

Abstract
The development of atherosclerotic lesions results from aberrant cell migration, proliferation, and extracellular matrix production. In advanced lesions, however, cellular apoptosis, leading to lesion remodeling, predominates. During lesion formation, the neurotrophins and the neurotrophin receptor tyrosine kinases, trks B and C, are induced and mediate smooth muscle cell migration. Here we demonstrate that a second neurotrophin receptor, p75NTR, is expressed by established human atherosclerotic lesions and late lesions that develop after balloon injury of the rat thoracic aorta. The p75NTR, a member of the tumor necrosis factor/FAS receptor family, can modulate trk receptor function as well as initiate cell death when expressed in cells of the nervous system that lack kinase-active trk receptors. p75NTR expression colocalizes to neointimal cells, which express smooth muscle cell α-actin and are expressed by cultured human endarterectomy-derived cells (HEDC). Areas of the plaque expressing p75NTR demonstrate increased TUNEL positivity, and HEDC undergo apoptosis in response to the neurotrophins. Finally, neurotrophins also induced apoptosis of a smooth muscle cell line genetically manipulated to express p75NTR, but lacking trk receptor expression. These studies identify the regulated expression of neurotrophins and p75NTR as an inducer of smooth muscle cell apoptosis in atherosclerotic lesions.
The neurotrophins are a family of highly conserved growth factors best characterized for their critical roles in the differentiation and survival of neurons, via activation of two classes of receptors. The trk receptor tyrosine kinases, including trk A, trk B, and trk C, bind selectively to distinct neurotrophins, with nerve growth factor (NGF) binding trk A, brain-derived neurotrophic factor (BDNF) binding trk B, and neurotrophin-3 (NT-3) binding trk C. Neurotrophin activation of trk receptors confers survival functions in neurons 1 and migration and proliferation functions in other cell types. 2-5 In addition to kinase-active isoforms, trk B and trk C genes encode truncated isoforms, generated by alternative splicing, 6,7 which inhibit intracellular signaling when coexpressed with the full length trk isoform. 8 This observation has been extended to in vivo studies, as transgenic mice overexpressing the truncated trk C receptor exhibit a phenotype similar to that observed with targeted deletion of kinase active trk C or NT-3. 9
All neurotrophins can also bind to the second class of neurotrophin receptor, p75NTR, a member of the tumor necrosis factor (TNF) receptor superfamily, whose members also include the p55 TNF receptor (TNF-R1) and FAS. Like the TNF and FAS receptors, p75NTR has a cysteine-rich extracellular domain and an intracellular domain that lacks enzymatic activity but encodes a so-called death domain, a structural domain necessary for TNF- and FAS-induced apoptosis. 10 Two opposing biological activities have been proposed for p75NTR. First, p75NTR can act as an accessory receptor for trk A, increasing the binding of NGF to trk A 11 and thereby enhancing NGF-mediated neuronal survival. Alternatively, ligand-induced activation of p75NTR can induce apoptosis, or programmed cell death, of certain classes of neuronal and glial cells, under conditions where trk activation is reduced or absent. 12-14
In addition to their well characterized expression and function in neurons, neurotrophins and trk receptors are also expressed in non-neuronal tissues, 2,15,16 where their biological actions are less well defined. Their activities have been best described in the developing cardiovascular system, where reduced trk C and trk B signaling results in abnormal cardiac 16 and intramyocardial vessel development, respectively. 49 Moreover, neurotrophins and trk receptors are expressed in human atherosclerotic lesions and in early lesions that develop after balloon de-endothelialization of the rat aorta, where they mediate chemotactic actions on vascular smooth muscle cells. 2,5 In the rat model of vascular injury, however, a switch from kinase-active trk isoforms to truncated trk isoforms is observed in later lesions, 2 which may limit the chemotactic activity of kinase-active trk receptors, and modulate neurotrophin responsiveness as the lesion continues to develop.
In response to vascular injury, medial smooth muscle cells migrate into the intima, where they proliferate and secrete extracellular matrix, resulting in the formation of a neointimal lesion. As the lesion continues to develop, however, areas of decreased cellularity are evident within the sclerotic regions of the plaque, with little appearance of cellular debris. 17 It is now recognized that apoptosis contributes to the extensive remodeling that occurs in vascular lesions; ultimately, the size and cellularity of vascular lesions will be dependent on the migration and proliferation of smooth muscle cells countered by their subsequent apoptosis. However, the molecular mechanisms regulating apoptosis in vascular lesions are incompletely understood.
Although the expression of trk receptors and the neurotrophins in vascular lesions has been examined, 2 the expression and biological actions of p75NTR have not been assessed. Thus, we sought to determine whether p75NTR is expressed in neointimal lesions and to define the biological consequences of p75NTR activation in vascular smooth muscle cells. Our results demonstrate that p75NTR is expressed exclusively in neointimal smooth muscle cells and that binding of the neurotrophins to p75NTR induces smooth muscle cell apoptosis. Thus, we have identified a new receptor regulating apoptosis of neointimal smooth muscle cells and contributing to the remodeling of atherosclerotic lesions.
Materials and Methods
Animal Surgery and Tissue Preparation
In Institutional Animal Care and Use Committee-approved studies, 3-month-old male Sprague-Dawley rats were anesthetized by intraperitoneal injection of ketamine/xylazine. Injury to the abdominal and thoracic aorta was performed using a balloon embolectomy catheter, as previously described, 2 using 3 to 5 animals per experimental group. In brief, a 2 French catheter was introduced into the left carotid, advanced to the abdominal aorta, inflated, and withdrawn. This procedure was repeated three times. Control animals were subjected to anesthesia and preparative surgery without vascular injury. After 5 and 14 days, the animals were sacrificed and tissues were fixed in 3% paraformaldehyde in phosphate buffered saline (PBS) for 30 minutes. Human endarterectomy specimens were obtained as waste surgical material, under Institutional Review Board-approved protocols, from patients undergoing surgical endarterectomy at the New York Presbyterian Hospital. Specimens were cryopreserved in 30% sucrose/OCT (1:1) within 1 hour of retrieval from the patient. Investigators were unaware of patient identifiers. Five specimens were analyzed and gave similar results.
Cell Culture
Mouse smooth muscle cells (TsTmSMC) were cultured from aortic explants of a transgenic mouse line expressing a temperature-sensitive SV40-T antigen, under the control of the promoter for smooth muscle cell α-actin. 5 TsTmSMC grown at 33°C were transfected, using the CaPO4 method, with pMex vector containing the cDNA for human p75NTR. 18,19 After selection in G418 (1 mg/ml; Gibco BRL, Rockville, MD), colonies were subcloned, expanded, and tested for stable expression of p75NTR by Western blot analysis (see below). Purified clonal populations of TsTmSMC expressing p75NTR were isolated by Dynal-bead selection using anti-p75NTR antisera. In brief, cells were incubated with a monoclonal antibody directed against the extracellular domain of p75NTR (ME 20.4 20 ) for 1 hour at 4°C. After washing, the cells were incubated with goat anti-mouse IgG bound to superparamagnetic polystyrene beads (M45; Dynal, Lake Success, NY) for 20 minutes at 4°C. p75NTR-expressing cells were selected using a Dynal magnetic particle concentrator. In all experimental protocols, the cells were seeded and cultured overnight at 33°C, after which the cells were cultured for 3 days at 39.5°C in media containing 10% fetal calf serum (FCS). A previously established cell line of TsTmSMCs stably expressing trk A (TrkA48-TsTmSMC), generated by transfection of TsTmSMC with the pMEX vector containing the cDNA for trk A, was cultured as described. 5
Human endarderectomy-derived cells (HEDC) were cultured from explants of primary human endarterectomy lesions as previously described. 21-23 Total RNA was isolated from cells within 2 to 4 passages (4–6 weeks) by extraction in guanidinium isothiocyanate followed by phenol/chloroform extraction. 24,25
Immunohistochemistry
Immunohistochemistry was performed after selected slides were stained with hematoxylin and eosin to identify lesions. Serial sections of rat aortic lesions were deparaffinized and endogenous peroxidase activity quenched with 3% H2O2 in methanol for 15 minutes at room temperature. Cryopreserved sections of human endarterectomy were air-dried onto microscope slides and prefixed with 3% paraformaldehyde for 5 minutes. Endogenous hydrogen peroxidase was quenched as above. Adjacent sections were then incubated with either the anti-p75NTR antibody (goat polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA), anti-full length trk B antibody (goat polyclonal; directed against the carboxy terminus of trk B); anti-truncated trk B antibody (rabbit polyclonal; Santa Cruz Biotechnology); anti-full length trk C antibody (rabbit polyclonal directed against carboxy terminus against trk C; Santa Cruz Biotechnology) anti-truncated trk C antisera (rabbit polyclonal 2 ), anti-smooth muscle cell α-actin antibody (monoclonal, Clone 1A4, Dako Corp., Carpinteria, CA), anti-CD31 antibody (Hec-7 26 kindly provided by Dr. William Muller, Weill Medical College of Cornell University) or anti-macrophage antibody, (HAM 56 mouse monoclonal; DAKO Corp.). Sections were incubated with antibody preabsorbed with blocking peptide (anti-p75NTR, anti-full length trk B and trk C and anti-truncated trk B), preimmune serum (anti-truncated trk C) or purified mouse IgG (anti-smooth muscle cell α-actin, anti-CD 31 and HAM 56; Santa Cruz Biochemicals) as a negative control. Following incubation with the appropriate biotinylated secondary antibody (Vector Laboratories, Burlingame, CA), immunoreactive proteins were detected using an avidin-biotin-based horseradish peroxidase system using Vector VIP solution as a chromogenic substrate (Vector Laboratories), followed by counterstain with hematoxylin.
Detection of Apoptotic Smooth Muscle Cells in Human Atherosclerosis
Cryopreserved sections of human endarterectomy were air-dried onto microscope slides and postfixed with 3% paraformaldehyde for 10 minutes, followed by incubation with 0.1% sodium citrate containing 0.1% Triton at 4°C. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL)-positive cells were detected using a kit purchased from Roche Biochemicals (Indianapolis, IN). Briefly, sections were incubated at 37°C for 1 hour with fluorescein-conjugated dUTP in the presence of terminal deoxynucleotidyl transferase (TdT). Control sections were incubated with fluorescenated dUTP in the absence of TdT. The sections were then incubated with anti-fluorescein antibody conjugated with alkaline phosphatase for 30 minutes at 37°C. TUNEL-positive cells were detected using Fast Red (Roche Biochemicals) as a chromogenic substrate.
Reverse Transcription Polymerase Chain Reaction (RT-PCR)
One microgram of total RNA isolated from HEDC was subjected to reverse transcription using murine leukemia virus-reverse transcriptase (Perkin-Elmer, Norwalk, CT). Total RNA not incubated with reverse transcriptase was used as a negative control. The cDNA products were incubated with AmpliTaq polymerase (Perkin-Elmer) and primers specific for human p75NTR (Whitehead Institute for Biomedical Research, Cambridge, MA). Primers were designed using the Primer 3 program and published sequences. 19
p75NTR (human) sense 5′ AGC CTT CAA GAG GTG GAA CA 3′
p75NTR (human) antisense 5′ CTG CAC AGA CTC TCC ACG AG 3′
After a 95°C hot start, cycling proceeded for 40 cycles of 1 minute at 95°C, 1 minute at 60°C, and 1 minute at 72°C. Products were resolved by electrophoresis in a 1% agarose gel, followed by visualization with ethidium bromide. The primer pair yields an oligonucleotide product of 463 bp. PCR using primer pairs against the unique exon expressed at the carboxy terminus of either truncated trk B, truncated trk C, or primer pairs directed against the kinase domains of trk B or trk C was performed as described. 27 RNA isolated from either p75-TsTmSMC or adult bone marrow cells was used as a positive control for p75NTR and trk receptors, 27 respectively.
Western Blot Analysis
Native or p75-expressing TsTmSMC were lysed in radioimmunoprecipitation assay buffer containing phenylmethylsulfonyl fluoride (1 mmol/L), aprotinin (1 mg/ml), and leupeptin (10 mg/ml). 28 Lysates were clarified by centrifugation at 14,000 rpm (Beckman Microfuge, Palo Alto, CA) at 4°C, and the protein content of the supernatant determined by Biorad protein assay (Bio-Rad Laboratories, Richmond, CA). Lysates containing equivalent amounts of protein were separated by 9% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, blotted onto nitrocellulose, and Western blot analysis was performed using either anti-p75NTR rabbit polyclonal antisera (Promega, Madison, WI), a rabbit polyclonal antiserum that recognizes all full length trk isoforms (203 antisera 29 ) or a rabbit polyclonal antibody specific for murine FAS receptor (M-20, Santa Cruz Biotechnology). Immunoreactive proteins were detected using enhanced chemiluminescence (ECL) detection (Amersham Corp., Arlington Heights, IL).
Annexin V Binding
Native TsTmSMC, trk A, or p75-expressing TsTmSMC (100,000 cells/well) or HEDC (30,000/well) were seeded in six-well tissue culture plates and cultured for 2 to 3 days in 10% FCS media at 39.5°C (TsTmSMC cell lines) or 37°C (HEDC). Native p75 and trk A expressing TsTmSMC were cultured overnight in 1% FCS media, then treated with either NGF, NT-3, BDNF (each at 2–4 nmol/L), or a monoclonal antibody which cross-links and activates the murine FAS receptor (Clone Jo2 100 ng/ml; PharMingen, San Diego, CA). 30 HEDC were cultured in 0.5% FCS in the presence or absence of either the neurotrophins or an antibody which binds and activates human FAS (Clone CH11 100 ng/ml; Upstate Biotechnology, Lake Placid, NY). 31 At the indicated time points, cells were suspended using PBS/EDTA and washed in serum-free Dulbecco’s modified Eagle’s medium (DMEM). Annexin V binding was determined by incubating the cells with fluorescein isothiocyanate (FITC)-conjugated annexin V (Immunotech, Miami, FL) in DMEM containing 1.5 mmol/L Ca2+ on ice for 10 minutes. After washing to remove unbound annexin V, the cells were incubated with propidium iodide and the cell samples were analyzed by flow cytometry using a Coulter Elite Flow Cytometer. Optimal parameters for flow cytometric analysis were set using control cells not incubated with FITC-annexin V and propidium iodide.
TUNEL Staining
Native TsTmSMC, p75-TsTmSMC, or trk A48-TsTmSMC were seeded on 8-chamber permanox slides and cultured for 3 days at 39.5°C. HEDC were seeded on 8-chamber glass slides and cultured overnight at 37°C. The cells were rinsed with PBS and treated with NGF, NT-3, or BDNF (each at 4 nmol/L) or anti-FAS activating antibody (Jo2; 100 ng/ml, murine cells only) in DMEM containing 0.5 to 1% FCS. After 24 hours, the cells were fixed in 3% paraformaldehyde and the TUNEL assay was performed by incubation with fluoroscein-labeled dUTP in the presence of TdT (Boehringer Mannheim, Chicago, IL). Using fluorescent microscopy, the results were quantified by counting the number of TUNEL-positive cells per high-power field in 10 to 15 fields, to assess 1000 cells per well.
Caspase Activation
p75-TsTmSMC were seeded and treated as for TUNEL staining. Immunodetection of activated caspase 3 was performed using a rabbit antibody directed against the p20 subunit 32 (provided by Idun Pharmaceuticals, La Jolla, CA) and a goat anti-rabbit IgG conjugated to Alexa 488 (Molecular Probes, Eugene, OR). The cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; 0.5 μg/ml; Sigma Chemical Co., St. Louis, MO) and fluorescence was detected at 488 nm (FITC; Alexa green) and 368 nm (DAPI).
Results
Expression of p75NTR in Neointimal Lesions
Although trk receptors and neurotrophins are up-regulated after vascular injury, 2 the expression of p75NTR has not been examined in neointimal lesions. No immunodetectable p75NTR was present in uninjured adult rat vessels (Figure 1A) ▶ . p75NTR was also not expressed in the small lesions which developed 5 days after balloon injury (1–2 cell layers composed of smooth muscle cells, as assessed by expression of smooth muscle cell α-actin; Figure 1, C and D ▶ ). However, at 2 weeks after balloon injury, significant expression of p75NTR was detectable in smooth muscle cells within a highly organized neointimal lesion (Figure 1, E and F) ▶ and in medial smooth muscle cells just adjacent to the lesion. No immunoreactive proteins were detected in adjacent sections incubated under control conditions (Figure 1, G and H) ▶ . Thus, in contrast to the early expression of trk receptors after balloon injury 2 p75NTR expression is up-regulated later in lesion development.
Figure 1.

Immunohistochemical analysis of p75NTR in vascular lesions of the rat thoracic aorta 2 weeks after balloon de-endothelialization. Sections of rat thoracic aorta from either control animals (A and B), or 5 (C and D) and 14 days (E and F) after balloon de-endothelialization were incubated with the indicated antibodies. Immunoreactive proteins were visualized using avidin-biotin-based horseradish peroxidase kit using Vector VIP as a chromogenic substrate; the sections were counterstained with hematoxylin. Original magnification, ×90.
To assess p75 expression following long-standing vascular injury, atherosclerotic lesions obtained from human endarterectomy specimens were examined. Figure 2A ▶ shows a section of endarterectomy lesion stained by hematoxylin and eosin, demonstrating the presence of the medial smooth muscle cell layer (as determined by morphological assessment and by immunohistochemistochemical analysis of smooth muscle cell α-actin; upper right inset labeled B-D of Figure 2, A and B ▶ ), surrounding a highly cellular plaque. Using serial sections, p75NTR immunoreactivity was present in regions of the plaque that exhibited prominent expression of smooth muscle cell α-actin; see Figure 2, E ▶ -G (anti-p75NTR) and K-M (anti-α-actin). Little or no immunoreactivity for the endothelial cell marker, PECAM (Hec-7) or HAM 56, a macrophage marker (results not shown) was detected in areas that stained for p75NTR or smooth muscle cell α-actin. Expression was highest in neointimal smooth muscle cells within the plaque, adjacent to the lumen area (lower inset labeled E-M in Figure 2A ▶ ), with very little expression within medial smooth muscle cells (Figure 2C) ▶ . Among populations of neointimal smooth muscle cells, there was variability in the expression of p75NTR, with some cells expressing high levels of p75NTR (arrows, Figure 2, F and G ▶ ), while others demonstrated less expression (Figure 2F) ▶ . No immunoreactive proteins were detected in adjacent sections incubated under control conditions (Figure 2F ▶ , inset for p75NTR and inset panel L for α-actin). Immunoreactivity for full length trkB and trkC was present in subpopulations of neointimal smooth muscle cells (results not shown) similar to our previously published results, 2 whereas immunostaining for either truncated trkB or truncated trkC showed little or no staining above sections incubated under control conditions (results not shown). Thus, similar to what was observed in the rat model of vascular injury, neointimal smooth muscle cells present in human atherosclerotic lesions express p75NTR.
Figure 2.

Immunohistochemical analysis of p75NTR expression and in situ TUNEL analysis in vascular lesions of human atherosclerotic lesions. A: Hematoxylin and eosin stain of atherectomy specimen derived from a carotid artery. Panels represented by the insets are indicated. The outer black line represents where the medial smooth muscle layer would extend to form the entire cross-section of the vessel. The inner black line represents the potential boundary of the lumen of the vessel. Panels B-M were treated as indicated. Immunoreactive proteins were detected as in Figure 1 ▶ . Areas marked off in E, H, and K are shown at higher magnification in F, I, and L, respectively. Open arrowheads in C are calcifications stained with hematoxylin. Arrows in F, cells expressing high levels p75NTR, shown at higher magnification in G; arrows in I, nuclei of TUNEL-positive cells, shown at higher maginification in J; arrowhead in I, nuclei of TUNEL-positive cells sectioned obliquely, shown at higher magnification in J. M, magnified area of L represented in G and J. Insets: F, control section incubated with anti-p75NTR preabsorbed with blocking peptide; J, control section incubated with fluorescenated-dUTP in the absence of TdT; L, control section incubated with mouse IgG.
Colocalization of p75NTR with TUNEL-Positive Neointimal Smooth Muscle Cells
To determine whether p75NTR activation could contribute to apoptosis of neointimal smooth muscle cells, we first assessed the localization of p75NTR expression with TUNEL reactivity as a marker of apoptotic cells in human endarterectomy samples. TUNEL-positive smooth muscle cells were detectable throughout the neointimal lesion, but the area with the highest concentration of TUNEL-positive cells was the neointimal smooth muscle cells adjacent to the lumen, where p75NTR was expressed (compare Figure 2, E ▶ -G, with Figure 2, H ▶ -J). In analysis of 10 sections of 5 lesions, TUNEL-positive cells were detected in areas where p75NTR was expressed. In contrast, very little TUNEL positivity was detected in medial smooth cells or in acellular areas of the plaque just below the medial smooth muscle cell layer (Figure 2D) ▶ . Sections incubated with fluorescentated dUTP in the absence of TdT confirmed the specificity of the TUNEL detection (Figure 2I ▶ , inset). These results suggest p75NTR is expressed in areas where neointimal smooth muscle cells are undergoing apoptosis in human atherosclerotic lesions.
Apoptosis of Atherectomy-Derived Smooth Muscle Cells
As our prior studies indicate that neurotrophins are expressed by human endarterectomy lesions, 2 we assessed whether ligand-induced activation of p75NTR induces apoptosis of neointimal cells, using cultured cells derived from explants of HEDC. 2,21 Previous studies demonstrated the expression of smooth muscle cell α-actin by these cells under culture conditions. 21 HEDC expressed mRNA for p75NTR, as well as truncated trk B and truncated trk C, as assessed by RT-PCR (Figure 3) ▶ . Neither cell line expressed mRNA for kinase active trk B, whereas one of two cell lines expressed mRNA for kinase active trk C. Apoptosis was assessed by two established techniques: annexin V binding to phosphatidyl serine exposed on the outer membrane of apoptotic cells (Figure 4A) ▶ or TUNEL staining for fragmented DNA (Figure 4B) ▶ . Flow cytometric analysis of annexin V binding revealed that HEDC cultured in media containing 0.5% FCS undergo low levels of spontaneous apoptosis, with 3% of the cells in the early stages (quadrant 4) and 10% in the later stages (quadrant 2) of the apoptotic pathway (Figure 4A) ▶ . Twenty-four hours after treatment with either NGF, BDNF, or NT-3, (at 2 nmol/L [50 ng/ml] concentration) the number of apoptotic cells increased three- to fourfold for both early and late stage populations of apoptotic cells. A similar response was observed on activation of the FAS receptor. In contrast, cells cultured in 10% FCS exhibit a similar level of annexin V binding as cells cultured under control conditions. TUNEL assays to detect fragmented DNA in situ confirmed the apoptotic activity of the neurotrophins on HEDC, where in the absence of the ligand, 2.5 ± 0.7% of HEDC were TUNEL-positive, and this increased by two- to threefold after treatment with NGF (Figure 4B) ▶ . These results demonstrate that nanomolar concentrations of neurotrophins can induce apoptosis of HEDC, potentially through binding and activation of p75NTR.
Figure 3.

Expression of p75NTR, truncated trk B, and truncated trk C mRNA in human atherectomy-derived smooth muscle cells. Total RNA was subjected to reverse transcription polymerase chain reaction and amplified products detected following cycling with p75NTR-, truncated trk B-, or truncated trk C-specific primer pairs and Taq polymerase. Sizes of amplified product: p75NTR, 463 bp; truncated trk B, 161 bp; 27 truncated trk C, 158 bp; 27 kinase-active trk B, 571 bp; 27 kinase-active trk C, 571 bp. 27 Lanes 1, 3–5, and 7, RNA subjected to reverse transcription. Lanes 2, 6, and 8, RNA not subjected to reverse transcription. Lanes 1 and 2, HEDC-12. Lanes 3 and 6, HEDC-201. Lane 4, p75-TsTmSMC (positive control, see below). Lane 5, TsTmSMC. Lanes 7 and 8, bone marrow.
Figure 4.
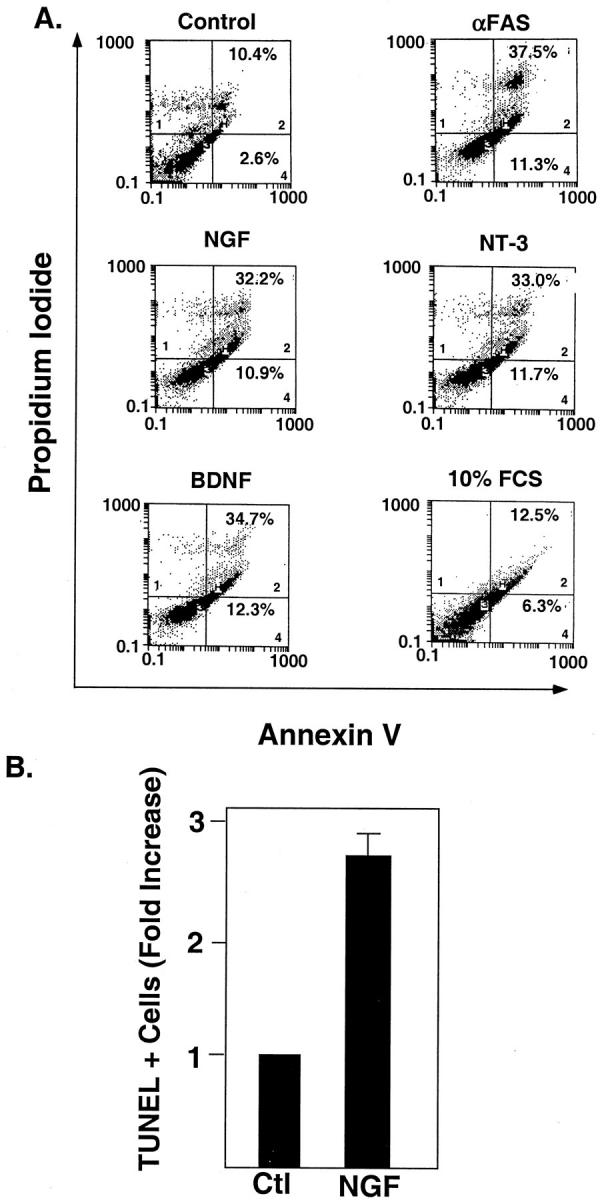
Neurotrophin-induced apoptosis of HEDC. A: Flow cytometric analysis of annexin V binding to HEDC. HEDC were treated with either NGF (2 nmol/L), NT-3 (2 nmol/L), BDNF (2 nmol/L), FAS-activating antibody (CH11; 100 ng/ml), or 10% FCS. After 24 hours, a cell suspension was prepared and the cells were stained with FITC-conjugated annexin V and propidium iodide as described in Methods, and flow cytometric analysis was performed. x axis, FITC-conjugated annexin V binding; y axis, propidium iodide incorporation. As has been previously demonstrated, 47,48 cells in quadrant 3 (lower left corner) represent live cells that do not bind annexin V or incorporate propidium iodide. Cells in quadrant 4 (lower right corner) represent cells in the early phases of apoptosis, in which annexin V binds to phosphatidyl serine exposed on the outer surface of apoptotic cells, but there is no incorporation of propidium iodide because membrane integrity is maintained. Cells in quadrant 2 (upper right corner) represent cells in the later stages of apoptosis, which bind annexin V and incorporate propidium iodide due to a loss of membrane integrity. Cells in quadrant 1 represent cells that have completed the apoptotic pathway. Histogram tracings from 1 of 3 duplicate experiments using 2 different HEDC culture lines is shown. Quadrants 1, 2, and 4 are labeled in the lower left (quadrant 1) and lower right (quadrants 2 and 4) corners. The percentage of the total cell population in either quadrant 2 (late stage, secondary necrotic) or quadrant 4 (early apoptotic phase) are given in the appropriate quadrant. B: Quantitative analysis of TUNEL for HEDC. In each experiment there were two replicates per treatment group. Data are the mean ± SE of 3 separate experiments using 3 different cell lines.
Derivation of p75NTR Expressing TsTmSMC
Although primary cultures of HEDC undergo neurotrophin-induced apoptosis while expressing p75NTR and truncated trk receptors, we sought to further analyze this activity in smooth muscle cells with known patterns of expression of neurotrophins and neurotrophin receptor isoforms. Thus, we used an established, conditionally immortalized temperature-sensitive mouse smooth muscle cell line (TsTmSMC). Previous studies from our laboratory demonstrated that TsTmSMC expressed mRNA for NGF, BDNF and NT 4/5, 5 but do not express full length or truncated trk receptors, or p75NTR, as assessed by Western blot analysis ( 5 and Figure 5 ▶ ). Additionally, native TsTmSMC expressed FAS (Figure 5) ▶ . After transfection with a cDNA encoding human p75NTR, 15 clones stably expressing p75NTR were isolated. Clonal populations expressing 20–70,000 p75NTR receptors/cell, were identified and expanded (Figure 5) ▶ . 28 Purified populations of p75-TsTmSMC were obtained by immunoseparation to yield populations in which greater than 95% of the cells expressed high levels of p75NTR, as assessed by immunohistochemistry (results not shown). The biological activity of the neurotrophins was examined using three stable-enriched clones expressing p75NTR cultured for 3 to 4 days at 39.5°C to reduce the expression of T antigen and induce a less transformed phenotype, as previously described. 5 All 3 clones yielded similar results.
Figure 5.

Western blot analysis of p75NTR, trk, and FAS in native TsTmSMC and p75-TsTmSMC. For FAS Western blot analysis African green monkey kidney cells and the murine lymphocyte cell line A20 are used as a negative and positive control, respectively. The pheochromocytoma cell line PC12, genetically manipulated to overexpress trk receptors, 28 was used as a positive control for trk and p75NTR Western blot analysis. This cell line expresses approximately 40,000 trk receptors and 40,000 p75NTR cell. Lane 1, TsTmSMC. Lane 2, A20 cells. Lane 3, COS cells. Lane 4, PC12. Lane 5, p75-TsTmSMC. Lane 6, p75-TsTmSMC-2.
Neurotrophins Induce Apoptosis of p75 TsTmSMC
Neurotrophin-induced apoptosis of p75-TsTmSMC was first assessed by annexin V binding. Cultured TsTmSMC, and clones stabilizing expressing either p75NTR or, as a control, trk A, exhibited variable but similar rates of spontaneous apoptosis (Figure 6 ▶ ; ranges, 3.3–25.4%, 4.0–12.7%, and 12.5–19.2%, respectively; in 3 separate experiments per cell line). Treatment of p75-TsTmSMC with nanomolar concentrations of NGF or NT-3 caused a dose dependent increase in annexin V binding, in a response comparable to that elicited by activation of FAS (Figure 6) ▶ . The addition of 4 nmol/L of either NGF or NT-3 resulted in an eight-to-ninefold increase in the number of cells in the early phases of apoptosis (quadrant 4), whereas 2 nmol/L caused a four- to fivefold increase. BDNF was less effective at inducing apoptosis of p75TsTmSMC, inducing only a four- to fivefold increase in annexin V binding in doses ranging from 1 to 4 nmol/L. All three neurotrophins increased by two- to threefold the number of cells in the later stages of apoptosis (quadrant 2). Treatment with 4 nmol/L of the neurotrophins for 24 hours increases the percentage of cells present in quadrant 1 (results not shown). In contrast, the neurotrophins did not induce an increase in annexin V binding to TsTmSMC (Figure 6) ▶ , whereas the FAS activating antibody induced a similar effect as observed in p75-expressing TsTmSMC. Thus, the expression of p75NTR results in neurotrophin-induced apoptosis of vascular smooth muscle cells.
Figure 6.

Flow cytometric analysis of annexin V binding to p75-TsTmSMC, native TsTmSMC, and trk48A-TsTmSMC. TsTmSMC cell lines were grown for 3 days at 39.5°C followed by treatment with the neurotrophins at the indicated doses. After 15 hours, a cell suspension was prepared and the cells were stained with FITC-conjugated annexin V and propidium iodide, as described in Methods, and flow cytometric analysis was performed. Histograms from one of three duplicate experiments for each cell line is shown. Quadrants 1, 2, and 4 are labeled in the lower left (quadrant 1) and lower right (quadrants 2 and 4) corners. The percentage of cells in the early and late stages of apoptosis are indicated in quadrants 4 and 2, respectively.
To differentiate between p75NTR-mediated apoptosis and the actions of neurotrophins on trk-expressing smooth muscle cells, we assessed the apoptotic activity of NGF using a previously established stable clone of trkA48-TsTmSMC. 5 TrkA48-TsTmSMC displayed a similar level of basal apoptotic activity as observed with either native or p75-TsTmSMC (Figure 6) ▶ . However, although FAS activation induced a two- to fourfold increase in annexin V binding, similar to the results with native TsTmSMC, the neurotrophins did not increase annexin V binding, suggesting that neurotrophin-induced vascular smooth muscle cell apoptosis is dependent on p75NTR expression and is not mediated by the trk receptor tyrosine kinase.
To further assess the apoptotic activity of the neurotrophins, TUNEL assays and immunofluorescence for detection of activated caspase 3 were performed. Treatment of p75-TsTmSMC with NGF, NT-3, or BDNF for 24 hours induced a two- to threefold increase in the number of TUNEL positive cells over control cells treated with media alone (Figure 7A) ▶ . In contrast, neither native TsTmSMC or trkA48-TsTmSMC showed increases in TUNEL positivity after treatment with the neurotrophins (Figure 7, A and B) ▶ , whereas FAS activation increased the number of TUNEL-positive cells three- to fourfold, in either native, trk A-, or p75-expressing TsTmSMC (results not shown). In separate experiments, similar increases in the number of apoptotic p75-TsTmSMC in response to the neurotrophins or anti-FAS activating antibody were observed using DAPI stain to assess chromatin condensation as index of apoptosis (Figure 8) ▶ .
Figure 7.
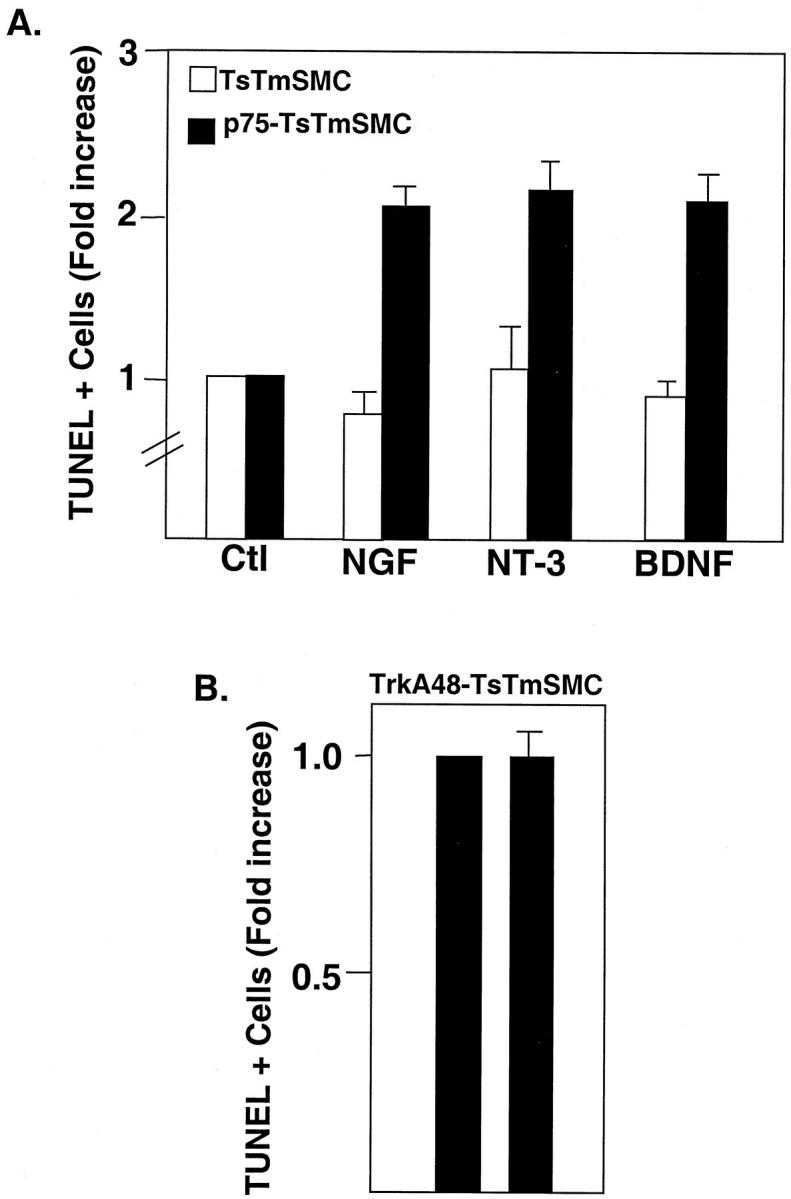
A: Quantitative analysis of the TUNEL assay for TsTmSMC and p75NTR-TsTmSMC after treatment with the neurotrophins (each 4 nmol/L). Data are presented as -fold increase of TUNEL-positive cells over control, nontreated cells and are the means ± SE of one of three duplicate experiments for both native TsTmSMC and p75-TsTmSMC. In each experiment, there were two replicates per treatment group. Percentage of TUNEL-positive cells in the absence of exogenous ligand: TsTmSMC = 2.1 ± 0.4%; p75TsTmSMC = 4.6 ± 0.9%. B: Quantitative analysis of TUNEL assay for trkA48-TsTmSMC. Data are presented as in A and are from one of two duplicate experiments, two replicates per experimental group. Percentage of TUNEL-positive cells in the absence of ligand = 1.9 ± 0.4%.
Figure 8.

Immunofluorescent analysis for expression of activated caspases in p75-TsTmSMC. Cells were counterstained with DAPI for detection of condensed chromatin as an index of apoptosis.
Activation of the caspases from inactive proenzymes mediates many of the morphological and biochemical changes associated with apoptosis. To assess if caspases are activated in neurotrophin-treated p75-TsTmSMC undergoing apoptosis, immunostaining was performed using the CM-1 antibody, which recognizes only the activated form of caspase-3 but not the inactive 32-kd proenzyme. 32 Treatment of p75-TsTmSMC with either the neurotrophins or FAS-activating antibody (Figure 8) ▶ increased CM-1 immunoreactivity compared to control, nontreated cells. Moreover, 90 to 95% of those cells expressing activated caspase 3 exhibit chromatin condensation, as detected by Hoescht staining (Figure 8) ▶ . These results confirm that neurotrophin activation of p75NTR induces apoptosis of smooth muscle cells through a signaling cascade that activates caspase 3.
Discussion
The development of atherosclerotic and restenotic lesions is a dynamic process regulated by competing forces: the migration and proliferation of medial smooth muscle cells in the intima balanced by plaque remodeling through apoptotic cell death. 17,33 Few factors initiating apoptosis of smooth muscle cells in vascular lesions have been identified to date, although FAS is expressed in advanced atherosclerotic lesions and cultured medial smooth muscle cells undergo FAS-induced apoptosis in vitro 34,35 (and current results).
Our current results identify the p75 neurotrophin receptor as a novel regulator of apoptosis in neointimal lesions. First, in the rat model of vascular injury, p75NTR is temporally expressed only in the later stages of lesion development, when apoptosis is prominent and trk receptor activity is diminished. 2,17 Second, in human atherosclerotic lesions, p75NTR spatially localized to regions of the neointima that demonstrate ongoing death. Moreover, neurotrophins at physiological concentrations induced p75NTR activation to initiate apoptosis of lesion-derived cells and stable cell lines that express p75NTR, but not kinase-active trk receptors.
The coexpression of neurotrophins 2 and p75NTR by neointimal muscle cells represents a local mechanism for induction of apoptosis in atherosclerotic lesions that contrasts with proposed models of FAS-induced apoptosis in atherosclerotic lesions. FAS-mediated apoptosis is dependent on the colocalization of smooth muscle cells with infiltrating macrophages and T cells, 34,36 which release cytokines to increase the expression of FAS by smooth muscle cells and are themselves a potential source of FAS ligand. 34 Not all apoptotic smooth muscle cells in atherosclerotic lesions express FAS receptors, 34,36 however, and other mechanisms must exist which initiate smooth muscle cell apoptosis in vascular lesions. Our data indicate that the neurotrophins represent an additional ligand receptor system to initiate smooth muscle cell apoptosis which is independent of lymphocyte/macrophage colocalization, as p75NTR colocalized to areas where apoptotic smooth muscle cells were present but macrophages were absent.
Alternatively, little is known about the signal transduction pathways regulating apoptosis of smooth muscle cells in response to ligand-induced activation of members of the TNF receptor superfamily and how these pathways are regulated in response to injury. It is well established, in Jurkat-T cells, that FAS-killing is mediated through activation of the caspase cascade. Binding of ligand to the FAS receptor leads to the recruitment of FAS-associated death domain (FADD), an adapter protein encoding a death domain, to the death domain of FAS. FADD then recruits and activates the initiator caspase, caspase 8, which, in turn, activates other caspases, eventually leading to activation of the effector caspase, caspase 3. In contrast, in oligodendrocytes, NGF activation of p75NTR induces the cleavage and activation of caspase 1 and caspase 3, but not caspase 8, 37 suggesting that p75NTR activates the caspase cascade by a mechanism distinct from that of FAS. The nuclear magnetic resonance (NMR) structure of the p75NTR death domain has distinctive differences from the death domain of FAS, 38 where, the death domain of p75NTR does not self-associate in solution, as the death domains of the FAS and TNFR1 receptors are known to do, nor does it bind to any of the known death domain adapter proteins. 38 These structural studies suggest that p75NTR may use alternative adapter proteins and signaling molecules to initiate caspase activation. The newly identified adapter protein neurotrophin receptor interacting factor (NRIF) has recently been described as a p75NTR-binding protein transducing apoptotic signals during embryonic development. 39 In addition, the adapter protein TRAF-6 interacts with the p75NTR-juxtamembrane domain and mediates p75-NTR induced nuclear factor κB activation. 40 Other studies have demonstrated that the increased expression of the pro-apoptotic molecules, p53 and BAX, mediates neurotrophin-induced apoptosis by p75NTR, although the signaling pathways leading to the increase in their expression in response to the neurotrophins are not completely understood. 41 Further studies will be necessary to determine whether there is regulation of expression of these or other, as yet unidentified signaling molecules in atherosclerotic lesions, which could regulate the apoptotic activity of p75NTR. Ultimately, however, although the upstream signals activated by p75NTR and FAS to initiate the caspase cascade may differ, both lead to the activation of the execution caspase, caspase 3, and our results demonstrate that a similar mechanism mediates smooth muscle cell apoptosis in response to p75NTR and FAS activation.
p75NTR activation has been implicated in mediating cell death in the nervous system, both during embryonic development and after neuronal injury. In mice carrying a deletion in the p75NTR gene, cell death is reduced in the retina and spinal cord, areas of the nervous system that express high levels of p75NTR during development. 42 In models of neuronal injury, p75NTR is expressed by hippocampal neurons undergoing apoptosis after seizures and by both oligodendrocytes and microglia in areas undergoing apoptosis in lesions from patients with multiple sclerosis. 43 NGF can induce apoptosis of cultured oligodendrocytes and of motor neurons in explants of rat embryonic spinal cords, 44 but only at high concentrations (>4 nmol/L). Neither BDNF nor NT3 however, has apoptotic activity on cultured oligodendrocytes, 13 and only BDNF has been reported to cause apoptosis of sympathetic neurons. 14 These results suggest that neurotrophin-induced apoptosis mediated by p75NTR may be a highly regulated process dependent on the cellular context in which it is induced. The ability of the neurotrophins to induce neuronal and glial cell death occurs in cells that express p75NTR, but not active trk receptors, 12,13,45 or in cells in which trk activity is reduced. 14,41 When both receptors are expressed and fully functional, signaling by trk receptors predominates, resulting in cell survival, mechanistically similar to the current study.
The present studies demonstrate a unique system, where in the presence of truncated and full length trk receptors, p75NTR mediates apoptosis of plaque-derived smooth muscle cells. Cultured HEDC expressed mRNA for p75NTR and kinase active trks, but in addition also expressed mRNA for truncated isoforms. This suggests that in HEDC, trk activity is reduced and neurotrophin-induced apoptosis is mediated by activation of p75NTR. This was confirmed in experiments where NGF induces apoptosis of p75-TsTmSMC but not trk A-TsTmSMC. Our results conflict with those of Bono et al, 46 who suggested that NGF-mediated apoptosis of cultured human medial smooth muscle cells was mediated by trk A. However, their conclusions were based on studies using pharmacological inhibitors of trk A receptor activation and downstream signaling components. Neither Trk A activation by NGF nor the expression of p75NTR was assessed. Thus, the role of trk A-mediated apoptosis in their studies is unclear. The expression of truncated trk isoforms by HEDC but the inability to detect them by immunohistochemistry in frozen tissue sections suggests that their expression in situ is below the limit of sensitivity of the antibodies.
Our studies are in agreement with those performed in neuronal cell systems demonstrating that the biological responsiveness of the neurotrophins is dependent on the class of receptor that is expressed (Figure 9) ▶ . Early in lesion development, in smooth muscle cells expressing full length, kinase active isoforms of trk, the neurotrophins confer a migratory response (Figure 9A) ▶ , 2,5 whereas in neointimal cells, which express p75NTR but in which kinase active trk activity is potentially reduced due to the coexpression of full length and truncated trk receptors, the apoptotic activity of the neurotrophins predominates (Figure 9B) ▶ . Thus, the ultimate responsiveness of neointimal smooth muscle cells to neurotrophins will depend on the dynamic regulation between the expression of full length isoforms, and potentially truncated isoforms of trk in the presence of p75NTR. Little is known about the regulation of trk receptor and p75NTR expression, or about how isoform switching from kinase-active to truncated trk receptors is modulated in response to injury. The coordinate regulation of both trk and p75NTR genes to promote neointimal lesion progression and later lesion regression suggests that complex regulatory signals are used to modulate lesion development in atherosclerosis, where reduced trk activation unmasks the apoptotic activity of the neurotrophins through p75NTR activation and may represent a mechanism for controlling apoptosis in vascular lesions.
Figure 9.

Model to assess the role of the neurotrophins in vascular lesion development. A: Migratory response of the neurotrophins on smooth muscle cells expressing full length, kinase-active trk receptors. B: Ligand-induced activation of p75NTR, in the presence of reduced trk activity, results in apoptosis of smooth muscle cells.
Acknowledgments
Special thanks to Hiep Nguyen, Michelle Lin, and A. J. Naiyer for technical assistance, to Drs. William Muller, Shahin Rafii, and Elaine Schattner for thoughtful review and discussion, and to Cherie Wieland for secretarial assistance.
Footnotes
Address reprint requests to Rosemary Kraemer, Ph.D., Department of Pathology, Weill Medical College of Cornell University, 1300 York Avenue, New York, NY 10021. E-mail: rtkraeme@mail.med.cornell.edu.
Supported by the American Heart Association grant-in-aid 9501510 (to R. K.) and Established Investigator Award (to B. L. H.), National Institutes of Health Public Service grants HL 58623 (to R. K.) and PO1HL46403 (to R. K. and B. H.), NIH National Heart, Lung and Blood Institute SCOR, HL56987 (to T. M.) and a VA Merit Review grant (to K. L. M.).
References
- 1.Chao MV, Hempstead BL: p75 and trk: a two-receptor system. Trends Neurosci 1995, 18:321-326 [PubMed] [Google Scholar]
- 2.Donovan MJ, Miranda RC, Kraemer R, Mccaffrey TA, Tessarollo L, Mahadeo D, Sharif S, Kaplan DR, Tsoulfas P, Parada L, Toran-Allerand D, Hajjar DP, Hempstead BL: Neurotrophin and neurotrophin receptors in vascular smooth muscle cells: regulation of expression in response to injury. Am J Pathol 1995, 147:309-324 [PMC free article] [PubMed] [Google Scholar]
- 3.Klein R, Lamballe F, Bryant S, Barbacid M: The trkB tyrosine protein kinase is a receptor for neurotrophin 4. Neuron 1992, 8:947-956 [DOI] [PubMed] [Google Scholar]
- 4.Cordon-Cardo C, Tapley P, Jing S, Nanduri V, O’Rourke E, Lamballe F, Kovary K, Klein K, Jones KR, Reichardt LF, Barbacid M: The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell 1991, 66:173-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kraemer R, Ngyuen H, March KL, Hempstead BL: NGF activates similar intracellular signaling pathways in vascular smooth muscle cells as PDGF-BB but elicits different biological respones. Arterioscler Thromb Vasc Biol 1999, 19:1041-1050 [DOI] [PubMed] [Google Scholar]
- 6.Klein R, Conway D, Parada LF, Barbacid M: The trk B tyrosine kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 1990, 61:647-656 [DOI] [PubMed] [Google Scholar]
- 7.Tsoulfas P, Soppet D, Escandon E, Tessarollo L, Mendoza-Rameriez J-L, Rosenthal A, Nikolics K, Parada LF: The rat trkC locus endodes multiple neurogenic receptors that exhibit differential response to neurotrophin-3 in PC12 cells. Neuron 1993, 10:975-990 [DOI] [PubMed] [Google Scholar]
- 8.Eide FF, Vining ER, Eide BL, Zang KL, Wang XY, Reichardt LF: Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci 1996, 16:3123-3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palko ME, Coppola V, Tessarollo L: Evidence for a role of truncated trk C receptor isoforms in mouse development. J Neurosci 1999, 19:775-782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ware CF, VanArsdale S, VanArsdale TL: Apoptosis mediated by the TNF-related cytokine and receptor families. J Cell Biochem 1996, 60:47-55 [DOI] [PubMed] [Google Scholar]
- 11.Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV: High affinity NGF binding requires coexpression of the trk proto-oncogene and the low affinity NGF receptor. Nature 1991, 350:678-683 [DOI] [PubMed] [Google Scholar]
- 12.Frade JM, Rodriguez-Tebar A, Barde Y-A, Rodrigueztebar A, Barde YA: Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 1996, 383:166-168 [DOI] [PubMed] [Google Scholar]
- 13.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV: Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 1996, 383:716-719 [DOI] [PubMed] [Google Scholar]
- 14.Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD: The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 1998, 140:911-923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber LJ, Hempstead B, Donovan MJ: Neurotrophin and neurotrophin receptors in human fetal kidney. Dev Biol 1996, 179:369-381 [DOI] [PubMed] [Google Scholar]
- 16.Donovan MJ, Hahn R, Tessarollo L, Hempstead BL: Identification of an essential nonneuronal function of neurotrophin 3 in mammalian cardiac development. Nat Genet 1996, 14:210-213 [DOI] [PubMed] [Google Scholar]
- 17.Han DKM, Haudenschild CC, Hong MK, Tinkle BT, Leon MB, Liau G: Evidence for apoptosis in human atherogenesis and in a rat vascular injury model. Am J Pathol 1995, 147:267-277 [PMC free article] [PubMed] [Google Scholar]
- 18.Chao MV, Bothwell M, Ross AH, Kowprowski H, Lanahan AA, Buck CR, Seghal A: Gene transfer and molecular cloning of the human NGF receptor. Science 1986, 232:518-521 [DOI] [PubMed] [Google Scholar]
- 19.Johnson D, Lanahan A, Buck CR, Seghal A, Morgan C, Mercer E, Bothwell M, Chao M: Expression and structure of the human NGF receptor. Cell 1986, 47:545-554 [DOI] [PubMed] [Google Scholar]
- 20.Ross AH, Grob P, Bothwell M, Elder DE, Ernst CS, Marano N, Ghrist FD, Slemp CC, Herlyn M, Atkinson B, Koproswki H: Characterization of nerve growth factor receptor in neural crest tumors using monoclonal antibodies. Proc Natl Acad Sci USA 1984, 81:6681-6685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mccaffrey TA, Du B, Consigli S, Szabo P, Bray PJ, Hartner L, Weksler BB, Sanborn TA, Bergman G, Bush HL, Jr: Genomic instability in the type II TGF-b1 receptor gene in atherosclerotic and restenotic vascular cells. J Clin Invest 1997, 100:2182-2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mccaffrey TA, Consigli S, Du B, Falcone DJ, Sanborn TA, Spokojny AM, Bush HL, Jr: Decreased type II/type I TGF-beta receptor ratio in cells derived from human atherosclerotic lesions: conversion from an antiproliferative to profibrotic response to TGF-beta1. J Clin Invest 1995, 96:2667-2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mccaffrey TA, Pomerantz KB, Sanborn TA, Spokojny AM, Du B, Park MH, Folk JE, Lamberg A, Kivirikko KI, Falcone DJ, Mehta SB, Hanauske-Abel HM: Specific inhibition of elF-5A and collagen hydroxylation by a single agent: antiproliferative and fibrosuppressive effects on smooth muscle cells from human coronary arteries. J Clin Invest 1995, 95:446-455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform-extraction. Anal Biochem 1987, 162:156-159 [DOI] [PubMed] [Google Scholar]
- 25.Puissant C, Houdebine L-M: An improvement of the single-step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Biotechniques 1990, 8:148-149 [PubMed] [Google Scholar]
- 26.Newman PJ, Berndt MC, Gorski J, White GS, II, Lyman S, Paddock C, Muller WA: PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science 1990, 247:1219-1222 [DOI] [PubMed] [Google Scholar]
- 27.Labouyrie E, Dubus P, Groppi A, Mahon FX, Ferrer J, Parrens M, Reiffers J, de Mascarel A, Merlio JP: Expression of neurotrophins and their receptors in human bone marrow. Am J Pathol 1999, 154:405-415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR: Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron 1992, 9:883-896 [DOI] [PubMed] [Google Scholar]
- 29.Huber LJ, Chao MV: A potential interaction of p75 and trk A NGF receptors revealed by affinity crosslinking and immunoprecipitation. J Neurosci Res 1995, 40:557-563 [DOI] [PubMed] [Google Scholar]
- 30.Ogasawara J, Watanabe-Fukanage R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S: Lethal effects of the anti-Fas antibody in mice. Nature 1993, 364:806-809 [DOI] [PubMed] [Google Scholar]
- 31.Yonehara S, Ishii A, Yonehara M: A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med 1989, 169:1747-1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ: In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differentiation 1998, 5:1004-1016 [DOI] [PubMed] [Google Scholar]
- 33.Geng Y-J, Libby P: Evidence for apoptosis in advanced human atheroma: colocalization with interleukin-1beta-converting enzyme. Am J Pathol 1995, 147:251-266 [PMC free article] [PubMed] [Google Scholar]
- 34.Geng YJ, Henderson LE, Levesque EB, Muszynski M, Libby P: Fas is expressed in human atherosclerotic intima and promotes apoptosis of cytokine-primed human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 1997, 17:2200-2208 [DOI] [PubMed] [Google Scholar]
- 35.Cai W, Devauz B, Schaper W, Schaper J: The role of Fas/APO 1 and apoptosis in the development of human atherosclerotic lesions. Atherosclerosis 1997, 131:177-186 [DOI] [PubMed] [Google Scholar]
- 36.Cai W-J, Devaux B, Schaper W, Schaper J: The role of Fas/APO 1 and apoptosis in the development of human atherosclerotic lesions. Atherosclerosis 1997, 131:177-186 [DOI] [PubMed] [Google Scholar]
- 37.Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV: Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci 1999, 19:3043-3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liepinsh E, Ilag L, Otting G, Ibanez C: NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J 1997, 16:4999-5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde Y-A: The zinc finger protein NRIF interacts with the neurotrophin receptor p75NTR and participates in programmed cell death. EMBO J 1999, 18:6050-6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khursigara G, Orlinick JR, Chao MV: Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem 1999, 274:2597-2600 [DOI] [PubMed] [Google Scholar]
- 41.Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD: P53 is essential for developmental neuron death as regulated by the trk A and p75 neurotrophin receptors. J Cell Biol 1998, 143:1691-1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frade JM, Barde Y-A: Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development 1999, 126:683-690 [DOI] [PubMed] [Google Scholar]
- 43.Dowling P, Ming X, Raval S, Husar W, Casaccia-Bonnefil P, Chao M, Cook S, Blumberg B: Up-regulated p75NTR neurotrophin receptor on glial cells in MS plaques. Neurology 1999, 53:1676-1682 [DOI] [PubMed] [Google Scholar]
- 44.Sedel F, Bechade C, Triller A: Nerve growth factor (NGF) induces motorneuron apoptosis in rat embryonic spinal cord in vitro. Eur J Neurosci 1999, 11:3904-3912 [DOI] [PubMed] [Google Scholar]
- 45.Weise S, Metzger F, Holtmann B, Sendtner M: The role of p75NTR in modulating neurotrophin survival effects in developing motorneurons. Eur J Neurosci 1999, 11:1668-1676 [DOI] [PubMed] [Google Scholar]
- 46.Bono F, Lamarche I, Herbert J-M: NGF exhibits a pro-apoptotic activity for human vascular smooth muscle cells that is inhibited by TGFb1. FEBS Lett 1997, 416:243-246 [DOI] [PubMed] [Google Scholar]
- 47.van Engeland M, Ramaekers FCS, Schutte B, Reutelingsperger CPM: A novel assay to measure loss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry 1996, 24:131-139 [DOI] [PubMed] [Google Scholar]
- 48.Castedo M, Hirsch T, Susin SA, Zamzami N, Marchetti P, Macho A, Kroemer G: Sequential acquisition of mitochondrial and plasma membrane alterations during early lymphocyte apoptosis. J Immunol 1996, 157:512-521 [PubMed] [Google Scholar]
- 49.Donovan MJ, Lin M, Wiegn P, Ringsted R, Kraemer R, Hahn R, Wang S, Ibanez C, Raffi SM, Hempstead BL: Brain-derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development (in press) [DOI] [PubMed]