

Abstract
Hepatoblastomas (HBs), representing malignant liver tumors of childhood, show frequent loss of heterozygosity (LOH) in the chromosomal region 11p15.5. This loss is of maternal origin suggesting the presence of a monoallelically expressed tumor suppressor gene in this region. p57KIP2 (KIP2) located at 11p15.5 is predominantly expressed from the maternal allele and encodes a cyclin-dependent kinase inhibitor. We screened a series of 56 HB tumors and five HB cell lines for allelic loss (LOH) of the KIP2 locus by microsatellite analysis and KIP2 coding sequence mutations by single-strand conformation polymorphism analysis. Although LOH at the KIP2 locus occurred in 25% of the cases, no mutations were found. Analysis of KIP2 mRNA expression by competitive reverse transcriptase-polymerase chain reaction revealed up-regulation in nine of 12 HBs compared to matching liver samples. In contrast, mRNA levels of the putative suppressor gene H19 on 11p15.5 were decreased in 10 of 12 tumors, indicating that KIP2 and H19 are not co-regulated in HBs. IGF2 mRNA expression was increased in 11 of 12 HB samples. All HBs showed monoallelic KIP2 expression. However, the overexpression of KIP2 in HBs with maternal loss of 11p15.5 suggests a reactivation of the paternal allele in these cases. Overexpression of KIP2 in HBs argues against a role as a HB suppressor gene.
Hepatoblastoma (HB) is the most common primary liver tumor in childhood with an incidence of 0.7 to 1 new case/million children 15 years of age or younger. 1 Although most cases are sporadic, an increased incidence is found in patients with familial adenomatous polyposis and Beckwith-Wiedemann syndrome (BWS). 2 Previous studies have revealed frequent loss of heterozygosity (LOH) in the chromosomal regions 1p and 11p15.5, 3,4 the latter region also being implicated in BWS and in several sporadic cancers, including other childhood tumors such as Wilms’ tumors (WTs) and some common adult cancers (eg, breast cancer). The lost alleles in HB at 11p15.5 are exclusively of maternal origin, 4 suggesting a tumor suppressor gene located there that is monoallelically expressed from the maternal allele. KIP2 is located in the 11p15.5 region 5 and belongs to the Cip/Kip-family of cyclin-dependent kinase inhibitors which share a common N-terminal domain for binding to and inhibiting the kinase activity of several cyclin-dependent kinase (CDK)-cyclin complexes active at the G1-checkpoint of the cell cycle. 3,6 As is the case for p21WAF1/CIP1, p57KIP2 has been reported to bind to proliferating cell nuclear antigen (PCNA) through its C-terminal domain and, thereby, to inhibit PCNA-dependent DNA replication in vitro and entry into S phase in vivo. 7
Mice lacking KIP2 function display phenotypes similar to those seen in BWS 8,9 and ∼5% of BWS patients carry germ line mutations in the KIP2 gene. 10-13 In addition, KIP2 is known to be expressed predominantly from the maternal allele 14,15 and its expression is significantly reduced in certain WTs and other embryonal tumors. 16 Thus, KIP2 is a candidate for the 11p15.5 tumor suppressor involved in HB. We, therefore, decided to screen a large series of HBs for KIP2 mutations and to determine the level of KIP2 mRNA expression in these tumors.
Two other important genes on 11p15.5 are imprinted, namely IGF2 and H19. IGF2 is expressed monoallelically from the paternal allele whereas H19 is expressed exclusively from the maternal allele. 17,18 IGF-II is a major fetal mitogen and growth factor whereas H19 codes for an apparently untranslated RNA whose function is still disputed. 19-21 Most WTs show increased IGF2 expression, because of loss of imprinting (LOI) with biallelic expression, along with markedly reduced H19 expression, either because of maternal LOH on 11p15.5 or because of biallelic hypermethylation of the gene, suggesting that there is a reciprocal regulation of these genes’ mRNA expressions that is disrupted in tumors. 22-24 We therefore also examined the expression levels of IGF2 and H19 in our tumors as well as the imprinting status of all three genes to assess if these genes show coordinate alterations in their expression or imprinting status.
Materials and Methods
Patients, Tumors, Cell Lines
We examined a total of 56 HB biopsies and the following continuous HB cell lines: HepT1, 25 HuH6, 26 HepT2, HepT3, and HepT4 (T Pietsch, and D von Schweinitz, unpublished observations). The cell line HUH6 was obtained from the Japanese Collection of Research Biosources (Osaka, Japan). Cell line HepT3 was established from tumor D204, HepT1 from DZ25, HepT2 from D166, and HepT4 from D161. Most of the HB patients were enrolled in the multicenter treatment study for pediatric malignant liver tumors of the German Society of Pediatric Hematology and Oncology. The age of the patients varied between 2 to 57 months. Constitutional DNA was available in all cases from peripheral blood or matching tumor-adjacent normal liver tissue (MNLT). For more detailed histological and clinical data on the subset of cases included in the study of expression and imprinting see Table 1 ▶ .
Table 1.
Clinical Data and Results of LOH Analysis for the Patients Involved in the Study of Expression and Imprinting
| Case | Age (months)/ sex | Histology | Chemo- therapy | LOH | D11S554 | WT1 | D11S1020 | D11S1323 | D11S1338 | HBB | HBE1 | BWR1A | D11S1318 | TH | IGF2 | H19 | DRD4 | D11S1363 | HRAS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D269 | 9 /F | Mixed | Yes | No | |||||||||||||||
| D272 | 12 /M | Mixed | No | No | |||||||||||||||
| D266 | 8 /F | Mixed | No | No | |||||||||||||||
| D263 | 12 /M | Epithelial | No | No | |||||||||||||||
| D166 | 48 /M | Epithelial, multifocal | Yes | Yes | • | • | ni | • | • | • | ni | • | • | ni | ni | ni | ni | ni | ni |
| D23II | 22 /M | Epithelial | No | No | |||||||||||||||
| D195 | 19 /M | Epithelial | Yes | No | |||||||||||||||
| D197 | 54 /M | Epithelial | Yes | No | |||||||||||||||
| DZ18 | 6 /M | Epithelial | No | Yes | ○ | ○ | ○ | ni | • | • | • | ni | ni | • | ni | ni | • | • | • |
| M12 | 13 /M | Epithelial | No | Yes | — | — | — | — | — | — | — | • | — | — | • | ni | — | — | — |
| M14 | 20 /M | Epithelial | Yes | No | |||||||||||||||
| D401 | 9 /F | Mixed | No | No | |||||||||||||||
| D104 | 27 /F | Epithelial | No | Yes | • | • | • | • | • | • | ni | ni | • | • | • | ni | ni | ○ | ○ |
| D199 | 25 /F | Epithelial | No | No | |||||||||||||||
| D497 | 6 /F | Epithelial | Yes | No | |||||||||||||||
| D268 | 6 /F | Mixed | No | Yes | ○ | ni | ○ | • | — | • | • | • | • | • | • | ni | ni | ni | ni |
| D161 | 11 /F | Epithelial | No | No | |||||||||||||||
| DZ29II | 13 /M | Epithelial | No | No | |||||||||||||||
| DZ20 | 27 /F | Epithelial, small cells | No | No | |||||||||||||||
| DZ25II | 34 /F | Epithelial, multifocal | No | Yes | • | • | ni | • | • | ni | ni | • | • | ni | • | — | ni | ni | • |
| D175 | 16 /M | Epithelial | No | No | |||||||||||||||
| D204 | 4 /F | Epithelial | No | No | |||||||||||||||
| D319 | 4 /M | Epithelial | No | No | |||||||||||||||
| D399II | 2 /M | Epithelial | No | No |
Information on the sample included in the examination of RNA expression and imprinting of p57KIP2, H19, and IGF2: clinical data, histological type, results of LOH studies on 11p15.5. Symbols and abbreviations: CL, cell line; ○, maintenance of heterozygosity; •, loss of heterozygosity; ni, noninformative; –, not done.
DNA Extraction
Tissue fragments selected for DNA extraction were checked by frozen section to ensure that they consisted either of tumor or of liver. Only fragments with a tumor cell content of at least 80% were included. DNA was extracted from the tissue fragments and peripheral blood by standard Proteinase K/sodium dodecyl sulfate digestion followed by phenol/chloroform extraction as described before. 27
LOH Analysis
All 56 HBs were analyzed for LOH on 11p15.5 by microsatellite analysis essentially as described previously. 4 Markers examined included the microsatellites HBB, HBE1, TH, D11S1318, DRD4, D11S1363, and HRAS which were amplified by polymerase chain reaction (PCR), separated by denaturating polyacrylamide gel electrophoresis, and visualized by silver staining. The restriction fragment length polymorphisms (RFLPs) of the IGF2 and H19 genes were amplified by PCR, digested with the appropriate restriction enzyme, and products were separated and visualized by single-strand conformation polymorphism (SSCP) and silver staining (IGF2) or agarose gel electrophoresis (H19). For BWR1A, sequence polymorphisms were PCR-amplified followed by SSCP and silver staining.
SSCP Analysis of the KIP2 Gene and DNA Sequencing
Fifty-two HBs and three cell lines (HepT1, HUH6, and HepT3) were examined by SSCP for KIP2 mutations. Ten to 50 ng of genomic DNA were used in each PCR reaction for amplification of six fragments spanning the coding region of the gene (Figure 1) ▶ . The reactions were all performed using a Uno Thermoblock cycler (Biometra, Göttingen, Germany), in a final volume of 10 μl, which contained 200 mmol/L of each deoxyribonucleoside triphosphate, 5 pmol of each primer, 0.25 unit of Taq polymerase (Life Technologies, Inc., Karlsrūhe, Germany), PCR buffer containing 1 mmol/L MgCl2 (Life Technologies, Inc.) and 10% dimethyl sulfoxide (Sigma Chemical Co., Deisenhofen, Germany). For amplification of fragment 4, we replaced dGTP with 7-deaza-dGTP (Boehringer Mannheim, Mannheim, Germany). The PCR products were mixed with an equal volume of loading buffer containing 90% formamide, denatured for 5 minutes at 95°C, and run out on nondenaturing polyacrylamide gels with different acrylamide:bisacrylamide ratios (Life Technologies, Inc.) and the single and double strands were visualized by silver staining as described previously. 28 To enhance sensitivity for all fragments but fragment 4, two different running conditions were used. For details, see Table 2 ▶ and Figure 1 ▶ .
Figure 1.

Schematic diagram to illustrate alternative splicing of the KIP2 gene 31 and the six PCR fragments analyzed.
Table 2.
Primers, PCR, and Gel Conditions Used for SSCP Analysis of the p57KIP2 Gene
| Primers forward (fw)-reverse (rv) | PCR conditions | Gel conditions (acrylamide:bis) | |
|---|---|---|---|
| Frag 1 | fw 5′GTCCACCACCGGACAGCCAG3′ | 1.0 mmol/L MgCl, 10% DMSO | 10% 29:1 22°C |
| rv 5′CGACAAGACGCTCCATCGTG3′ | 35 cycles 94°C× 35 seconds/62°C× 40 seconds/72°C× 40 seconds | 10% 79:1 4°C | |
| Frag 2 | fw 5′CGTCCCTCCGCAGCACATCC3′ | 1.0 mmol/L MgCl, 10% DMSO | 10% 29:1 4°C |
| rv 5′CCTGCACCGTCTCGCGGTAG3′ | 35 cycles 94°C× 35 seconds/60°C× 40 seconds/72°C× 40 seconds | 10% 29:1 10% glycerol 22°C | |
| Frag 3 | fw 5′TGGACCGAAGTGGACAGCGA3′ | 1.0 mmol/L MgCl, 10% DMSO | 10% 29:1 22°C |
| rv 5′GGGGCCAGGACCGCGACC3′ | 35 cycles 94°C× 35 seconds/62°C× 40 seconds/72°C× 40 seconds | 10% 79:1 4°C | |
| Frag 4 | fw 5′CGGAATTCCGGAGCAGCTGCCTAGTGTC3′ | 1.0 mmol/L MgCl, 10% DMSO, 7-deaza dGTP | 10% 49:1 4°C |
| rv 5′CGTCCCGAAATCCCCGAGTGC3′ | 40 cycles 94°C× 35 seconds/63°C× 30 seconds/72°C× 30 seconds | ||
| Frag 5 | fw 5′CCGAGCAGGGCGCGAACCAG3′ | 1.0 mmol/L MgCl, 10% DMSO | 10% 29:1 22°C |
| rv 5′CACGGGAGGAGGCGGGAACC3′ | 35 cycles 94°C× 35 seconds/62°C× 40 seconds/72°C× 40 seconds | 10% 29:1 10% glycerol 4°C | |
| Frag 6 | fw 5′TGTCGCCCGCAGATTTCTTC3′ | 1.0 mmol/L MgCl, 10% DMSO | 10% 29:1 10% glycerol 22°C |
| rv 5′CCCAGGTGCGCTGTACTCAC3′ | 35 cycles 94°C× 35 seconds/60°C× 40 seconds/72°C× 40 seconds | 10% 29:1 10% glycerol 4°C |
Abbreviations: Frag, fragment; DMSO, dimethyl sulfoxide; MgCl, magnesium chloride (MgCl2).
In cases showing a mobility shift compared to a normal control, we performed direct sequencing of DNA from the tumor tissue and the corresponding normal tissue with the same primers and PCR conditions as for the SSCP analysis. PCR products were purified using spin columns (QIAquick PCR purification kit; Qiagen, Inc., Hilden, Germany) and sequencing of 20 ng of PCR product was done applying the ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Weiterstadt, Germany) on a TC 9600 Thermocycler (Perkin-Elmer-Cetus, Emeryville, CA). Sequencing products were analyzed on an ABI 373A sequencer (Applied Biosystems).
RNA Extraction, cDNA Preparation, and Semiquantitative RT-PCR
Twenty-three of the tumors were included in the analysis of expression; for 12 of them, RNA from MNLT was also available. Four normal liver samples from children (newborn to an age of 11 years) and one fetal liver (14-weeks gestational age), were also included in the analysis.
Before RNA extraction, tissue samples were first examined by frozen section to verify their composition. Total cellular RNA was extracted by lysis in guanidinium thiocyanate and ultracentrifugation through a cesium chloride cushion 29 or by extraction with the Trizol reagent (Life Technologies, Inc.), according to the manufacturer’s instructions. To remove any contaminating DNA, RNA was digested with DNase I (Boehringer Mannheim). To evaluate the amount of KIP2, H19, and IGF2 transcripts by competitive RT-PCR, exogenous RNA standards for these genes and β2-microglobulin, a housekeeping gene, were generated with internal deletions by in vitro mutagenesis and in vitro transcription. 30 RNA (250 ng) from each tumor or normal liver sample were reverse-transcribed together with known amounts of exogenous standards covering the equimolar range of the corresponding RNA transcripts. Reverse transcription was performed using the Superscript II Preamplification System (Life Technologies, Inc.) with random hexamers as primers in a final volume of 10 μl. Absence of contaminating genomic DNA was confirmed by amplification of a PCR fragment of the CDK4 gene with primers spanning the intronic sequence.
cDNA (0.5 μl) was used as template in RT-PCR amplification of the target and the housekeeping gene transcripts. Primers, fragment sizes, and PCR conditions were for KIP2 5′-GTCCACCACCGGACAGCCAG-3′ and 5′-AGAACGCGGGCACCGAGTCG-3′, product size wild type 268 bp, standard 235 bp, PCR conditions: 1.0 mmol/L MgCl2, 10% dimethyl sulfoxide, 40 cycles of 94°C × 40 seconds, 63°C × 45 seconds, 72°C × 50 seconds; for H19 5′-AAATGGTGCTACCCAGCTC-3′ and 5′-GGTCTGGTTCCTCTAGCTTC-3′, product size wild type 145 bp, standard 126 bp, PCR conditions: 1.0 mmol/L MgCl2, 10% dimethyl sulfoxide, 40 cycles of 94°C × 40 seconds, 52°C × 50 seconds, 72°C × 50 seconds; for IGF2 5′-TGCTGCATTGCTGCTTACCG-3′ and 5′-ACAGCACTCCTCAACGATGC-3′, product size wild type 153 bp, standard 145 bp, PCR with 1.0 mmol/L MgCl2, 40 cycles of 94°C × 35 seconds, 52°C × 40 seconds, 72°C × 40 seconds; for β2-microglobulin 5′-TGTCTTTCAGCAAGGACTGG-3′ and 5′-GATGCTTACATGTCT-CG-3′, product size wild type 148 bp, standard 130 bp, PCR conditions: 1.5 mmol/L MgCl2, 40 cycles of 94°C × 30 seconds, 57°C × 40 seconds, 72°C × 40 seconds. All PCR reactions were performed using a Uno Thermoblock cycler (Biometra), in a volume of 10 μl, with the same reagents as above. One primer for each gene was labeled with a fluorescent dye. Primers were chosen from adjacent exons spanning intronic sequences to exclude signals from residual genomic DNA. The KIP2 mRNA shows alternative splicing variants that include intronic sequences within the main transcript (Figure 1) ▶ . However, these products, as already reported by Tokino et al, 31 constituted <10% of total transcript in all of our samples. In the following we always refer to the main transcript. Amplicons were separated on a 6% denaturating acrylamide gel on the DNA sequencer (as above) using the Genescan software (ABI). The expression levels of target RNAs were calculated as the ratio of the target’s and its standard’s signal intensities, which was then normalized by dividing it by the β2-microglobulin level: (TARGETsample/TARGETstandard) / (β2-microglobulinsample/β2-microglobulinstandard). Constant amounts of standards allowed for the comparative analysis of the expression of our target genes among the samples and the pairs of tumor tissue and matching liver.
RFLP Analysis for KIP2, H19, and IGF2
Imprinting of KIP2, IGF2, and H19 was examined in 24 of the samples for which RNA was available and in three cell lines (HepT1, HepT3, HUH6). For this purpose, we first analyzed gDNA from these cases to determine which ones were informative for known transcribed exonic RFLPs for each of the three genes. For heterozygotes, the analysis was then repeated with cDNA to assess the allelic expression pattern.
For KIP2 we examined the PAPA polymorphism in exon 2 15 using nested PCR. First round primers were 5′-ACCGTCGGGATTACGACTTCCA-3′ and 5′-CAAAACCGAACGCTGCTCTGC-3′. PCR was performed with 20 ng of constitutional genomic DNA or 1 μl of the RT reaction product in a final volume of 10 μl with 2.0 mmol/L MgCl2, 5% dimethyl sulfoxide, and 7-deaza-dGTP instead of dGTP (other components as above). The PCR protocol was 95°C × 30 seconds, 54°C × 40 seconds, 72°C × 120 seconds for 20 cycles. The second round, performed with the primers 5′-GTTCTACCGCGAGACGGTGCAG-3′ and 5′-CTCTTGCGCTTGGCGAAGAAAT-3′, using 1 μl of the product of the first PCR as template, was identical to the first round except for an MgCl2 concentration of 1.5 mmol/L and an annealing temperature of 55°C. The products of the second round were digested in a volume of 20 μl with 20 units of PvuII (Boehringer Mannheim) for 18 hours. Products were run out on nondenaturating polyacrylamide gels (8%, 29:1 acrylamide:bisacrylamide) which were silver stained. 32 The imprinting status of H19 was analyzed using a transcribed RsaI polymorphism located in exon 5, 33 using primers and PCR conditions, as described previously. 27 Allele-specific expression of the IGF2 gene was examined with the ApaI RFLP in exon 9, 34 as described elsewhere. 27 Products were digested with the appropriate restriction enzyme (Boehringer Mannheim) and separated on 10% denaturating polyacrylamide gels which were processed as described above.
Results
LOH Analysis
In agreement with results reported previously, 4 we detected LOH on 11p15.5 in 14 of 56 cases (25%). As has been shown before, for HBs as well as other embryonal tumors, the lost allele was found to be of maternal origin in those cases in which parental DNA was available (data not shown). Detailed LOH data concerning the subset of cases included in the study of expression are summarized in Table 1 ▶ .
SSCP Analysis of KIP2
No mutations in the coding sequence of the gene were found. One band shift detected represents a rare polymorphism in the nontranslated region located in intron 1 of the main splicing variant, 60 bases after the last base of exon 1, 31 changing G to A; it was found only once in our set of 52 tumors. The other band shifts found were all located in the known polymorphic regions of the gene covered by SSCP fragments 3 and 4 and their polymorphic background was confirmed by demonstration of the same band pattern with constitutional DNA.
Examination of Expression of KIP2, H19, and IGF2
In our set of tumors, KIP2 transcription was up-regulated in the tumors compared to the MNLT in nine of the 12 cases for which MNLT was available (Figure 2b ▶ and Table 3 ▶ ). The increase (expressed as ratios of tumor/MNLT) in the level of mRNA transcript ranged from 2.12 to 29.17 (mean, 8.19). In seven of these nine cases, the increase ranged from 2.12 to 5.57 with a mean of 3.24 whereas two samples (D263 and D197) had a highly increased KIP2 expression of 21.83 and 29.17. Three samples (D166, M14, and D401) showed slight to clear reduction of KIP2 RNA ranging from 0.26 to 0.81 (mean, 0.50). Three (DZ18, D166, and M12) of the total 12 cases displayed LOH on 11p15.5 (DZ18 and D166, proven to be of maternal origin) but only D166 showed a reduction of expression whereas DZ18 and M12 had increases of 2.21 and 3.40, respectively.
Figure 2.
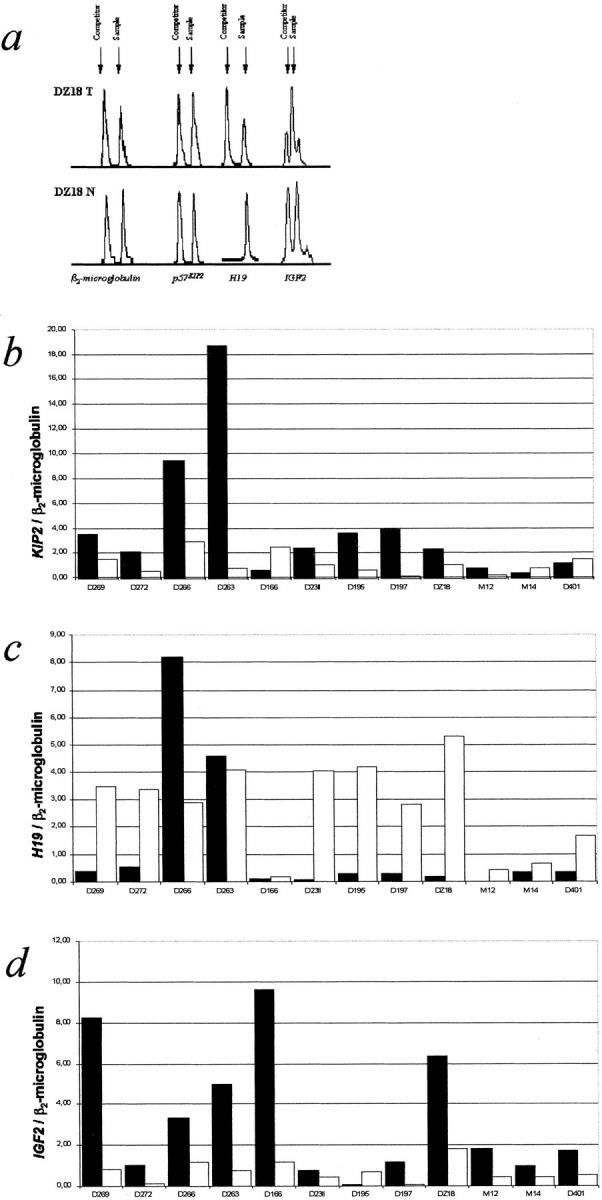
Analysis of RNA expression of KIP2, H19, and IGF2. a: Graphs obtained for the sample DZ18T and the corresponding normal liver tissue. For each gene the peaks shown here are derived from samples with equal amounts of competitor in tumor and liver RNA. The amount of H19 in the normal liver sample suppresses amplification of the competitor. This sample displays two peaks in another RT-sample with more competitor added (not shown). Results of the analysis of mRNA expression for KIP2 (b), H19 (c), and IGF2 (d) in 12 HB for which matching normal liver tissue was available: ▪, tumor; □, matching normal liver tissue.
Table 3.
Expression of KIP2, H19, and IGF2 mRNA in Hepatoblastomas and Liver Samples
| RNA | KIP2 | H19 | IGF2 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor | Liver | Ratio | Tumor | Liver | Ratio | Tumor | Liver | Ratio | ||||||||||
| Tumors with matching normal liver tissue | ||||||||||||||||||
| D269 | 3.58 | 1.54 | 2.33 | 0.42 | 3.52 | 0.12 | 8.27 | 0.82 | 10.14 | |||||||||
| D272 | 2.13 | 0.55 | 3.88 | 0.59 | 3.38 | 0.17 | 1.00 | 0.14 | 6.98 | |||||||||
| D266 | 9.50 | 2.98 | 3.19 | 8.24 | 2.93 | 2.81 | 3.36 | 1.19 | 2.83 | |||||||||
| D263 | 18.72 | 0.86 | 21.83 | 4.61 | 4.11 | 1.12 | 5.01 | 0.75 | 6.71 | |||||||||
| D166 | 0.65 | 2.55 | 0.26 | 0.12 | 0.19 | 0.60 | 9.64 | 1.18 | 8.15 | |||||||||
| D23II | 2.44 | 1.15 | 2.12 | 0.09 | 4.08 | 0.02 | 0.76 | 0.44 | 1.72 | |||||||||
| D195 | 3.67 | 0.66 | 5.57 | 0.31 | 4.21 | 0.07 | 0.07 | 0.70 | 0.10 | |||||||||
| D197 | 3.89 | 0.13 | 29.17 | 0.33 | 2.84 | 0.12 | 1.15 | 0.06 | 19.73 | |||||||||
| DZ18 | 2.37 | 1.07 | 2.21 | 0.20 | 5.33 | 0.04 | 6.34 | 1.83 | 3.47 | |||||||||
| M12 | 0.82 | 0.24 | 3.40 | 0.02 | 0.45 | 0.05 | 1.80 | 0.45 | 4.00 | |||||||||
| M14 | 0.38 | 0.86 | 0.44 | 0.38 | 0.70 | 0.54 | 0.98 | 0.42 | 2.33 | |||||||||
| D401 | 1.23 | 1.52 | 0.81 | 0.37 | 1.68 | 0.22 | 1.72 | 0.57 | 3.03 | |||||||||
| Tumors without matching normal liver tissue | ||||||||||||||||||
| D104 | 0.43 | 0.32 | 6.34 | 2.14 | 5.85 | 6.51 | ||||||||||||
| D199 | 0.61 | 0.45 | 4.61 | 1.56 | 1.28 | 1.42 | ||||||||||||
| D497 | 0.40 | 0.30 | 3.46 | 1.17 | 0.46 | 0.51 | ||||||||||||
| D268 | 2.77 | 2.07 | 1.05 | 0.36 | 6.99 | 7.77 | ||||||||||||
| D161 | 4.55 | 3.40 | 1.87 | 0.63 | 1.42 | 1.58 | ||||||||||||
| DZ29II | 6.20 | 4.64 | 3.49 | 1.18 | 1.23 | 1.37 | ||||||||||||
| DZ20 | 32.90 | 24.61 | 0.01 | 0.00 | 0.01 | 0.01 | ||||||||||||
| DZ25II | 49.90 | 37.32 | 1.30 | 0.44 | 7.58 | 8.43 | ||||||||||||
| D175 | 6.42 | 4.80 | 1.04 | 0.35 | 2.90 | 3.23 | ||||||||||||
| D204 | 8.18 | 6.12 | 2.43 | 0.82 | 1.00 | 1.12 | ||||||||||||
| D319 | 0.95 | 0.71 | 1.83 | 0.62 | 3.54 | 3.94 | ||||||||||||
| Cell lines | ||||||||||||||||||
| DZ27II CLHepT1 | 3.67 | 2.75 | 0.30 | 0.10 | 0.08 | 0.08 | ||||||||||||
| D501 CL HepT3 | 0.00 | 0.00 | 1.87 | 0.63 | 13.17 | 14.65 | ||||||||||||
| D339 CL HUH6 | 0.00 | 0.00 | 0.01 | 0.00 | 1.22 | 1.35 | ||||||||||||
| CL HepT2 | 0.00 | 0.00 | 0.00 | 0.00 | 23.40 | 26.03 | ||||||||||||
| CL Hep T4 | 0.00 | 0.00 | 0.00 | 0.00 | 6.10 | 6.79 | ||||||||||||
| Control tissues | ||||||||||||||||||
| F17 fetal liver (GA 14w) | 4.47 | 3.35 | 9.43 | 3.19 | ↑↑↑ | ↑↑↑ | ||||||||||||
| newborn liver | 0.61 | 0.46 | 2.71 | 0.91 | 0.09 | 0.10 | ||||||||||||
| D400 liver 10m | 2.59 | 1.94 | — | — | 2.69 | 2.99 | ||||||||||||
| D162 liver 19m | 2.78 | 2.08 | 1.46 | 0.49 | 1.92 | 2.14 | ||||||||||||
| M15 liver 36m | 0.33 | 0.25 | — | — | 0.34 | 0.38 | ||||||||||||
| liver 11y | 1.59 | 1.19 | 0.44 | 0.15 | 0.74 | 0.82 | ||||||||||||
Results of the examination of expression of p57KIP2, H19, and IGF2. For 12 cases with matching normal liver tissue, the mRNA expression levels and the tumor/matching normal liver tissue ratio is indicated; for the other cases, the cell lines and the controls, the mRNA expression level and the sample/average normal liver ratio is indicated. Averages in normal liver are: KIP2, 1.337; H19, 2.96; and IGF2, 0.899. For fetal liver IGF2 mRNA levels see text.
Most of the tumors for which no MNLT was available had a similar KIP2 expression pattern compared to the average KIP2 expression level in normal liver tissue (Table 3) ▶ . Interestingly, DZ25II, a case with LOH on 11p15.5, showed high KIP2 mRNA expression. DZ20 also had an increased KIP2 expression and RFLP analysis showed biallelic expression. The cell lines examined did not show KIP2 expression except HepT1, which is derived from the highly expressing tumor DZ25II. There was no correlation between KIP2 RNA levels and either tumor histology or pre-operative therapy (pre-operative chemotherapy or not).
Expression of H19 was reduced in eight of the 12 cases with MNLT, the reduction in the amount of transcript ranging from 0.02 to 0.22 (mean, 0.10) (Figure 2c ▶ and Table 3 ▶ ). Two of these eight (DZ18 and M12) had LOH on 11p15.5 and showed low H19 RNA levels (0.04 and 0.05). Two cases (D166 and M14) of the 12 showed only a moderate reduction of transcript levels, 0.60 and 0.54. One of these, D166, had maternal LOH at all informative markers on 11p15.5. The last two cases (D266 and D263) of the 12 differed in so far as they displayed not a diminished H19 RNA content in the tumor tissue compared with the MNLT, with ratios of 2.81 and 1.12. Interestingly, these cases also displayed a high absolute amount of KIP2 RNA compared to the other cases examined (Figure 2b ▶ and Table 3 ▶ ).
A majority of the tumors for which no MNLT was available, compared with the average of H19 expression in normal liver tissue (mean age, 31 months), confirmed the general trend of reduction of H19 RNA (Table 3) ▶ . Variation in normal liver was obvious, which probably reflects interindividual variation of H19 expression. Except for D104, the cases with LOH at the H19 locus showed reduced H19 expression compared with the average of H19 RNA in normal liver. We found a 3.5-fold decrease of H19 RNA from fetal to newborn tissue which fits well with previously reported age-dependent expression. 35 Four of the cell lines examined tended to be low in H19 expression ranging from 0.0001 to 0.1 (mean, 0.026) compared to the average of normal liver tissue H19 expression. Only the HepT3 cell line had detectable H19 mRNA expression (0.63).
Eleven of the 12 HBs with MNLT had increased levels of IGF2 transcript that ranged from 1.72 to 19.73 (mean, 6.28) (Figure 2d ▶ and Table 3 ▶ ). The three cases with LOH on 11p15.5 showed an increase ranging from 3.47 to 8.15 (mean, 5.21). One case (D195) displayed the inverse ratio of IGF2 mRNA between tumor and MNLT (0.10). H19 and KIP2, however, were significantly up-regulated in this case (Figure 2, b and c ▶ , and Table 3 ▶ ).
Most of the tumors for which no MNLT was available had a similar trend toward increased IGF2 expression using the average of IGF2 expression in normal liver as a point of reference: nine of the 11 had increased IGF2 expression ranging from 1.12 to 8.43, one (D497) showed reduced expression (0.51), and one (DZ20) showed extreme reduction in IGF2 expression (0.01) (Table 3) ▶ . The cases with LOH in the IGF2/H19 region all revealed augmented levels of IGF2 mRNA: 6.51 to 8.43 (mean, 7.57). Fetal liver had such an abundant IGF2 expression that it was not covered by the titration of our competitive RT-PCR approach. However, we have identified the vast majority of IGF2 mRNA as being derived from the P3 promoter of the gene whereas P1, P2, and P4 contributed detectably but on a much lower level (W. Hartmann and T. Pietsch, data not shown). Newborn liver, which displayed a clearly lower IGF2 expression compared with fetal liver tissue, for the most part also recruited its IGF2 transcripts from the P3 promoter, P1 and P4 contributing only marginally, whereas P2 did not show significant activity (data not shown). All of the cell lines except for HepT1 expressed high amounts of IGF2 mRNA; thus HepT1 completely changed the expression profile for IGF2 found in its original tumor, DZ25II. Compared with the other cell lines, HUH6 showed only moderate expression of IGF2 (Table 3) ▶ .
Analysis of the Imprinting Status of KIP2, H19, and IGF2
Fourteen of 24 cases (58%) analyzed for the KIP2 polymorphism were informative, five of 25 (20%) were informative for the H19 polymorphism, and five of 27 (19%) were informative for the IGF2 polymorphism; three cases were informative for KIP2 and H19, or KIP2 and IGF2, whereas only one sample was informative for all three (Table 4) ▶ .
Table 4.
Imprint Status of KIP2, H19, and IGF2 in Hepatoblastomas and HB Cell Lines
| RNA | Informativity KIP2# | Imprint KIP2 | Informativity H19 RsaI | Imprint H19 RsaI | Informativity IGF2 | Imprint IGF2 |
|---|---|---|---|---|---|---|
| D269 | No | — | No | — | No | — |
| D272 | No | — | No | — | No | — |
| D266 | No | — | No | — | Yes | MOI |
| D263 | Yes | MOI | No | — | No | — |
| D166 | Yes | LOH | No | — | No | — |
| D23II | — | — | — | — | No | — |
| D195 | No | — | Yes | MOI | No | — |
| D197 | Yes | MOI | Yes | MOI | No | — |
| DZ18 | — | — | — | — | No | — |
| M12 | No | — | No | — | No | — |
| M14 | Yes | MOI* | No | — | Yes | LOI |
| D401 | No | — | No | — | No | — |
| D104 | Yes | LOH | No | — | No | — |
| D199 | No | — | No | — | Yes | MOI |
| D497 | No | — | No | — | No | — |
| D268 | Yes | LOH | No | — | No | — |
| D161 | Yes | MOI* | No | — | Yes | LOI |
| DZ29II | No | — | Yes | MOI | No | — |
| DZ20 | Yes | MOI* | No | — | No | — |
| DZ25II | Yes | LOH | No | — | No | — |
| D175 | Yes | MOI | Yes | MOI | No | — |
| D204 | No | — | No | — | No | — |
| D319 | Yes | MOI | No | — | No | — |
| D399II | Yes | MOI | No | — | No | — |
| DZ27II CL HepT1 | Yes+ | LOH | No | — | No | — |
| D501 CL HepT3 | — | — | No | — | No | — |
| D339 CL HUH6 | Yes | MOI | Yes | MOI | Yes | LOI |
Status of imprinting for p57KIP2, H19, and IGF2. Informativity of this polymorphism was assessed by the amplification of genomic DNA from normal tissue if available. Abbreviations: MOI, maintenance of imprint; LOI, loss of imprint; MOI*, leaky expression from the second allele visible on silver-stained polyacrylamide gel electrophoresis gel; +, genomic DNA of the original tumor’s adjacent original liver tissue has been used for comparison; LOH, loss of heterozygosity (monoallelic expression).
All tumors and cell lines examined displayed monoallelic expression of KIP2, although three showed small amounts of detectable transcript from the second allele, which is in accordance with previous observations on the somewhat leaky character of this imprint (Figure 3 ▶ , Table 4 ▶ , and data not shown). However, for the cases with maternal LOH at the KIP2 (and H19) locus, monoallelic expression cannot be regarded as maintenance of the imprint because loss of the active allele requires activation of the previously silenced paternal allele to explain the continued expression. All of the informative cases for H19 turned out to express the mRNA monoallelically (Figure 3b ▶ and data not shown). In the case of IGF2, we observed biallelic expression in three of the five informative cases, one being the cell line HUH6. The ages of the two patients with LOI, M14, and D161 were 20 months and 11 months, respectively (Figure 3 ▶ , Table 1 ▶ , and data not shown).
Figure 3.

Status of imprinting of KIP2, H19, and IGF2. a: KIP2: informativity was determined with constitutional genomic DNA. MOI was found in D175, leaky expression from the second allele was detected in sample DZ20. b: H19 was found to be monoallelically expressed in D175 and all other informative cases. c: Sample M14 displayed LOI of IGF2, the case D199 retained monoallelic expression. Abbreviations: gDNA, genomic DNA; cDNA, complementary DNA.
Discussion
Frequent LOH on 11p15.5 with a clear maternal bias has been observed in several embryonal tumors, including WT, rhabdomyosarcoma, and HB. 4,36 This suggests the presence of at least one maternally expressed, ie, paternally imprinted, tumor suppressor gene at this location. We found LOH on 11p15.5 in 25% of our 56 cases of HB, which is the largest LOH study performed on HB to date. This region contains several imprinted genes, of which KIP2 and H19 are mainly expressed from the maternal allele, whereas transcripts of the mitogen IGF-II are preferentially of paternal origin. Transcriptional activity of IGF2 is increased not only in several cancers but also in the overgrowth syndrome, BWS. Duplication of the paternal allele, referred to as paternal uniparental disomy, which occurs during mitotic recombination, has been observed in WTs. There is also evidence for paternal uniparental disomy in our HB cases. We have performed comparative genomic hybridization analysis on HB with LOH 11p15.5 63 and there is no net loss of genetic material in this region, indicating that the observed maternal LOH is because of duplication of the paternal alleles, ie, paternal uniparental disomy. Loss of the maternally-expressed putative tumor suppressor genes, H19 and KIP2, and duplication of the IGF2-expressing paternal allele, assuming maintenance of the imprint, would lead to an imbalance between growth-inhibiting and growth-promoting forces and, thus, might contribute to the development and/or progression of the tumor.
Although, in the case of the KIP2 gene, the paternal allele is transcribed at a low level, a major reduction in the amount of transcript should be seen in the cases having lost the maternal allele. H19 expression has been shown to be drastically reduced in embryonal tumors due not only to loss of the maternal allele but also by extensive hypermethylation of both alleles’ promoters in those tumors with maintenance of heterozyosity (MOH). 37,38 Hatada et al, 14 Thompson et al, 39 as well as Chung et al, 16 all reported clearly reduced expression of KIP2 in WTs, with Chung et al 16 even suggesting coordinate disruption of imprinting in WTs, having found H19 and KIP2 simultaneously reduced. However, our own results differ significantly from these findings.
In most of our HBs, KIP2 showed increased rather than diminished transcriptional activity and loss of the maternal allele was not consistently associated with a significant reduction of KIP2 transcriptional activity: only two of six cases with LOH on 11p15.5 showed decreased expression, whereas the others displayed normal or elevated transcriptional activity, following the general trend of all samples with or without LOH. Furthermore, in those samples with LOH and reduced expression, the decrease was not as strong as would be expected for tumors having lost their main transcriptionally active allele. All informative cases tested for LOI had MOI at the KIP2 locus. A low level of expression from the second allele, which represents the slightly leaky character of this imprint in normal tissues, was detectable in a few cases and not regarded as LOI. 15 Therefore, LOI of KIP2 in HB is a rare event, with a frequency lower than that observed in other tumors and BWS. 15,39 Consequently, in the cases with MOH, the presence of KIP2 transcript is unlikely to be because of disruption of imprinting. Interestingly, the cases with LOH do not show any significant trend toward lower expression of KIP2 compared to the cases with MOH. Duplication of the paternal leaky allele might lead to a somewhat higher KIP2 expression but cannot explain an increase of KIP2 expression to levels well beyond those of normal liver, as seen in some of our cases with LOH.
Although there are several studies showing reduction of expression of KIP2 in WTs, there are also results similar to ours. Thompson et al 39 did not see a clear correlation between LOH and reduction of KIP2 mRNA in WT samples. Chung et al 16 found a residual level of KIP2 expression of up to 40% in WT cases with LOH. The finding of a lack of effect of LOH on 11p15.5 on KIP2 mRNA levels has further support from the observations of Overall et al, 40 who reported that LOH at 11p15.5 did not affect KIP2 expression in WTs and who even found increased amounts of KIP2 transcript compared to matching normal kidney tissue in three of five WT samples with LOH. These data and our data on HB argue against a tumor suppressor function of KIP2 in certain embryonal tumors.
Similar to previous observations, 41 most HBs in our series had high amounts of IGF2 mRNA and clearly diminished amounts of H19 transcripts. There is a subgroup of HB with concordant increase of H19 and KIP2 transcriptional activity. This expression pattern has already been observed in a subgroup of WTs suggesting that these genes may sometimes be co-regulated in HB cells. Furthermore, we characterized one sample showing low H19 RNA but almost undetectable IGF2 expression, a finding with implications for the still ongoing discussion on the relationship of these genes. This expression pattern cannot be explained by the enhancer/competition model of H19/IGF2 regulation. 42 In agreement with previous reports, we found MOI of H19 in most HBs. 41,43 For IGF2, we detected biallelic expression in two of four informative cases and one cell line, a figure similar to those reported by Rainier et al 43 (one of five) and Li et al 41 (one of three). The expression pattern of H19 and IGF2 displayed in these cases did not differ consistently from the cases with MOI and the amount of IGF2 transcript was not increased compared to the latter.
Consequently, LOI is not the cause for IGF2 up-regulation in HBs or for the increase of KIP2 expression. H19 also remains monoallelically expressed in HBs and shows a clear reduction of transcription. The only feature in common between the maternally expressed H19 and KIP2 is the relative stability of the imprint, yet, in HB with LOH on 11p15.5, the loss of the active maternal KIP2 allele seems to be overcompensated for by the previously imprinted allele. To explain the expression of mRNA from the normally silenced allele of an imprinted gene requires revision of the imprint. The mechanism of this revision is unclear as Chung et al 16 reported that both KIP2 alleles are hypomethylated. To get an impression of the methylation status in HB, we performed genomic sequencing after bisulfite treatment of the DNA in a randomly selected HB case (D266) and the corresponding liver. Similar to WT, we found the majority of CpG dinucleotides in the 5′-promoter region and exon 1 unmethylated in both the tumor and the corresponding liver tissue. This argues against a role of demethylation in up-regulation of the KIP2 transcription, at least for the regions scanned (position −877 to −468 5′ of the transcription start, as well as to position −127 to +434). Dao et al 44 also found that the imprint of the KIP2 gene remains stable in DNA methyltransferase-hypomorphic mice. One might speculate about the existence of a second promoter for the gene which lacks imprinting and which takes over control of the paternal KIP2 allele after maternal LOH. Another possibility, at least for the samples with maternal LOH, is the loss of potential inhibitors which are maternally transcribed and which act on the paternal allele, controlling specific enhancing elements. However, most samples retain heterozygosity and most show a monoallelic KIP2 up-regulation. Although the majority of HBs and WTs share a similar pattern of IGF2/H19 expression, they differ in their biological behavior concerning the KIP2 gene. In this regard, in contrast to results on KIP2 and H19 reported for WTs, 16 our data strongly argue against the idea of a coordinate disruption of imprinting for HB. We suggest that there are different and independent ways of regulating these genes. This idea has support from the findings of Caspary et al 45 in mice, where loss of the h19 gene and 10 kb of its 5′-flanking DNA had no effect on kip2 expression. However, mice whose maternal h19 allele has been knocked out have somatic overgrowth and biallelic rather than monoallelic igf2 expression in their tissues. 46 Therefore, at least in the murine model, h19 seems to regulate the imprint of igf2, but not that of kip2, and our results in HB suggest that the same applies to the human system.
The hypothesis of KIP2 and H19 being regulated independently of each other gets additional support from recent findings on the genomic structure of 11p15.5. Lee et al 47 postulate two separate imprinted domains, the centromeric one harboring KIP2 and the telomeric ones IGF2 and H19. These authors found two genes in the center of the 11p15.5 region, TSSC4 and TSSC6, which escape imprinting whereas the flanking genes on both sides are imprinted. 48 Furthermore, a paternally expressed transcript, LIT1 (strictly speaking its CpG island), located within KvLQT1 and transcribed in antisense orientation to it, is suspected to play a role in KIP2 regulation similar to that of H19 in IGF2 regulation. 47 The authors suggest a more important role of the centromeric domain in BWS whereas the telomeric one is supposed to be more frequently affected in certain tumors such as WT, HB, and RMS. Nevertheless, some overlap of the phenotypes caused by alterations in either domain is proposed. Given the somewhat surprising finding of up-regulated KIP2 expression in HB, a thorough analysis of LIT1 will be interesting.
As already mentioned above, our data do not suggest a tumor-suppressing function of KIP2 in HB whereas some evidence argues against such a role even in WT. Zhang et al 8 showed that lack of kip2 in mice does cause some features of BWS and demonstrated its involvement in negative regulation of cell proliferation in the adrenal cortex and in the developing lens. However, mice lacking kip2 did not develop tumors, either in the kidney or in the liver. Yan et al 9 reported similar results and they even found increased apoptosis among epithelial cells and surrounding mesenchymal cells when examining defects in palate formation. Lee et al 6 reported that overexpression of KIP2 in R-1B/L17 cells caused accumulation of the cells in G1 but they did not observe changes in apoptotic activity. These results are not in keeping with the idea of KIP2 as a tumor suppressor gene.
As has already been reported for WT and RMS, 13,49 where no mutations in the KIP2 gene could be detected, we did not find sequence mutations of the coding KIP2 regions in HB. KIP2, thus, seems to share the low mutation rate in human tumors which has been shown for the other members of the Cip/Kip family, p21 and p27. 50-53
The seemingly paradoxical expression pattern of the tumor suppressor KIP2 in HBs might make sense if KIP2 can assume a growth promoting function under certain conditions. It has been suggested that retention of the related p27-cyclin-dependent kinase inhibitors might constitute a survival advantage for tumors in certain contexts, such as adhesion-dependent resistance to DNA-damaging agents. 54 It is not yet known whether KIP2 has a comparable protective action. However, in our series, there was no correlation between the levels of KIP2 transcript detected and clinical outcome.
Recent reports make it probable that the Cip/Kip-proteins, which were originally described only as inhibitors of cell-cycle promoters, also have an important role as assembly factors for CDK4-cyclin D complexes. 55-57 Furthermore all three known proteins of the family have been reported to direct the accumulation of CDK4 and cyclin D1 in the nucleus during G1 55 where the complex eventually becomes activated by CDK-activating kinase and inactivates the transcriptional-repressor retinoblastoma protein by phosphorylation, thus allowing progression of the cell cycle. 58 This feature of the Cip/Kip-CKIs gains even more importance in view of the demonstration of the cyclin D1 gene as a target of the β-catenin/LEF-1 pathway. Shtutman et al 59 reported that high levels of β-catenin lead to LEF-1-mediated increased transcriptional activity of the cyclin D1 gene. β-catenin mutations were found in 48% of sporadic HBs, all of which affected its degradation targeting box, and nuclear accumulation of the mutated protein was detected. 60 Overexpression of cyclin D1 in HBs has been reported previously 61 and this may well be because of β-catenin mutations or mutations of its degrading compounds like APC. 62 One requirement for the biological activity of cyclin D1 is its assembly with CDK4 and its transfer into the cell nucleus. The increased levels of KIP2 mRNA point to a role in cyclin D1 trafficking, as has been recently described for other members of the Cip/Kip family in murine fibroblasts, in which p21 and p27 were found to be assembling factors for cyclin D-CDK complexes and nuclear directors for cyclin D proteins. 56 It is conceivable that KIP2 in HBs has a growth-promoting rather than an inhibiting character in that its presence may positively influence the effectiveness of overexpressed cyclin D1. In this scenario, the inhibitor KIP2 may gain growth promoting properties and it is no longer enigmatic why a tumor increases transcription of a growth-inhibiting gene. This may also explain the low frequency of KIP2 mutations.
The frequency of involvement of the chromosomal domain 11p15.5 in HBs and other human cancers suggests that it still hides tumor suppressing gene(s). Our data indicate that KIP2 is not a candidate for this role in HB. Further work is necessary to understand its function in HB.
Acknowledgments
We thank B. Meyer-Puttlitz for sequencing, D. Denkhaus for cell culture, S.G. Gray for critical comments, and U. Klatt for photographic work.
Footnotes
Address reprint requests to Torsten Pietsch, Department of Neuropathology, University of Bonn Medical Center, Sigmund-Freud-St. 25, D-53105 Bonn, Germany. E-mail: pietsch-t@uni-bonn.de.
Supported by Deutsche Forschungsgemeinschaft (grant Pi 191/9-1).
References
- 1.Weinberg AG, Finegold MJ: Primary hepatic tumors of childhood. Hum Pathol 1983, 14:512-537 [DOI] [PubMed] [Google Scholar]
- 2.Byrne JA, Simms LA, Little MH, Algar EM, Smith PJ: Three non-overlapping regions of chromosome arm 11p allele loss identified in infantile tumors of adrenal and liver. Genes Chromosom Cancer 1993, 8:104-111 [DOI] [PubMed] [Google Scholar]
- 3.Kraus JA, Albrecht S, Wiestler OD, von Schweinitz D, Pietsch T: Loss of heterozygosity on chromosome 1 in human hepatoblastoma. Int J Cancer 1996, 67:467-471 [DOI] [PubMed] [Google Scholar]
- 4.Albrecht S, von Schweinitz D, Waha A, Kraus JA, von Deimling A, Pietsch T: Loss of maternal alleles on chromosome arm 11p in hepatoblastoma. Cancer Res 1994, 54:5041-5044 [PubMed] [Google Scholar]
- 5.Matsuoka S, Edwards MC, Bai C, Parker S, Zhang P, Baldini A, Harper JW, Elledge SJ: p57KIP2, a structurally distinct member of the p21CIP1Cdk family, is a candidate tumor suppressor gene. Genes Dev 1995, 9:650-662 [DOI] [PubMed] [Google Scholar]
- 6.Lee MH, Reynisdottir I, Massague J: Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev 1995, 9:639-649 [DOI] [PubMed] [Google Scholar]
- 7.Watanabe H, Pan Z-H, Schreiber-Agus N, DePhino RA, Hurwitz J, Xiong Y: Suppression of cell transformation by the cyclin-dependent kinase inhibitor p57KIP2 requires binding to proliferating cell nuclear antigen. Proc Natl Acad Sci USA 1998, 95:1392-1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang P, Liegois N, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePhino RA, Elledge SJ: Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 1997, 387:151-158 [DOI] [PubMed] [Google Scholar]
- 9.Yan Y, Frisen J, Lee M-H, Massague J, Barbacis M: Ablation of the CDK inhibitor p57KIP2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 1997, 11:973-983 [DOI] [PubMed] [Google Scholar]
- 10.Lee MP, DeBraun M, Randhawa G, Reichard BA, Elledge SJ, Feinberg AP: Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am J Hum Genet 1997, 61:304-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatada I, Ohashi H, Fukushima Y, Kaneko Y, Inoue M, Komoto Y, Okada A, Ohishi S, Nabetani A, Morisaki H, Nakayama M, Niikawa M, Mukai T: An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat Genet 1996, 14:171-173 [DOI] [PubMed] [Google Scholar]
- 12.Hatada I, Nabetani A, Morisaki H, Xin Z, Ohishi S, Tonoki H, Niikawa N, Inoue M, Komoto Y, Okada A, Steichen E, Ohashi H, Fukushima Y, Nakayama M, Mukai T: New p57KIP2 mutations in Beckwith-Wiedemann syndrome. Hum Genet 1997, 100:681-683 [DOI] [PubMed] [Google Scholar]
- 13.O’Keefe D, Dao D, Zhao L, Sanderson R, Wartburton D, Weiss L, Anaye-Yeboa K, Tycko B: Coding mutations in p57KIP2 are present in some cases of Beckwith-Wiedemann syndrome but are rare or absent in Wilms tumors. Am J Hum Genet 1997, 61:295-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatada I, Inazawa J, Abe T, Nakayama M, Kaneko Y, Jinno Y, Niikawa N, Ohashi H, Fukushima Y, Iida K, Yutani C, Takahashi S, Chiba Y, Ohishi S, Mukai T: Genomic imprinting of human p57KIP2 and its reduced expression in Wilms tumors. Hum Mol Genet 1996, 5:783-788 [DOI] [PubMed] [Google Scholar]
- 15.Matsuoka S, Thompson JS, Edwards MC, Barletta JM, Grundy P, Kalikin LM, Harper JW, Elledge SJ, Feinberg AP: Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc Natl Acad Sci USA 1996, 93:3026-3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung WY, Yuan L, Feng L, Hensle T, Tycko B: Chromosome 11p15.5 regional imprinting: comparative analysis of KIP2 and H19 in human tissues and Wilms’ tumors. Hum Mol Genet 1996, 5:1101-1108 [DOI] [PubMed] [Google Scholar]
- 17.Giannoukakis N, Deal C, Paquette J, Goodyer CG, Polychronakos C: Parental genomic imprinting of the human IGF2 gene. Nat Genet 1993, 4:98-101 [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Tycko B: Monoallelic expression of the human H19 gene. Nat Genet 1992, 1:40-44 [DOI] [PubMed] [Google Scholar]
- 19.Brannan CI, Dees EC, Ingram RS, Tilghman SM: The product of the H19 gene may function as an RNA. Mol Cell Biol 1990, 10:28-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones BK, Levorse JM, Tilghman SM: Igf2 imprinting does not require its own DNA methylation or H19 RNA. Genes Dev 1998, 12:2200-2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B: Tumor suppressor activity of the H19 RNA. Nature 1993, 365:764-767 [DOI] [PubMed] [Google Scholar]
- 22.Steenman MJC, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP: Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms tumor. Nat Genet 1994, 7:433-439 [DOI] [PubMed] [Google Scholar]
- 23.Cui H, Hedborg F, He L, Nordenskjold A, Sandstedt B, Pfeifer-Ohlsson S, Ohlsson R: Inactivation of H19, an imprinted and putative tumor repressor gene, is a preneoplastic event during Wilms’ tumorigenesis. Cancer Res 1997, 57:4469-4473 [PubMed] [Google Scholar]
- 24.Bartolomei MS, Tilghman SM: Genomic imprinting in mammals. Annu Rev Genet 1997, 31:493-525 [DOI] [PubMed] [Google Scholar]
- 25.Pietsch T, Fonatsch C, Albrecht S, Maschek H, Wolf HK, von Schweinitz D: Characterization of the continuous cell line HepT1 derived from a human hepatoblastoma. Lab Invest 1996, 74:809-818 [PubMed] [Google Scholar]
- 26.Doi I: Establishment of a cell line and its clonal sublines from a patient with hepatoblastoma. Gann 1976, 67:1-10 [PubMed] [Google Scholar]
- 27.Albrecht S, Waha A, Koch A, Kraus JA, Goodyer CG, Pietsch T: Variable imprinting of H19 and IGF2 in fetal cerebellum and medulloblastoma. J Neuropathol Exp Neurol 1996, 55:1270-1276 [DOI] [PubMed] [Google Scholar]
- 28.Budowle B, Chakraborty R, Giusti AM, Eisenberg AJ, Allen RC: Analysis of the VNTR locus D1S80 by the PCR followed by high-resolution PAGE. Am J Hum Genet 1991, 48:137-144 [PMC free article] [PubMed] [Google Scholar]
- 29.Koch A, Tonn J, Kraus JA, Sorensen N, Albrecht NS, Wiestler OD, Pietsch T: Molecular analysis of the lissencephaly gene 1 (LIS-1) in medulloblastomas. Neuropathol Appl Neurobiol 1996, 22:233-242 [PubMed] [Google Scholar]
- 30.Waha A, Watzka M, Koch A, Pietsch T, Przkora R, Peters N, Wiestler OD, von Deimling A: A rapid and sensitive protocol for competitive reverse transcriptase (cRT) PCR analysis of cellular genes. Brain Pathol 1998, 8:13-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tokino T, Urano T, Furuhata T, Matsushima M, Miyatsu T, Sasaki S, Nakamura Y: Characterization of the human p57KIP2 gene: alternative splicing, insertion/deletion polymorphisms in VNTR sequences in the coding region, and mutational analysis. Hum Genet 1996, 97:625-631 [DOI] [PubMed] [Google Scholar]
- 32.Bender B, Wiestler OD, von Deimling A: A device for processing large acrylamide gels. Biotechniques 1994, 16:204-205 [PubMed] [Google Scholar]
- 33.Zhang Y, Tycko B: Monoallelic expression of the human H19 gene. Nat Genet 1992, 1:40-44 [DOI] [PubMed] [Google Scholar]
- 34.Tadokoro K, Fujii H, Inoue T, Yamada M: Polymerase chain reaction (PCR) for detection of ApaI polymorphism at the insulin like growth factor II gene (IGF2). Nucleic Acids Res 1991, 19:6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pachnis V, Belayew A, Tilghman SM: Locus unlinked to alpha-fetoprotein under the control of the murine raf and Rif genes. Proc Natl Acad Sci USA 1984, 81:5523-5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tycko B: Genomic imprinting: mechanism and role in human pathology. Am J Pathol 1994, 144:431-443 [PMC free article] [PubMed] [Google Scholar]
- 37.Moulton T, Crenshaw T, Hao Y, Moosikasuwan J, Lin N, Dembitzer F, Hensle T, Weiss L, McMorrow L, Loew T, Kraus W, Gerald W, Tycko B: Epigenetic lesions at the H19 locus in Wilms’ tumour patients. Nat Genet 1994, 7:440-447 [DOI] [PubMed] [Google Scholar]
- 38.Taniguchi T, Sullivan MJ, Ogawa O, Reeve AE: Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc Natl Acad Sci USA 1995, 92:2159-2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson JS, Reese KJ, DeBraun M, Perlman EJ, Feinberg AP: Reduced expression of the cyclin-dependent kinase inhibitor p57KIP2 in Wilms’ tumor. Cancer Res 1996, 56:5723-5727 [PubMed] [Google Scholar]
- 40.Overall ML, Spencer J, Bakker M, Dziadek M, Smith PJ: p57KIP2 is expressed in Wilms’ tumors with LOH of 11p15.5. Genes Chromosom Cancer 1996, 17:56-59 [DOI] [PubMed] [Google Scholar]
- 41.Li X, Adam G, Sandstedt B, Ohlsson R, Ekstrom TJ: Expression, promoter usage and parental imprinting status of insulin-like growth factor (IGF2) in human hepatoblastoma: uncoupling of IGF2 and H19 imprinting. Oncogene 1995, 11:221-229 [PubMed] [Google Scholar]
- 42.Barlow DP: Competition—a common motif for the imprinting mechanism? EMBO J 1997, 16:6899-6905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rainier S, Dobry CJ, Feinberg AP: Loss of imprinting in hepatoblastoma. Cancer Res 1995, 55:1836-1838 [PubMed] [Google Scholar]
- 44.Dao D, Walsh CP, Yuan L, Gorelov D, Feng L, Hensle T, Nisen P, Yamashiro DJ, Bestor TH, Tycko B: Multipoint analysis of human chromosome 11p15/mouse distal chromosome 7: inclusion of H19/IGF2 in the minimal WT2 region, gene specificity of H19 silencing in Wilms’ tumorigenesis and methylation hyper-dependence of H19 imprinting. Hum Mol Genet 1999, 8:1337-1352 [DOI] [PubMed] [Google Scholar]
- 45.Caspary T, Cleary MA, Baker CC, Guan X-J, Tilghman SM: Multiple mechanisms regulate imprinting of the mouse distal chromosome 7 cluster. Mol Cell Biol 1998, 18:3466-3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leighton PA, Ingram RS, Eggenschwilger J, Efstratiadis A, Tilghman SM: Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 1995, 375:34-39 [DOI] [PubMed] [Google Scholar]
- 47.Lee MP, DeBraun M, Mitsuya K, Galonek HL, Brandenburg S, Oshimura M, Feinberg AP: Loss of imprinting of a paternally expressed transcript, with antisense orientation to KvLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci USA 1999, 96:5203-5208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee MP, Brandenburg S, Landes GM, Adams M, Miller G, Feinberg AP: Two novel genes in the center of the 11p15.5 imprinted domain escape genomic imprinting. Hum Mol Genet 1999, 8:683-690 [DOI] [PubMed] [Google Scholar]
- 49.Orlow I, Iavarone A, Crider-Miller SJ, Bonilla F, Latres E, Lee MH, Gerald WL, Massague J, Weissman BE, Cordon-Cardo C: Cyclin-dependent kinase inhibitor p57KIP2 in soft tissue sarcomas and Wilms’ tumors. Cancer Res 1996, 56:1219-1221 [PubMed] [Google Scholar]
- 50.Ponce-Castaneda MV, Lee MH, Latres E, Polyak K, Lacombe L, Montgomery K, Mathew S, Krauter K, Sheinfeld J, Massague J, Cordon-Cardo C: p27KIP1: chromosomal mapping to 12p12–12p13.1 and absence of mutations in human tumors. Cancer Res 1995, 55:1211-1214 [PubMed] [Google Scholar]
- 51.Shiohara M, el-Deiry WS, Wada M, Nakamaki T, Takeuchi S, Yang R, Chen DL, Vogelstein B, Koeffler HP: Absence of WAF1 mutations in a variety of human malignancies. Blood 1994, 84:3781-3784 [PubMed] [Google Scholar]
- 52.Ferrando AA, Balbin M, Pendas AM, Vizoso F, Velasco G, Lopez-Otin C: Mutational analysis of the human cyclin-dependent kinase inhibitor p27KIP1 in primary breast carcinomas. Hum Genet 1996, 97:91-94 [DOI] [PubMed] [Google Scholar]
- 53.Kawamata N, Morosetti R, Miller CW, Park D, Spirin KS, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilcyznski S, Lee YY, Bartram CR, Koeffler HP: Molecular analysis of the cyclin-dependent kinase inhibitor gene p27/KIP1in human malignancies. Cancer Res 1995, 55:2266-2269 [PubMed] [Google Scholar]
- 54.St Croix BS, Florenes VA, Rak JW, Flanagan M, Bhattacharya N, Slingerland JM, Kerbel RS: Impact of the cyclin-dependent kinase inhibitor p27KIP1 on resistance of tumor cells to anticancer therapy. Nat Med 1996, 2:1204-1210 [DOI] [PubMed] [Google Scholar]
- 55.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E: New functional activities for the p21 family of CDK inhibitors. Genes Dev 1997, 11:847-862 [DOI] [PubMed] [Google Scholar]
- 56.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ: The p21CIP1 and p27 KIP1 CDK inhibitors are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 1999, 18:1571-1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parry D, Mahony D, Wills K, Lees M: Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol Cell Biol 1999, 19:1775-1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinberg RA: The retinoblastoma protein and cell cycle control. Cell 1995, 81:323-330 [DOI] [PubMed] [Google Scholar]
- 59.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A: The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 1999, 96:5522-5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T: Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the b-catenin gene. Cancer Res 1999, 59:269-273 [PubMed] [Google Scholar]
- 61.Kim H, Ham EK, Kim YI, Chi JG, Lee HS, Park SH, Jung YM, Myung NK, Lee MJ, Jang J-J: Overexpression of cyclin D1 and cdk4 in tumorigenesis of sporadic hepatoblastoma. Cancer Lett 1998, 131:177-183 [DOI] [PubMed] [Google Scholar]
- 62.Kurahashi H, Takami K, Oue T, Kusafuka T, Okada A, Tawa A, Okada S, Nishisho I: Biallelic inactivation of the APC gene in hepatoblastoma. Cancer Res 1995, 56:5007-5011 [PubMed] [Google Scholar]
- 63.Weber RG, Pietsch T, von Schweinitz D, Lichter P: Characterization of genomic alterations in hepatoblastomas: a role for gains on chromosomes 8q and 20 as predictors of poor outcome. Am J Pathol 2000, 157:571-578 [DOI] [PMC free article] [PubMed] [Google Scholar]