantigens of pemphigus foliaceus and pemphigus vulgaris. N Engl J Med 1989, 321:631-635 [DOI] [PubMed] [Google Scholar]
- 8.Dmochowski M, Hashimoto T, Chidgey MAJ, Yue KKM, Wilkinson RW, Nishikawa T, Garrod DR: Demonstration of antibodies to bovine desmocollin isoforms in certain pemphigus sera. Br J Dermatol 1995, 133:519-525 [DOI] [PubMed] [Google Scholar]
- 9.Schumann H, Baetge J, Tasanen K, Wojnarowska F, Schacke H, Zillikens D, Bruckner-Tuderman L: The shed ectodomain of collagen XVII/BP180 is targeted by autoantibodies in different blistering skin diseases. Am J Pathol 2000, 156:685-695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashimoto T, Amagai M, Watanabe K, Dmochowski M, Chidgey MAJ, Yue KKM, Garrod DR, Nishikawa T: A case of pemphigus vulgaris showing reactivity with pemphigus antigens (Dsg1 and Dsg3) and desmocollins. J Invest Dermatol 1995, 104:541-544 [DOI] [PubMed] [Google Scholar]
- 11.Bastian BC, Nuss B, Romisch J, Kraus M, Brocker EB: Autoantibodies to annexins: a diagnostic marker for cutaneous disorders? J Dermatol Sci 1994, 8:194-202 [DOI] [PubMed] [Google Scholar]
- 12.Fiebiger E, Hammerschmid F, Stingl G, Maurer D: Anti-FcεRIα autoantibodies in autoimmune-mediated disorders. Identification of a structure-function relationship. J Clin Invest 1998, 101:243-251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amagai M, Klaus-Kovtun V, Stanley JR: Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 1991, 67:869-877 [DOI] [PubMed] [Google Scholar]
- 14.Korman NJ: Pemphigus. Dermatol Clin 1990, 8:689-700 [PubMed] [Google Scholar]
- 15.Amagai M: Autoimmunity against desmosomal cadherins in pemphigus. J Dermatol Sci 1999, 20:92-102 [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto T: Cadherins and blistering skin diseases. Curr Opin Dermatol 1993, 2:244-249 [Google Scholar]
- 17.Stanley JR: Defective cell-cell adhesion in the epidermis. Ciba Found Symp 1995, 189:107-120 [DOI] [PubMed] [Google Scholar]
- 18.Anhalt GJ: Making sense of antigens and antibodies in pemphigus. J Am Acad Dermatol 1999, 40:763-766 [DOI] [PubMed] [Google Scholar]
- 19.Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR: Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J Clin Invest 1992, 90:919-926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T: Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest 1998, 102:775-782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Memar O, Christensen B, Rajaraman S, Goldblum R, Tyring SK, Brysk MM, McCormick DJ, el-Zaim H, Fan JL, Prabhakar BS: Induction of blister-causing antibodies by a recombinant full-length, but not the extracellular, domain of the pemphigus vulgaris antigen (desmoglein 3). J Immunol 1996, 157:3171-3177 [PubMed] [Google Scholar]
- 22.Memar OM, Rajaraman S, Thotakura R, Tyring SK, Fan JL, Seetharamaiah GS, Lopez A, Jordon RE, Prabhakar BS: Recombinant desmoglein 3 has the necessary epitopes to adsorb and induce blister-causing antibodies. J Invest Dermatol 1996, 106:261-268 [DOI] [PubMed] [Google Scholar]
- 23.Amagai M, Hashimoto T, Shimizu N, Nishikawa T: Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. J Clin Invest 1994, 94:59-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grando SA, Dahl MV: Activation of keratinocyte muscarinic acetylcholine receptors reverses pemphigus acantholysis. J Eur Acad Dermatol Venereol 1993, 2:72-86 [Google Scholar]
- 25.Grando SA: Biological functions of keratinocyte cholinergic receptors. J Invest Dermatol Symp Proc 1997, 2:41-48 [DOI] [PubMed] [Google Scholar]
- 26.Grando SA, Horton RM, Pereira EFR, Diethelm-Okita BM, George PM, Albuquerque EX, Conti-Fine BM: A nicotinic acetylcholine receptor regulating cell adhesion and motility is expressed in human keratinocytes. J Invest Dermatol 1995, 105:774-781 [DOI] [PubMed] [Google Scholar]
- 27.Nguyen VT, Hall LL, Gallacher G, Ndoye A, Jolkovsky DL, Webber RJ, Buchli R, Grando SA: Choline acetyltransferase, acetylcholinesterase, and nicotinic acetylcholine receptors of human gingival and esophageal epithelia. J Dent Res 2000, 79:939-949 [DOI] [PubMed] [Google Scholar]
- 28.Wessler I: New Use of Active Ingredients which Affect Non-Neuronal Acetylcholine Functions. PCT Gazette WO 98/00119 January 8 Germany patent EP97/ 1998, 03415
- 29.Robinson ND, Hashimoto T, Amagai M, Chan LS: The new pemphigus variants. J Am Acad Dermatol 1999, 40:649-671 [DOI] [PubMed] [Google Scholar]
- 30.Udey MC, Stanley JR: Pemphigus—diseases of antidesmosomal autoimmunity. JAMA 1999, 282:572-576 [DOI] [PubMed] [Google Scholar]
- 31.Nousari HC, Anhalt GJ: Pemphigus and bullous pemphigoid. Lancet 1999, 354:667-672 [DOI] [PubMed] [Google Scholar]
- 32.Curtis CA, Wheatley M, Bansal S, Birdsall NJ, Eveleigh P, Pedder EK, Poyner D, Hulme EC: Propylbenzilylcholine mustard labels an acidic residue in transmembrane helix 3 of the muscarinic receptor. J Biol Chem 1989, 264:489-495 [PubMed] [Google Scholar]
- 33.Horton RM, Lee TX, Grando SA: Cloning of the human α9 AChR subunit from normal epidermis and investigations into its physiological role. FASEB J 1996, 10:A688 [Google Scholar]
- 34.Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S: α9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 1994, 79:705-715 [DOI] [PubMed] [Google Scholar]
- 35.Ndoye A, Buchli R, Greenberg B, Nguyen VT, Zia S, Rodriguez JG, Webber RJ, Lawry MA, Grando SA: Identification and mapping of keratinocyte muscarinic acetylcholine receptor subtypes in human epidermis. J Invest Dermatol 1998, 111:100-106 [DOI] [PubMed] [Google Scholar]
- 36.Peralta EG, Winslow JW, Peterson GL, Smith DH, Ashkenazi A, Ramachandran J, Schimerlik MI, Capon DJ: Primary structure and biochemical properties of an M2 muscarinic receptor. Science 1987, 236:600-605 [DOI] [PubMed] [Google Scholar]
- 37.Bonner TI, Young AC, Brann MR, Buckley NJ: Cloning and expression of the human and rat m5 muscarinic acetylcholine receptor genes. Neuron 1988, 1:403-410 [DOI] [PubMed] [Google Scholar]
- 38.Bonner TI: New subtypes of muscarinic acetylcholine receptors. Trends Pharmacol Sci 1989, (Suppl):11-15 [PubMed] [Google Scholar]
- 39.Ohata Y, Hashimoto T, Nishikawa T: Comparative study of autoantigens for various bullous skin diseases by immunoblotting using different dermo-epidermal separation techniques. Clin Exp Dermatol 1995, 20:454-458 [DOI] [PubMed] [Google Scholar]
- 40.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 41.Oda D, Dale BA, Bourekis G: Human oral epithelial cell culture. II. Keratin expression in fetal and adult gingival cells. In Vitro Cell Dev Biol 1990, 26:596-603 [DOI] [PubMed] [Google Scholar]
- 42.Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA: Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med 1982, 306:1189-1196 [DOI] [PubMed] [Google Scholar]
- 43.Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, Murphy GF, Whitaker-Menezes D, Stanley JR: Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol 1997, 137:1091-1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wikstrom MA, Lawoko G, Heilbronn E: Cholinergic modulation of extracellular ATP-induced cytoplasmic calcium concentrations in cochlear outer hair cells. J Physiol Paris 1998, 92:345-349 [DOI] [PubMed] [Google Scholar]
- 45.Rothlin CV, Katz E, Verbitsky M, Elgoyhen AB: The α9 nicotinic acetylcholine receptor shares pharmacological properties with type A gamma-aminobutyric acid, glycine, and type 3 serotonin receptors. Mol Pharmacol 1999, 55:248-254 [DOI] [PubMed] [Google Scholar]
- 46.Cox TC: Calcium and ATP regulation of ion transport in larval frog skin. J Comp Physiol [B] 1999, 169:344-350 [DOI] [PubMed] [Google Scholar]
- 47.Cox TC: Low-affinity mixed acetylcholine-responsive receptors at the apical membrane of frog tadpole skin. Am J Physiol 1993, 264:C552-C558 [DOI] [PubMed] [Google Scholar]
- 48.Wilcox ER, Fex J: Construction of a cDNA library from microdissected guinea pig organ of Corti. Hear Res 1992, 62:124-126 [DOI] [PubMed] [Google Scholar]
- 49.Park HJ, Niedzielski AS, Wenthold RJ: Expression of the nicotinic acetylcholine receptor subunit, α9, in the guinea pig cochlea. Hear Res 1997, 112:95-105 [DOI] [PubMed] [Google Scholar]
- 50.Simmons DD, Morley BJ: Differential expression of the α9 nicotinic acetylcholine receptor subunit in neonatal and adult cochlear hair cells. Brain Res Mol Brain Res 1998, 56:287-292 [DOI] [PubMed] [Google Scholar]
- 51.Cuevas J, Berg DK: Mammalian nicotinic receptors with α7 subunits that slowly desensitize and rapidly recover from α-bungarotoxin blockade. J Neurosci 1998, 18:10335-10344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo L, Bennett T, Jung HH, Ryan AF: Developmental expression of α9 acetylcholine receptor mRNA in the rat cochlea and vestibular inner ear. J Comp Neurol 1998, 393:320-331 [PubMed] [Google Scholar]
- 53.Grando SA, Horton RM: The keratinocyte cholinergic system with acetylcholine as an epidermal cytotransmitter. Curr Opin Dermatol 1997, 4:262-268 [Google Scholar]
- 54.Sherer Y, Bar-Dayan Y, Shoenfeld Y: Thymoma, thymic hyperplasia, thymectomy and autoimmune diseases. Review. Int J Oncol 1997, 10:939-943 [DOI] [PubMed] [Google Scholar]
- 55.Kaplan RP, Callen JP: Pemphigus associated diseases and induced pemphigus. Clin Dermatol 1983, 1:42-71 [DOI] [PubMed] [Google Scholar]
- 56.Lyubimov H, Goldshmit D, Michel B, Oron Y, Milner Y: Pemphigus—identifying the autoantigen and its possible induction of epidermal acantholysis via Ca2+ signalling. Isr J Med Sci 1995, 31:42-48 [PubMed] [Google Scholar]
- 57.Grando SA, Glukhenky BT, Romanenko AB, Demidov SV: Pemphigus antibody-induced intercellular separation of cultivated murine epidermocytes is accompanied by change in ratio of intraepidermocytic cAMP/cGMP. The IV International Congress of Cell Biology. Ottawa, 1988, p 69
- 58.Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y: Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol 1995, 104:33-37 [DOI] [PubMed] [Google Scholar]
- 59.Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y: Pharmacologic evidence for involvement of phospholipase C in pemphigus IgG-induced inositol 1,4,5-trisphosphate generation, intracellular calcium increase, and plasminogen activator secretion in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol 1995, 105:329-333 [DOI] [PubMed] [Google Scholar]
- 60.Osada K, Seishima M, Kitajima Y: Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J Invest Dermatol 1997, 108:482-487 [DOI] [PubMed] [Google Scholar]
- 61.Aoyama Y, Owada MK, Kitajima Y: A pathogenic autoantibody, pemphigus vulgaris-IgG, induces phosphorylation of desmoglein 3, and its dissociation from plakoglobin in cultured keratinocytes. Eur J Immunol 1999, 29:2233-2240 [DOI] [PubMed] [Google Scholar]
- 62.Szonyi M, Csermely P, Sziklai I: Acetylcholine-induced phosphorylation in isolated outer hair cells. Acta Otolaryngol 1999, 119:185-188 [DOI] [PubMed] [Google Scholar]
- 63.Mareel MM, Behrens J, Birchmeier W, De Bruyne GK, Vleminckx K, Hoogewijs A, Fiers WC, Van Roy FM: Down-regulation of E-cadherin expression in Madin Darby canine kidney (MDCK) cells inside tumors of nude mice. Int J Cancer 1991, 47:922-928 [DOI] [PubMed] [Google Scholar]
- 64.Joly P, Thomine E, Gilbert D, Verdier S, Delpech A, Prost C, Lebb C, Lauret P, Tron F: Overlapping distribution of autoantibody specificities in paraneoplastic pemphigus and pemphigus vulgaris. J Invest Dermatol 1994, 103:65-72 [DOI] [PubMed] [Google Scholar]
- 65.Williams CL, Hayes VY, Hummel AM, Tarara JE, Halsey TJ: Regulation of E cadherin-mediated adhesion by muscarinic acetylcholine receptors in small cell lung carcinoma. J Cell Biol 1993, 121:643-654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vetter DE, Liberman MC, Mann J, Barhanin J, Boulter J, Brown MC, Saffiote-Kolman J, Heinemann SF, Elgoyhen AB: Role of α9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron 1999, 23:93-103 [DOI] [PubMed] [Google Scholar]
- 67.Montagutelli X, Lalouette A, Boulouis HJ, Guenet JL, Sundberg JP: Vesicle formation and follicular root sheath separation in mice homozygous for deleterious alleles at the balding (bal) locus. J Invest Dermatol 1997, 109:324-328 [DOI] [PubMed] [Google Scholar]
- 68.Lever WF: Pemphigus and Pemphigoid. 1965:pp 1-266 Charles C. Thomas, Springfield
- 69.Nguyen VT, Ndoye A, Grando SA: Pemphigus vulgaris antibody identifies pemphaxin: a novel keratinocyte annexin-like molecule binding acethylcholine. J Biol Chem 2000, 275:29466-29476 [DOI] [PubMed] [Google Scholar]
- 70.Nguyen VT, Ndoye A, Grando SA: A pool of disease-causing pemphigus vulgaris IgGs contains an autoantibody to pemphaxin, a calcium-dependent keratinocyte annexin-like molecule that can be upregulated by methylprednisolone. J Invest Dermatol 1999, 112:250A [Google Scholar]
- 71.Chung CY, Murphy-Ullrich JE, Erickson HP: Mitogenesis, cell migration, and loss of focal adhesions induced by tenascin-C interacting with its cell surface receptor, annexin II. Mol Biol Cell 1996, 7:883-892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vasioukhin V, Bauer C, Yin M, Fuchs E: Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell 2000, 100:209-219 [DOI] [PubMed] [Google Scholar]
- 73.Grando SA, Glukhenky BT, Drannik GN, Epshtein EV, Kostromin AP, Korostash TA: Mediators of inflammation in blister fluids from patients with pemphigus vulgaris and bullous pemphigoid. Arch Dermatol 1989, 125:925-930 [PubMed] [Google Scholar]
- 74.Grando SA, Glukhenky BT, Drannik GN, Kostromin AP, Boiko Y, Senyuk OF: Autoreactive cytotoxic T lymphocytes in pemphigus and pemphigoid. Autoimmunity 1989, 3:247-260 [DOI] [PubMed] [Google Scholar]
- 75.Grando SA, Glukhenky BT, Drannik GN, Kostromin AP, Chernyavsky AI, Barabash TM: Pemphigus and pemphigoid epidermocytes interfere with interleukin cascade reactions. J Clin Lab Immunol 1990, 32:137-141 [PubMed] [Google Scholar]
- 76.Toto P, Feliciani C, Amerio P, Suzuki H, Wang B, Shivji GM, Woodley D, Sauder DN: Immune modulation in pemphigus vulgaris: role of CD28 and IL-10. J Immunol 2000, 164:522-529 [DOI] [PubMed] [Google Scholar]
- 77.Judd KP, Lever WF: Correlation of antibodies in skin and serum with disease severity in pemphigus. Arch Dermatol 1979, 115:428-432 [PubMed] [Google Scholar]
- 78.Mehta JN, Martin AG: A case of pemphigus vulgaris improved by cigarette smoking. Arch Dermatol 2000, 136:15-17 [DOI] [PubMed] [Google Scholar]
- 79.Chaffins ML, Collison D, Fivenson DP: Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol 1993, 28:998-1000 [DOI] [PubMed] [Google Scholar]
- 80.Romanenko AV: The action of nicotinamide on neuromuscular transmission. Fiziologicheskii Zhurnal (Kiev) 1987, 33:51–56 [PubMed]
- 81.Koeppen A, Klein J, Erb C, Loeffelholz K: Acetylcholine release and choline availability in rat hippocampus: effects of exogenous choline and nicotinamide. J Pharmacol Exp Ther 1997, 282:1139-1145 [PubMed] [Google Scholar]
- 82.Stoytcheva M, Zlatev R: Bioelectrocatalytical studies of the effect of some pharmaceuticals on the acetylcholinesterase activity. Electroanalysis 1996, 8:676-679 [Google Scholar]