

Abstract
The pathogenesis of sporadic endocrine pancreatic tumors (EPTs) is still primarily unknown. Comparative genomic hybridization studies revealed loss of 10q in a significant number (nine of 31) of EPTs. The tumor suppressor gene PTEN lies on 10q23, and so, is a candidate to play some role in EPT pathogenesis. Germline PTEN mutations are found in Cowden and Bannayan-Riley-Ruvalcaba syndromes, whereas somatic mutations and deletions are found in a variety of sporadic cancers. The mutation and expression status of PTEN in EPTs has not yet been examined. Mutation analysis of the entire coding region of PTEN including splice sites was performed in 33 tumors, revealing one tumor with somatic L182F (exon 6). Loss of heterozygosity of the 10q23 region was detected in eight of 15 informative malignant (53%) and in none of seven benign EPTs. PTEN expression was assessed in 24 available EPTs by immunohistochemistry using a monoclonal anti-PTEN antibody. Of these 24, 23 tumors showed strong immunoreactivity for PTEN. Only the EPTs with PTEN mutation lacked PTEN protein expression. Although normal islet cells always exhibited predominantly nuclear PTEN immunostaining, 19 of 23 EPTs had a predominantly cytoplasmic PTEN expression pattern. Exocrine pancreatic tissue was PTEN-negative throughout. PTEN mutation is a rare event in malignant EPTs and PTEN protein is expressed in most (23 of 24) EPTs. Thus, intragenic mutation or another means of physical loss of PTEN is rarely involved in the pathogenesis of EPTs. Instead, either an impaired transport system of PTEN to the nucleus or some other means of differential compartmentalization could account for impaired PTEN function. Loss of heterozygosity of the 10q23 region is a frequent event in malignant EPTs and might suggest several hypotheses: a different tumor suppressor gene in the vicinity of PTEN might be principally involved in EPT formation; alternatively, 10q loss, including PTEN, seems to be associated with malignant transformation, but the first step toward neoplasia might involve altered subcellular localization of PTEN.
The etiology and pathogenesis of sporadic endocrine pancreatic tumors (EPTs) remain primarily unknown. Mutations of the MEN1 gene responsible for the autosomal dominantly inherited MEN 1 syndrome are found only in ∼15 to 30% of sporadic EPTs. 1,2 Oncogenes (such as FOS, C-MYC, M-MYC, and SIS) or tumor suppressor genes (such as TP53 or RB1) which are frequently activated or mutated in other human tumors seem not to be involved in the neoplastic transformation of EPTs. 3,4 Comparative genomic hybridization analysis of EPTs revealed losses of Y, 6q, 11q, 3p, 3q, 11p, 6p, 10q, and Xq. The frequency of 10q loss was 25% of all EPTs with a frequency as high as seven of nine of nonfunctioning EPTs. 5 The tumor suppressor gene PTEN maps to 10q23.3. 6-8
Germline PTEN mutations are responsible for the autosomal dominantly inherited Cowden and Bannayan-Riley Ruvalcaba syndromes as well as a Proteus-like syndrome. 9-13 Relatively high frequencies of somatic intragenic PTEN mutations and deletions are found in noncultured endometrial carcinomas 14-17 and malignant gliomas. 18,19 Despite the frequency of structural PTEN mutations in noncultured endometrial carcinomas, we have shown that epigenetic silencing of PTEN not only plays a prominent role in its pathogenesis, but also in the pathogenesis of the earliest endometrial precancers. 17 Further, we have demonstrated that loss of PTEN protein expression in the absence of mutations occurs in breast carcinogenesis. 20 To examine whether 10q loss in EPTs points to involvement of the tumor suppressor PTEN, we analyzed a series of 33 EPTs for intragenic mutations and deletions of PTEN and PTEN protein expression.
Materials and Methods
Tumor Samples
Thirty-three EPTs were drawn from the files of the Department of Pathology, University Hospital Zürich, Switzerland. The tumors were classified according to the most recent World Health Organization classification. 21 They comprised 19 insulinomas (six malignant, 12 benign, one MEN 1-associated, and one of uncertain clinical behavior), two malignant glucagonomas, three malignant VIP-omas, three malignant gastrinomas (one MEN 1-associated), and six nonfunctioning (five malignant, one benign) EPTs. Except for one gastrinoma and one insulinoma as noted, all of the EPTs were sporadic and not associated with MEN 1 or von Hippel-Lindau (VHL) syndrome. Comparative genomic hybridization analysis of these tumors has been performed previously. 5
Fresh-frozen tissue was snap-frozen in liquid nitrogen and stored at −80°C. Paraffin samples were fixed by immersion in 4% buffered formalin and embedded in paraffin according to standard procedures.
DNA Extraction
Genomic DNA from fresh-frozen tissue was isolated using the D-5000 Purgene DNA Isolation Kit (Gentra Systems, Minneapolis, MN) according to the manufacturer’s instructions. DNA from these fresh-frozen tissues was used for mutation analysis. Where no nonneoplastic fresh-frozen tissue was available, DNA was extracted from paraffin blocks for loss of heterozygosity (LOH) analysis. For this purpose, 10-μm sections of formalin-fixed paraffin-embedded tumor specimens were microdissected and DNA extraction was performed as described. 2,22
LOH Analysis
To assess LOH of the PTEN region at 10q23, we used the centromeric marker D10S579, the intragenic markers AFMa086wg9 and D10S2491, and the telomeric marker D10S1735. Polymerase chain reaction (PCR) was performed according to standard procedures and the products were electrophoresed through polyacrylamide gels containing 7 mol/L urea followed by silver staining as previously described. 2 LOH was defined as a complete absence or reduced signal of one of the constitutional alleles in the tumor tissue compared to the corresponding nonneoplastic tissue.
Mutation Analysis
PCR amplification was performed in a 50-μl mixture 1× PCR buffer (Perkin Elmer Europe, Rotkreuz, Switzerland) containing 400 ng of template DNA, 200 μmol/L of dNTP (Roche Diagnostics, Rotkreuz, Switzerland), 1 μmol/L each of intronic-based primers flanking each exon (Table 1) ▶ , and 0.2 μl of Taq polymerase (AmpliTaq Gold; Perkin Elmer Europe). A touchdown PCR was performed with denaturation at 95°C for 1 minute, annealing at 55 to 48°C (with 1°C decrements per cycle) for 1 minute, and extension at 72°C for 1 minute followed by additional 30 cycles at 48° annealing temperature and a final extension at 72° for 10 minutes. Varying concentrations of MgCl2 and dimethylsulfoxide were used (Table 1) ▶ .
Table 1.
Primers and PCR Conditions
| Primer | Position AH007803 | Sequence | Length (bp) | Condition |
|---|---|---|---|---|
| PTEN 1 | 2229–2379 | TCTGCCATCTCTCTCTCCT | 151 | 2.0 mmol/L MgCl2, 10% DMSO |
| AGAGGAGCAGCCGCAGAAATG | ||||
| PTEN 2 | 10242–10412 | TTTCAGATATTTCTTTCCTTA | 171 | 2.5 mmol/L MgCl2 |
| PTEN 3 | 18382–18528 | TAATTTCAAATGTTAGCTCAT | 147 | 2.5 mmol/L MgCl2 |
| AAGATATTTGCAAGCATACAA | ||||
| PTEN 4 | 24099–24248 | GTTTGTTAGTATTAGTACTTT | 150 | 2.5 mmol/L MgCl2 |
| ACAACATAGTACAGTACATTC | ||||
| PTEN 5a | 26126–26283 | TATTCTGAGGTTATCTTTTA | 158 | 2.5 mmol/L MgCl2, 10% DMSO |
| CTTTCCAGCTTTACAGTGAA | ||||
| PTEN 5b | 26232–26417 | GCTAAGTGAAGATGACAATCA | 186 | 2.5 mmol/L MgCl2, 10% DMSO |
| AGGAAAAACATCAAAAAATAA | ||||
| PTEN 6 | 32123–32324 | TTGGCTTCTCTTTTTTTTCTG | 202 | 2.5 mmol/L MgCl2 |
| ACATGGAAGGATGAGAATTTC | ||||
| PTEN 7 | 37865–38093 | CCTGTGAAATAATACTGGTATG | 229 | — |
| CTCCCAATGAAAGTAAAGTACA | ||||
| PTEN 8b | 40894–41137 | TTAAATATGTCATTTCATTTCTTTTTC | 244 | — |
| CTTTGTCTTTATTTGCTTTGT | ||||
| PTEN 8b | 41084–41200 | GTGCAGATAATGACAAGGAATA | 117 | — |
| ACACATCACATACATACAAGTC | ||||
| PTEN 9 | 45308–45549 | TTCATTTTAAATTTTCTTTCT | 242 | 2 mmol/L MgCl2 |
| TGGTGTTTTATCCCTCTTGAT |
DMSO, dimethylsulfoxide.
For the single-strand conformation polymorphism analysis, 10 μl of denatured PCR products in stop buffer (95% formamide, 20 mmol/L ethylenediaminetetraacetic acid, 0.05% xylene cyanol, 0.05% bromophenol blue) were loaded onto nondenaturing polyacrylamide gels. Electrophoresis was performed at 40 W for 5 hours at room temperature. After electrophoresis, the DNA was visualized by silver staining as described. 23
Immunohistochemistry
The monoclonal anti-human PTEN antibody 6H2.1 raised against the last 100 C-terminal amino acids was used in all immunohistochemical analysis. 20,24 Specificity and characterization of 6H2.1 has previously been demonstrated by Western blot, immunohistochemistry on cell lines with known PTEN expression status as well as the ability of cold peptide to compete off immunostaining on paraffin-embedded sections. 17,20,25
Twenty-four cases where paraffin blocks were available were subjected to immunohistochemistry (Table 2) ▶ . Four-μm sections were cut and mounted on Superfrost Plus slides (Fischer Scientific, Pittsburgh, PA). Immunostaining was performed as described. 20 A semiquantitative score was given to the nuclear and cytoplasmic staining of tumor and normal tissue: −, +, and ++.
Table 2.
Clinical Data and Results
| Clinical data | Tumor type | CGH† | ICH‡ | LOH - Analysis§ | Mutation analysis | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Sex | Age | D10S579 | AFM86 | D10S2491 | D10S1735 | ||||
| 1 | M | 93 | ins | neg | ||||||
| 2 | M | 36 | ins | C | * | ni | □ | ni | neg | |
| 3 | M | 58 | ins | ni | ni | □ | ni | neg | ||
| 4 | F | 81 | ins | neg | ||||||
| 5 | F | 26 | ins | ni | ni | □ | ni | neg | ||
| 6 | ? | ? | Ins | ni | ni | □ | □ | neg | ||
| 7 | M | 62 | ins | * | ni | * | ni | neg | ||
| 8 | F | 22 | ins | neg | ||||||
| 9 | F | 59 | ins | C | neg | |||||
| 10 | F | 35 | ins | N/C | neg | |||||
| 11 | F | 42 | ins | C | ni | ni | □ | ni | neg | |
| 12 | M | 53 | ins | C | ni | ni | □ | ni | neg | |
| 13 | F | 52 | non f | 10q- | neg | |||||
| 14 | M | 61 | ins, mal | C/M | * | ni | ni | ▪ | neg | |
| 15 | M | 62 | ins, mal | N/C | ni | □ | □ | □ | neg | |
| 16 | M | 52 | ins, mal | C | * | ni | ni | ni | neg | |
| 17 | M | 58 | ins, mal | 10q rg- | C/M | ni | □ | ▪ | ni | neg |
| 18 | M | 56 | ins, mal | C | * | ni | ▪ | ni | neg | |
| 19 | F | 64 | ins, mal | 10q- | N/C | ni | ni | ni | * | neg |
| 20 | F | 56 | gluc, mal | 10q- | C | ni | ▪ | ▪ | ni | neg |
| 21 | F | 48 | gluc, mal | 10q rg- | C | ▪ | ni | ▪ | ni | neg |
| 22 | M | 44 | non f, mal | 10q rg- | C | * | ni | ▪ | ▪ | neg |
| 23 | F | 33 | non f, mal | 10q- | neg | ni | * | ni | ▪ | Exon 6 L182F |
| 24 | M | 51 | non f, mal | 10q- | N/C | ni | ni | ni | ni | neg |
| 25 | F | 34 | non f, mal | 10q- | C/M | neg | ||||
| 26 | M | 53 | non f, mal | 10q- | C | neg | ||||
| 27 | F | 46 | vip, mal | C | * | ni | * | ni | neg | |
| 28 | F | 59 | vip, mal | C | ▪ | ▪ | ▪ | ▪ | neg | |
| 29 | F | 36 | vip, mal | C | neg | |||||
| 30 | M | 57 | vip, mal | C/M | * | ni | ni | ni | neg | |
| 31 | M | 63 | gast, mal | C | □ | □ | □ | ni | neg | |
| 32 | M | 20 | ins, MEN1 | neg | ||||||
| 33 | M | 41 | gast, MEN1 | C | neg | |||||
ins, insulinoma; gluc, glucogonoma; vip, VIPoma; gast, gastrinoma; non F, non-functioning; mal, malignant
† 10q rg-, partial 10q deletion by CGH (5)
‡ C, cytoplasmic expression; N, nuclear expression; M, cytoplasmic membrane staining; neg, no expression; blanks, no paraffin blocks available
§ ▪ LOH; □ ROH; ni, not informative; *, no PCR amplification; blanks, no germline tissue available.
Results
LOH Analysis
For 22 tumor samples, paired normal tissue was available and therefore, could be used for LOH analysis (Table 2) ▶ . Sixteen of these EPTs were informative for at least one of four markers within and flanking PTEN. Among these 16, eight tumors exhibited loss of one allele at the 10q23 region, and eight retained heterozygosity for all of the informative markers (Table 2 ▶ and Figure 1 ▶ ). Case 17 showed retention of heterozygosity at AFMa086wg9 and LOH of the adjacent marker D10S2491 indicating partial loss of one PTEN allele. LOH at 10q23 seemed to be associated with malignant phenotype: whereas eight of 10 (80%) informative malignant and none of six informative benign EPTs showed evidence of loss of one allele, two of 10 (20%) malignant and six of six (100%) informative-benign EPTs retained both 10q23 alleles (P < 0.05, Fischer’s exact test).
Figure 1.

LOH analysis of EPT-nontumor DNA from cases 28 and 31 using microsatellite markers within and flanking PTEN. ▪, LOH; □, ROH; ni, not informative.
Of the eight informative tumors with LOH, five have been shown by comparative genomic hybridization to have large 10q losses.
Mutation Analysis
Single-strand conformation polymorphism analysis revealed an additional band in exon 6 in one tumor (case 23, Figure 2 ▶ ). Sequencing of exon 6 confirmed that the aberrant single-strand conformation polymorphism band reflected a sequence variant (546A>T) which was absent in the corresponding germline DNA. This 546 A>T transition represents a somatic missense mutation resulting in an amino acid change at codon 182, L182F. This tumor also showed LOH at 10q23, at least involving the 3′ part of PTEN (Table 1) ▶ .
Figure 2.
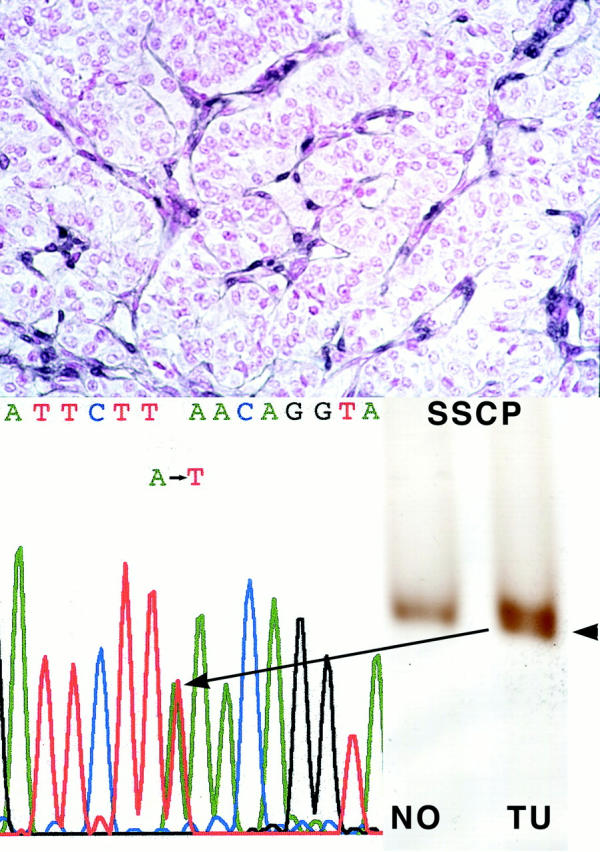
Case 23 EPT with somatic PTEN mutation, 546A>T and loss of wild-type allele, resulting in negative PTEN immunoreactivity. Top: Negative PTEN immunostaining; note internal positive control of strongly staining neo-vessels. Single-strand conformation polymorphism and sequencing of exon 6 reveals a somatic mutation (546A>T). Note that the chromatogram shows the mutation as an apparent heterozygote although by both immunohistochemical and LOH analysis, this tumor has loss one allele and is mutant in the remaining. This pseudo-heterozygote appearance is because of some admixture with DNA from contaminating normal cells.
Immunohistochemistry
Twenty-four paraffin-embedded tumor samples were available for immunohistochemical analysis. Fifteen of these had adjacent normal pancreatic tissue containing the islets of Langerhans, the normal counterpart of EPTs. All of the islets showed strong immunoreactivity (++) to the antibody 6H2.1. With the exception of one case which was fixed in Bouin’s solution (case 33), all normal islets exhibited homogenous predominant nuclear PTEN expression (Figure 3a) ▶ . As internal positive controls, we used predominantly nuclear staining of endothelial cells (especially within neo-vessels) and cytoplasmic staining of Schwann cells in peripheral nerves (graded ++ staining). Exocrine pancreatic acini were PTEN immunostain-negative throughout and were used as internal negative controls.
Figure 3.
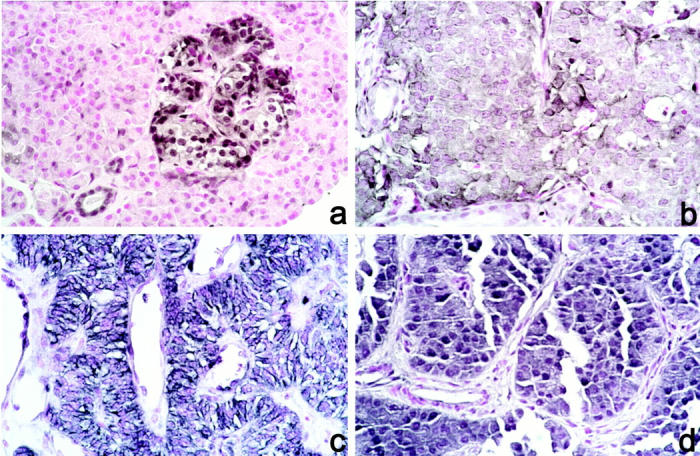
Immunohistochemistry with anti-PTEN antibody. Original magnification, ×100. a: Normal islet (case 11). b: Cytoplasmic staining of tumor cells (case 11). c: Cytoplasmic staining of tumor cells with membrane pattern (case 25). d: Nuclear and cytoplasmic staining (case 24).
Among 24 EPTs that were subjected to immunohistochemistry, one (4%) was completely negative for PTEN immunostaining (Figure 2) ▶ . This malignant, nonfunctioning EPT had a somatic PTEN mutation (as described above) and loss of the remaining allele (Table 2 ▶ , case 23). In the remaining 23 EPTs, all showed strong PTEN expression, graded + to ++. Nineteen of these 23 (83%) tumors exhibited a predominantly cytoplasmic PTEN staining (Figure 3b ▶ and Table 2 ▶ ); the staining pattern of four of these 19 tumors was suggestive of PTEN localization at the cell membrane (Figure 3c) ▶ . In addition, four of these 19 tumors were found to have cytoplasmic staining as well as strong nuclear PTEN staining (case 10, 15, 19, and 24; Figure 3d ▶ ). We found no association between staining pattern and clinical behavior in this relatively small series.
Discussion
We have detected a novel somatic mutation in exon 6 of PTEN. The nucleotide change 546A>T results in an amino acid change at codon 182, L182F. As expected for a tumor suppressor gene, this mutation was accompanied by LOH of the wild-type allele. The structural two-hits in a malignant nonfunctioning EPT (case 23) resulted in loss of immunoreactivity to the monoclonal anti-PTEN antibody recognizing a C-terminal epitope. One would have expected that loss of one allele with a missense mutation in the remaining allele would result in a full-length protein, with subsequent decreased PTEN immunoreactivity. Our observations of complete loss of immunoreactivity may be explained by decreased transcription or translation, increased degradation of the mutant protein, or conformational change in the C-terminal 100 amino acids induced by the missense mutation. Most likely, the missense mutation results in decreased transcript stability. In sum, somatic intragenic PTEN mutations do occur in a small percentage, one of 33 (3%), of EPTs. The patient with this widely invasive nonfunctioning EPT underwent a Whipple procedure and remains disease-free 11 years after the surgery.
Although somatic intragenic PTEN mutation is infrequent in EPT, we have shown that half of all informative EPTs harbor deletions of the 10q23 region, specifically involving PTEN. On the one hand, LOH analysis confirmed the large losses of 10q detected by comparative genomic hybridization analysis; on the other, we detected three additional malignant EPTs (cases 14, 18, and 28) with loss of the 10q23 region solely detected by PCR-based analysis of microsatellite markers. Interestingly, all of the samples that had LOH were malignant EPTs. This finding suggests that allelic loss of this region could be associated with malignant behavior. Apart from the single malignant EPT with two structural hits and no PTEN expression, all of the EPTs with LOH remained PTEN-immunopositive. These observations are in contrast to those made in breast cancer, thyroid neoplasia, and endometrial cancer, where loss of one PTEN allele is strongly associated with decreased PTEN protein level or complete loss of PTEN expression (XP Zhou and C Eng, unpublished observations). 17,20,25 In these tumors, therefore, either one or both inactivational events can be epigenetic. In EPTs, however, hemizygous loss of 10q and PTEN do not seem to be associated with decreased expression, an observation that may generate several hypotheses. It may be argued that PTEN is not the primary target of 10q23 deletion and that other tumor suppressor genes in the region are the major targets. Fine deletion mapping of the 10q22–24 region in thyroid adenomas and carcinomas, for example, pointed to two distinct critical intervals of LOH in this region. 26 An alternative postulate might be supported by our immunohistochemical data. Although PTEN protein is localized mainly in the nucleus in nonneoplastic islets, it is localized predominantly in the cytoplasm and cell membrane in 19 of 24 (80%) EPTs. Differential subcellular localization of PTEN has been observed previously in a large series ranging from normal thyroid to anaplastic thyroid carcinoma. 25 In the thyroid series, exclusion of nuclear staining was associated with increasing malignant potential. The percentage of tumors with decreased nuclear PTEN was lowest in breast cancer (Perren and Eng, unpublished observations), up to 50% in thyroid carcinomas 25 and is highest in EPTs (19 of 24, 80%). In contrast to the thyroid tumors, however, this shifting of PTEN from nucleus to cytoplasm is not associated with increasing malignant behavior in EPTs but instead, is associated with the neoplastic state in general.
The observation of nuclear staining remains a puzzle but clearly has been observed by others. 27,28 PTEN lacks a clear nuclear localization signal, and so, if it does traffic in and out of the nucleus, a shuttle must be involved. Our observations in vivo together with circumstantial preliminary evidence demonstrating positive PTEN signal by Western blot analysis of nuclear fractions 28 (L-P Weng and C Eng, unpublished observations) argue that this phenomenon is not an artifact. Although PTENs major substrate PtdIns(3,4,5)P3 as well as its antagonist phosphatidyl inositol 3-kinase (PI3-K) normally interact with PTEN in the cytoplasm, more specifically, at the cytoplasmic membrane, the phospholipids and PI3-K have been found in the nucleus as well although their role in the nucleus is still unknown. 29-31 Whether PTENs nuclear localization is significant for its interaction with the phospholipids or whether it best controls proper cell cycling in that location are still speculative. Therefore, based on our series of observations 20,25 (this report; LP Weng and C Eng, unpublished observations), and independent data from other groups, 27,28 we hypothesize that inappropriate qualitative or quantitative subcellular compartmentalization of PTEN could be a frequent initiating event in EPTs, which results in neoplasia, whereas physical loss of 10q leads to progression to malignancy.
Footnotes
Address reprint requests to Charis Eng, Human Cancer Genetics Program, The Ohio State University Comprehensive Cancer Center, 420 W. 12th Avenue, Room 690C Medical Research Facility, Columbus, OH 43210. E-mail: eng-1@medctr.osu.edu.
Supported in part by the American Cancer Society RPG98–211-01CCE (to C. E.) and the National Cancer Institute (P30 CA16058 to The Ohio State University Comprehensive Cancer Center).
References
- 1.Zhuang Z, Vortmeyer AO, Pack S, Huang S, Pham TA, Wang C, Park WS, Agarwal SK, Debelenko LV, Kester M, Guru SC, Manickam P, Olufemi SE, Yu F, Heppner C, Crabtree JS, Skarulis MC, Venzon DJ, Emmert-Buck MR, Spiegel AM, Chandrasekharappa SC, Collins FS, Burns AL, Marx SJ, Lubensky IA: Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res 1997, 57:4682-4686 [PubMed] [Google Scholar]
- 2.Gortz B, Roth J, Krahenmann A, de Krijger RR, Muletta-Feurer S, Rutimann K, Saremaslani P, Speel EJ, Heitz PU, Komminoth P: Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol 1999, 154:429-436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lohmann DR, Funk A, Niedermeyer HP, Haupel S, Hofler H: Identification of p53 gene mutations in gastrointestinal and pancreatic carcinoids by nonradioisotopic SSCA. Virchows Arch B Cell Pathol 1993, 64:293-296 [DOI] [PubMed] [Google Scholar]
- 4.Hofler H, Ruhri C, Putz B, Wirnsberger G, Hauser H: Oncogene expression in endocrine pancreatic tumors. Virchows Arch B Cell Pathol 1988, 55:355-361 [DOI] [PubMed] [Google Scholar]
- 5.Speel EJ, Richter J, Moch H, Egenter C, Saremaslani P, Rutimann K, Zhao J, Barghorn A, Roth J, Heitz PU, Komminoth P: Genetic differences in endocrine pancreatic tumor subtypes detected by comparative genomic hybridization. Am J Pathol 1999, 155:1787-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelen MR, Padberg GW, Peeters EAJ, Lin AY, van den Helm B, Frants RR, Coulon V, Goldstein AM, van Reen MMM, Easton DF, Eeles RA, Hodgson S, Mulvihill JJ, Murday VA, Tucker MA, Mariman ECM, Starink TM, Ponder BAJ, Ropers HH, Kremer H, Longy M, Eng C: Localization of the gene for Cowden disease to 10q22–23. Nat Genet 1996, 13:114-116 [DOI] [PubMed] [Google Scholar]
- 7.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang S, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittman M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast and prostate cancer. Science 1997, 275:1943-1947 [DOI] [PubMed] [Google Scholar]
- 8.Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DHF, Tavtigian SV: Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997, 15:356-362 [DOI] [PubMed] [Google Scholar]
- 9.Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R: Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 1997, 16:64-67 [DOI] [PubMed] [Google Scholar]
- 10.Marsh DJ, Dahia PLM, Zheng Z, Liaw D, Parsons R, Gorlin RJ, Eng C: Germline mutations in PTEN are present in Bannayan-Zonana syndrome. Nat Genet 1997, 16:333-334 [DOI] [PubMed] [Google Scholar]
- 11.Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PLM, Zheng Z, Liaw D, Caron S, Duboué B, Lin AY, Richardson A-L, Bonnetblanc J-M, Bressieux J-M, Cabarrot-Moreau A, Chompret A, Demange L, Eeles RA, Yahanda AM, Fearon ER, Fricker J-P, Gorlin RJ, Hodgson SV, Huson S, Lacombe D, LePrat F, Odent S, Toulouse C, Olopade OI, Sobol H, Tishler S, Woods CG, Robinson BG, Weber HC, Parsons R, Peacocke M, Longy M, Eng C: Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet 1998, 7:507-515 [DOI] [PubMed] [Google Scholar]
- 12.Marsh DJ, Kum JB, Lunetta KL, Bennett MJ, Gorlin RJ, Ahmed SF, Bodurtha J, Crowe C, Curtis MA, Dazouki M, Dunn T, Feit H, Geraghty MT, Graham JM, Hodgson SV, Hunter A, Korf BR, Manchester D, Miesfeldt S, Murday VA, Nathanson KA, Parisi M, Pober B, Romano C, Tolmie JL, Trembath R, Winter RM, Zackai EH, Zori RT, Weng LP, Dahia PLM, Eng C: PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet 1999, 8:1461-1472 [DOI] [PubMed] [Google Scholar]
- 13.Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C: Germline and germline mosaic mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arterio-venous malformations and lipomatosis. Hum Mol Genet 2000, 9:765-768 [DOI] [PubMed] [Google Scholar]
- 14.Kong D, Suzuki A, Zou T-T, Sakurada A, Kemp LW, Wakatsuki S, Yokohama T, Yamakawa H, Furukawa T, Sato M, Ohuchi N, Sato S, Yin J, Want S, Abraham JM, Souza RF, Smolinksi KN, Meltzer SJ, Horii A: PTEN1 is frequently mutated in primary endometrial carcinomas. Nat Genet 1997, 17:143-144 [DOI] [PubMed] [Google Scholar]
- 15.Levine RL, Cargile CB, Blazes MS, van Rees B, Kurman RJ, Ellenson LH: PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res 1998, 58:3254-3258 [PubMed] [Google Scholar]
- 16.Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI, Li J, Parsons R, Ellenson LH: Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res 1997, 57:3935-3940 [PubMed] [Google Scholar]
- 17.Mutter GL, Lin M-C, Fitzgerald JT, Kum JB, Baak JPA, Lees JA, Weng L-P, Eng C: Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst 2000, 92:924-931 [DOI] [PubMed] [Google Scholar]
- 18.Dürr E-M, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von Deimling A: PTEN mutations in gliomas and glioneuronal tumours. Oncogene 1998, 16:2259-2264 [DOI] [PubMed] [Google Scholar]
- 19.Boström J, Ludwig-Cobbers JMJ, Wolter M, Tabatabai G, Weber RG, Lichter P, Collins VP, Reifenberger G: Mutation of the PTEN (MMAC1) tumour suppressor gene in a subset of glioblastomas but not in meningiomas with loss of chromosome arm 10q. Cancer Res 1998, 58:29-33 [PubMed] [Google Scholar]
- 20.Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PLM, Komminoth P, Less JA, Mulligan LM, Mutter GL, Eng C: Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol 1999, 155:1253-1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solcia E, Klöppel G, Sobin LH, (Eds): Histological Typing of Endocrine Tumours. Heidelberg, Springer, 2000
- 22.Perren A, Roth J, Muletta-Feurer S, Saramaslani P, Speel EJ, Heitz PU, Komminoth P: Clonal analysis of sporadic pancreatic endocrine tumours. J Pathol 1998, 186:363-371 [DOI] [PubMed] [Google Scholar]
- 23.Komminoth P, Kunz E, Hiort O, Schöder S, Matias-Guiu X, Christiansen G, Roth J, Heitz PU: Detection of RET proto-oncogene point mutations in paraffin-embedded pheochromocytoma specimens by nonradioactive single-strand conformation polymorphism analysis and direct sequencing. Am J Pathol 1994, 145:922-929 [PMC free article] [PubMed] [Google Scholar]
- 24.Weng L-P, Smith WM, Dahia PLM, Ziebold U, Gil E, Lees JA, Eng C: PTEN suppresses breast cancer cell growth by phosphatase function-dependent G1 arrest followed by apoptosis. Cancer Res 1999, 59:5808-5814 [PubMed] [Google Scholar]
- 25.Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, Gil E, Hinze R, Delbridge L, Lees JA, Robinson BG, Komminoth P, Dralle H, Eng C: Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol 2000, 156:1693-1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeh JJ, Marsh DJ, Zedenius J, Dwight T, Delbridge L, Robinson BG, Eng C: Fine structure deletion analysis of 10q22–24 demonstrates novel regions of loss and suggests that sporadic follicular thyroid adenomas and follicular thyroid carcinomas develop along distinct parallel neoplastic pathways. Gene Chromsom Cancer 1999, 26:322-328 [DOI] [PubMed] [Google Scholar]
- 27.Tamura M, Gu J, Matsumoto K, Aota S-I, Parsons R, Yamada KM: Inhibition of cell migration, spreading and focal adhesions by tumor suppressor PTEN. Science 1998, 280:1614-1617 [DOI] [PubMed] [Google Scholar]
- 28.Lachyankar MB, Sultana N, Schonhoff CM, Mitra P, Poluha W, Lambert S, Quesenberry PJ, Litofsky NS, Recht LD, Nabi R, Miller SJ, Ohta S, Neel BG, Ross AH: A role for nuclear PTEN in neuronal differentiation. J Neurosci 2000, 20:1404-1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Santos CS, Clarke JH, Divecha N: Phospholipid signalling in the nucleus. Biochim Biophys Acta 1998, 1436:201-232 [DOI] [PubMed] [Google Scholar]
- 30.Metjian A, Roll RL, Ma AD, Abrams CS: Agonists cause nuclear translocation of phosphotidylinositol 3-kinase gamma. A Gbeta-gamma-dependent pathway that requires p110-gamma amino terminus. J Biol Chem 1999, 274:27943-27947 [DOI] [PubMed] [Google Scholar]
- 31.Marchisio M, Bertagnolo V, Colamussi ML, Capitani S, Neri LM: Phosphotidylinositol 3-kinase in HL-60 nuclei is bound to the nuclear matrix and increases during granulocyte differentiation. Biochem Biophys Res Commun 1998, 253:346-351 [DOI] [PubMed] [Google Scholar]