

Abstract
Many studies suggest a role for endogenous opioid peptides and their receptors in regulation of ethanol intake. It is commonly accepted that the κ-opioid receptors and their endogenous ligands, dynorphins, produce a dysphoric state and therefore may be responsible for avoidance of alcohol. We used mutant mice lacking preprodynorphin in a variety of behavioral tests of alcohol actions. Null mutant female, but not male, mice showed significantly lower preference for alcohol and consumed lower amounts of alcohol in a two-bottle choice test as compared with wild-type littermates. In the same test, knockout mice of both sexes showed a strong reduction of preference for saccharin compared to control mice. In contrast, under conditions of limited (4 hours) access (light phase of the light/dark cycle), null mutant mice did not show any differences in consumption of saccharin but they showed significantly reduced intake of sucrose. To determine the possible cause for reduction of ethanol preference and intake, we studied other ethanol-related behaviors in mice lacking the preprodynorphin gene. There were no differences between null mutant and wild type mice in ethanol-induced loss of righting reflex, acute ethanol withdrawal, ethanol-induced conditioned place preference or conditioned taste aversion to ethanol. These results indicate that deletion of preprodynorphin leads to substantial reduction of alcohol intake in female mice, and suggest thath this is caused by decreased orosensory reward of alcohol (sweet taste and/or palatability).
Keywords: Dynorphin, Mutant mouse, Ethanol intake, Palatability, Sweet taste
1. Introduction
Pharmacological and clinical studies have implicated the opioid system in various aspects of alcoholism (see Gianoulakis, 2004; Koob et al., 2003; Oswald & Wand, 2005 for rev.). There are three major groups of endogenous opioid peptides, each derived from a specific precursor hormone: the endorphins from the β-endorphin/ACTH precursor proopiomelanocortin (POMC) (Nakanishi et al., 1979); the enkephalins from the precursor proenkephalin (Noda et al., 1982); and the dynorphins and neoendorphins from the precursor prodynorphin (Kakidani et al., 1982). Each prohormone is the product of a distinct gene and a separate messenger RNA. The opioid peptides may function as neurotransmitters or neuromodulators and regulate a variety of behaviors such as psychomotor stimulation, positive reinforcement, drinking, eating, sexual behaviors, thermoregulation, nociception, and mood (Amalric et al., 1987; Mezey et al., 1985; Spanagel et al., 1991; Wilcox et al., 1986).
The three classes of opioid peptides described above interact with at least three opioid receptor subtypes: mu (μ), delta (δ), and kappa (κ) (Reisine & Bell, 1993). β-Endorphin binds with about equal affinity to μ- and δ-opioid receptors, whereas the enkephalins bind with approximately 20-fold greater affinity to δ- compared with μ-opioid receptors. The prodynorphin peptide products tend to bind selectively to κ-opioid receptors. The activation of μ- or δ-receptors often leads to similar patterns of neurotransmitter release, whereas the activation of κ-receptors often produces opposing patterns (Herz & Spanagel, 1995). For example, in the mesolimbic system, β-endorphin and enkephalin peptides increase DA release within the nucleus accumbens through their interactions with the μ- and δ-opioid receptors and are, therefore, an intricate part of the process associated with reward and reinforcement (Koob, 1992). Dynorphin, on the other hand, has been shown to decrease DA release secondary to activation of κ-opioid receptors, thereby producing aversive states.
There is extensive evidence that all three opioid receptors regulate ethanol drinking. Nonspecific opioid receptor antagonists, such as naloxone and naltrexone, reduce ethanol intake in rats (Coonfield et al., 2002; Froehlich et al., 1990) and mice (Middaugh et al., 1999). Naltrindole, a selective δ-opioid receptor antagonist, was found to suppress the ethanol self-administration in a similar manner in AA and Wistar rats (Hyytia & Kiianmaa, 2001). In contrast, the activation of κ-opioid receptors by a selective agonist, U50,488H, dose-dependently decreased ethanol intake in rats (Lindholm et al., 2001). Evidence that complements the findings from the opioid receptor blockade studies comes from studies that have disrupted opioid receptor and opioid peptide expression. For example, the microinjection of antisense oligonucleotides targeted to the μ-opioid receptor in the nucleus accumbens disrupted ethanol drinking by the ethanol-preferring (HEP) rats (Myers & Robinson, 1999). Targeted gene mutation (knockout) strategies provided mice that lack opioid peptides or receptors. Thus, mice lacking μ-opioid receptors, generated from three distinct constructs, do not self-administer ethanol under various conditions, including oral self-administration and two-bottle choice, and do not display conditioned place preference to ethanol (Becker et al., 2002; Hall et al., 2001; Roberts et al., 2000). Surprisingly, significantly reduced ethanol consumption was also shown in mice lacking κ-opioid receptors (Kovacs et al., 2005). In contrast, δ-knockout mice showed a greater preference for ethanol and consumed more ethanol than their wild type counterparts (Roberts et al., 2001). Lack of preproenkephalin also leads to reduction of ethanol consumption (Blednov et al., 2004), whereas ethanol oral self-administration is increased in mutant mice with decreased β-endorphin expression (Grisel et al., 1999). Consistent with these data, Grahame et al. (1998) found that β-endorphin deficient mice acquired reliable intravenous self-administration of ethanol, while the wild type mice did not.
Based on these studies, there are two possible, and contradictory, roles for dynorphin peptides in regulation of alcohol consumption. First, peptides derived from preprodynorphin could reduce the reinforcing actions of alcohol and mice lacking these peptides should show increased alcohol consumption. Second, deletion of preprodynorphin could reduce activation of κ-opioid receptors and this should recapitulate the phenotype of the κ-opioid receptor null mutants which is less consumption of alcohol. Our studies with preprodynorphin knockout mice demonstrate that lack of this peptide reduces alcohol self-administration and suggest a mechanism for this decreased consumption.
2. Materials and methods
2.1. Animals
Generation of preprodynorphin deficient mice was described earlier (Sharifi et al., 2001). Breeding pairs were kindly provided by Dr. Toni Shippenberg. They were maintained on the mixed genetic background C57Bl/6J x 129/SvEv-Tac. All behavioral analyses were performed on homozygous knockout (−/−) and wild type (+/+) littermates generated from crosses between heterozygous animals. The genotype was confirmed by multiple primer PCR performed on tail biopsy samples from offspring. Mice were group-housed three-five per cage based on sex. Food and water were available ad libitum. The vivarium was maintained on a 12:12 h light:dark cycle with light on at 7.00 AM. The temperature and humidity of the room were controlled. All experiments were performed during the light phase of the light/dark cycle. Behavioral testing began when the mice were at least two months-of-age. Only naive mice of both sexes were used in all behavioral experiments. All experiments were conducted in the isolated behavioral testing rooms in the animal facility to avoid external distractions. All experiments were approved by the Institutional Animal Care and Use Committee and adhered to NIH Guidelines. The University of Texas facility is AAALAC accredited.
2.2. Alcohol preference drinking
The two-bottle choice protocol was carried out as previously described (Blednov et al., 2001). Briefly, mice were allowed to acclimate for 1 week to individual housing. Two drinking tubes were continuously available to each mouse, and tubes were weighed daily. One tube always contained water. Food was available ad libitum, and mice were weighed every 4 d. After 4 d of water consumption (both tubes), mice were offered 3% ethanol (v/v) versus water for 4 d. Tube positions were changed every day to control for position preferences. Quantity of ethanol consumed (g/kg body weight/24 h) was calculated for each mouse and these values were averaged for every concentration of ethanol. Immediately following 3% ethanol, a choice between 6% (v/v) ethanol and water was offered for 4 d, then 9% (v/v) ethanol for 4 d and finally 12% (v/v) ethanol vs. water for 4 d. Throughout the experiment, evaporation/spillage estimates were calculated every day from two bottles placed on an empty cage, one containing water and the other containing the appropriate ethanol solution. All together, 10 animals of each sex and genotype were used.
2.3. Preference for non-alcohol tastants
Wild-type or knockout mice were also tested for saccharin and quinine consumption. One tube always contained water and the other contained the tastant solution. Mice were serially offered saccharin (0.033% and 0.066%) and quinine hemisulfate (0.03 mM and 0.06 mM) and intakes were calculated. Each concentration was offered for 4 d, with bottle position changed every day. Within each tastant, the low concentration was always presented first, followed by the higher concentrations. Between tastants mice had two bottles with water for three weeks. All together, 10 animals of each sex and genotype were used.
2.4. Saccharin and sucrose consumption under conditions of limited access
Nocturnal rodents, such as mice and rats feed primarily during the dark hours (Erickson et al., 1996) and characteristically eat their largest meal shortly after “lights off” (Green et al., 1992). To distinguish between consumption and response to a palatable stimulus, mice are usually provided with limited access to a highly palatable stimulus at a time different than their normal consumption period – e.g., during “lights on”. This approach has been used for comparison of palatable responses in wild type and mutant mice (Sindelar et al., 2005). In our experiments, mice were acclimated for 1 week to individual housing with free access to food and water. To initiate the first approach to the drinking tube after acclimation, water was removed from cage at 8.00 am (1 hour after lights on) every morning and water intake was measured for 4 hours (from 11.00 am till 3.00 pm). After 3.00 pm until 8.00 am of the next day mice had free access to water. Every bottle was weighed before and after each drinking session. The initial measurement of water intake was repeated for 4 days and mean of 4 hours water intake was taken as 100% of basal fluid intake. The same procedure was repeated with consequent presentation of one bottle of saccharin (0.033% and 0.066%) or sucrose (2.5%, 5.0% and 7.5%). Each concentration of both tastants was presented for 3 days and means from 3 days intake were calculated in percent from basal water intake. Saccharin was used to distinguish between response to sweet taste and caloric consumption of sucrose. All together, 20 wild type mice (10 per sex) and 27 knockout mice (14 males and 13 females) were used.
2.5. Ethanol-Induced Acute Withdrawal
Mice were scored for handling-induced convulsion (HIC) severity 30 min before and immediately before i.p. ethanol administration. The two predrug base-line scores (PRE) were averaged. A dose of 4 g/kg of ethanol in saline was injected i.p. and the HIC score was tested every hour until the HIC level reached base-line. Acute withdrawal was quantified as the area under the curve but above PRE level (Crabbe et al., 1991). Briefly, each mouse is picked up gently by the tail and, if necessary, gently rotated 1800, and the HIC is scored as follows: 5, tonic-clonic convulsion when lifted; 4, tonic convulsion when lifted; 3, tonic-clonic convulsion after a gentle spin; 2, no convulsion when lifted, but tonic convulsion elicited by a gentle spin; 1, facial grimace only after a gentle spin; 0, no convulsion. The scorer was blind to the genotype of the mice. All together, 14 wild type mice (7 per sex) and 16 knockout mice (8 per sex) were used.
2.6. Conditioned Place Preference
The conditioned place preference protocol was carried out as previously described (Blednov et al., 2003). Four identical acrylic boxes (30 × 15 × 15 cm) were separately enclosed in ventilated, light, and sound-attenuating chambers (Med Associates, St. Albans, VT). Each box has two compartments separated by wall with a door. The two compartments each have a different type of floor (either bars – [GRID-] or wire mesh – [GRID+]). Infrared light sources and photodetectors were mounted opposite each other at 2.5-cm intervals along the length of each box, 2.2 cm above the floor. Occlusion of the infrared light beams was used to measure general activity and location of the animal (left or right) within the box. Total activity counts and location of the animal (left or right compartment) within the box were recorded by computer. The floors and the inside of the boxes were wiped with a damp sponge and the litter paper beneath the floors was changed between animals. The main principles of conditioned place preference procedure have been described earlier (Cunningham et al., 1993). Ethanol was injected at dose 2.0 g/kg (i.p.) 5 min before session. All together, 41 wild type mice (20 males and 21 females) and 42 knockout mice (21 per sex) were used.
2.7. Conditioned Taste Aversion
Subjects were adapted to a water-restriction schedule (2 h of water per day) over a 7-day period. At 48-h intervals over the next 10 days, all mice received 1-h access to a solution of saccharin (0.15% w/v sodium saccharin in tap water) or NaCl (0.2 M). Immediately after 1-h access to tastant, mice received injections of saline or ethanol (2.5 g/kg i.p.). All mice also received 60-min access to tap water 5 hours after injection of ethanol to prevent dehydration. Two-hour access to tap water was given during intervening days. All together, 30 wild type mice (15 per sex) and 29 knockout mice (14 males and 15 females) were used.
2.8. Loss of Righting Reflex
Sensitivity to ethanol was determined using the standard sleep time assay. Ethanol was diluted in 0.9% saline (20 % v/v) and administered in doses 3.4 g/kg and 3.8 g/kg (i.p.) adjusted by injected volumes. These doses were selected to give LORR in all mice, but not to give a prohibitively long sleep time because the dependence between dose of alcohol and duration of LORR is remarkably steep. Mice were injected with ethanol and when they became ataxic, they were placed in the supine position in V-shaped plastic troughs until they were able to right themselves three times within 30 s. Sleep time was defined as the time from being placed in the supine position until they regained their righting reflex. During all sleep time assays, room temperature was 22°C. Mice that failed to lose the righting reflex (misplaced injections) or had a sleep time greater than two standard deviations from the group mean were excluded from the analysis. All together, 37 wild type mice (12 males and 25 females) and 39 knockout mice (16 males and 23 females) were used.
2.9. Ethanol metabolism
Animals were given a single dose of ethanol (3.8 g/kg i.p.) and blood samples were taken from the retro-orbital sinus in 30, 60, 120, 180 and 240 min after injection. Blood alcohol concentration (BEC) values, expressed as mg ethanol per ml blood were determined spectrophotometrically by an enzyme assay (Lundquist, 1959). All together, 5 mice per sex and genotype were used.
2.10. Statistical analysis
Data are reported as the mean ± S.E.M value. The statistics software program GraphPad Prizm (Jandel Scientific, Costa Madre, CA) was used throughout. Data were analyzed by one-way ANOVA and two-way ANOVA with Bonferroni post-hoc analyses.
3. Results
3.1. Ethanol consumption
In a two-bottle free-choice paradigm in which mice could drink either water or an ascending series of ethanol concentrations (3, 6, 9 and 12%), mutant female mice lacking preprodynorphin displayed reduced preference for ethanol [F(1,71) = 11.3, P < .0001, main effect of genotype and F(3,71) = 3.1; P < .05, main effect of concentration). No genotype x concentration interaction [F(3,71) = 1.2, P > .05) was found (Fig.1 a). The amount of ethanol consumed by null female mice showed significant dependence on genotype (F1,71 = 5.1; P< .05), concentration (F3,71 = 6.1; P< .001) but no genotype x concentration interaction (F3,71 = 1.6; P> .05) (Fig.1 b). Total fluid intake was significantly increased in null female mice (F1,71 = 9.5; P< .01, main effect of genotype) (Fig.1 c). There was no main effect of concentration (F3,71 = 0.2; P> .05) as well as genotype x concentration interaction (F3,71 = 0.2; P> .05). In male mice only main effect of concentration was found for preference for ethanol (F3,72 = 20.5; P< .001) as well as for amount of consumed ethanol (F3,72 = 4.5; P< .01) (Fig.1 d,e). There was no difference between total intake between wild type and null male mice (Fig.1 f).
FIG.1.
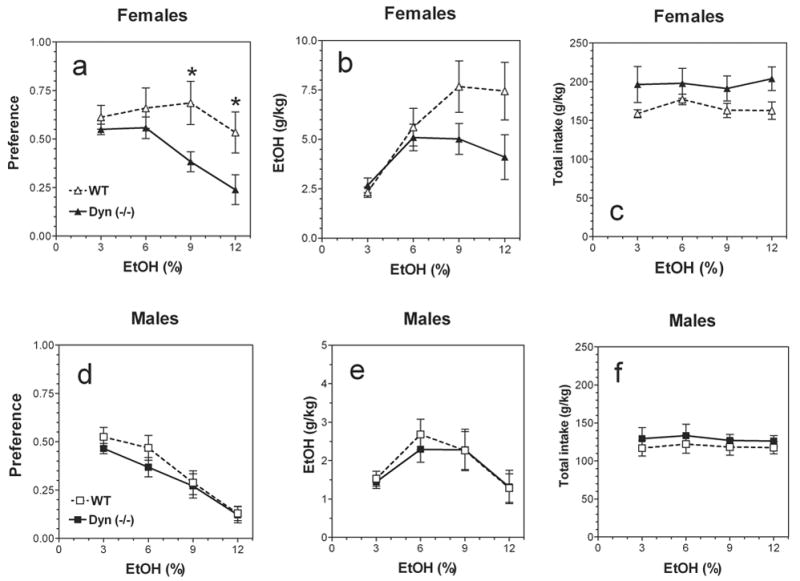
Voluntary ethanol consumption in prodynorphin deficient mice.
Females. Preference for ethanol. B. Females. Amount of ethanol consumed. C. Females. Total fluid intake. D. Males. Preference for ethanol. E. Males. Amount of ethanol consumed. F. Males. Total fluid intake. n=10 for all groups. * - p<0.05 – significant differences relative to wild type mice for similar concentration of ethanol (two-way ANOVA, Bonferroni post-hoc analyses).
3.2. Preference for non-alcohol tastants
Null female mice showed a strong reduction of preference for saccharin compared to wild type females (F1,36 = 14.2; P< .001, main effect of genotype) (Fig.2 a). Neither a main effect of concentration (F1,36 = 0.5; P> .05) nor genotype x concentration interaction (F1,36 = 0.01; P> .05) were found. Only main effect of concentration (F1,36 = 4.6; P< .05) but not genotype (F1,36 = 0.05; P> .05) as well as genotype x concentration interaction (F1,36 = 1.6; P> .05) were revealed for total intake of saccharin solutions (saccharin + water) in female mice (Fig.2 b). Null male mice also showed a strong reduction of preference for saccharin compared to wild type males (F1,36 = 27.3; P< .001, main effect of genotype and F1,36 = 4.2; P< .05, main effect of concentration) (Fig.2 c). No genotype x concentration interaction (F1,36 = 0.8; P> .05) was found. Neither main effect of concentration (F1,36 = 2.3; P> .05), genotype (F1,36 = 3.9; P> .05) as well as genotype x concentration interaction (F1,36 = 0.2; P> .05) were revealed for total intake of saccharin solutions (saccharin + water) in male mice (Fig.2 d).
FIG.2.
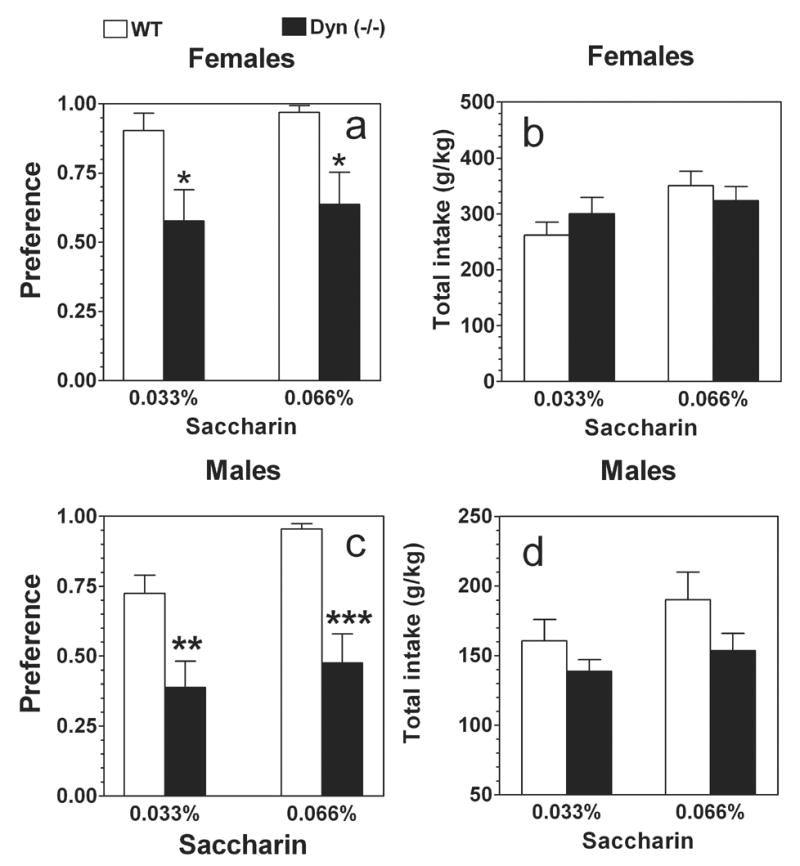
Voluntary saccharin consumption in prodynorphin deficient mice.
Females. Preference for saccharin. B. Females. Total intake (saccharin + water). C. Males. Preference for saccharin. D. Males. Total intake (saccharin + water). n=10 for all groups. * - p<0.05 – significant differences relative to wild type mice for similar concentration of tastants (two-way ANOVA, Bonferroni post-hoc analyses).
No main effects of concentration (F1,36 = 0.01; P> .05), genotype (F1,36 = 0.02; P> .05) or genotype x concentration interaction (F1,36 = 0.01; P> .05) were revealed for preference of quinine solutions in female mice (Fig.3 a). However, null female mice consumed significantly greater amount of quinine solution and water than wild type females (F1,36 = 9.4; P< .01, main effect of genotype). No main effects of concentration (F1,36 = 2.9; P> .05) and genotype x concentration interaction (F1,36 = 0.003; P> .05) were revealed (Fig.3 b). On the contrary, null male mice showed marginally significant reduced avoidance of quinine solutions compare with wild type males (F1,36 = 6.7; P< .05, main effect of genotype; F1,36 = 0.2; P> .05, main effect of concentration; F1,36 = 1.4; P> .05, genotype x concentration interaction) (Fig.3 c). No differences in total intake of quinine solutions or water were found in male mice (F1,36 = 2.8; P> .05, main effect of genotype; F1,36 = 0.4; P> .05, main effect of concentration; F1,36 = 0.1; P> .05, genotype x concentration interaction) (Fig.3 d).
FIG.3.
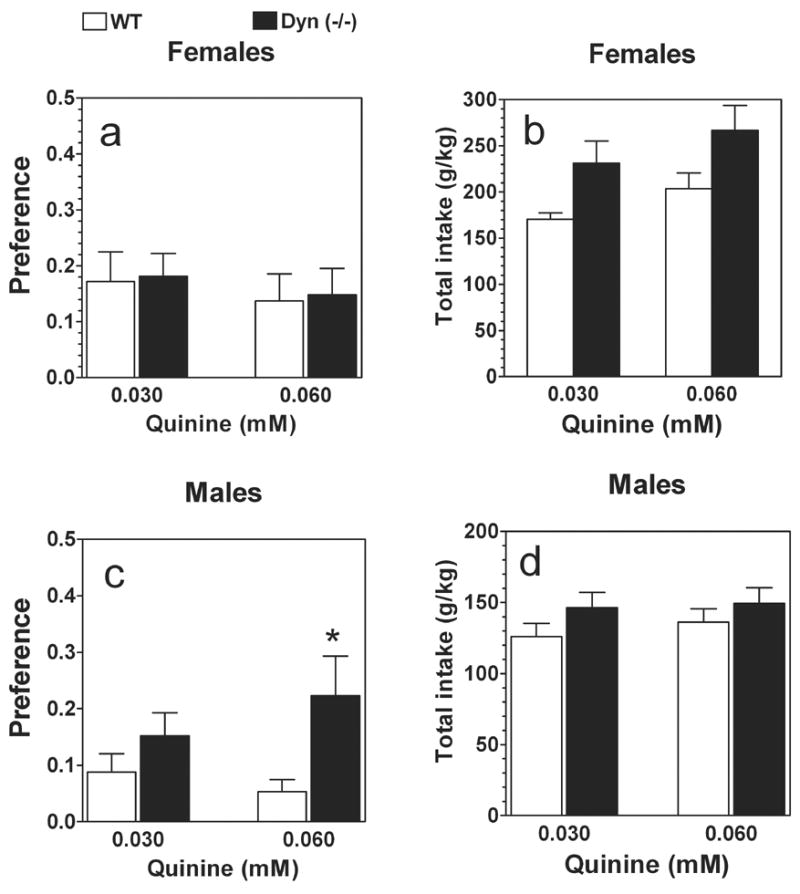
Voluntary quinine consumption in prodynorphin deficient mice.
A. Females. Preference for quinine. B. Females. Total intake (quinine + water). C. Males. Preference for quinine. D. Males. Total intake (quinine + water). n=10 for all groups. * - p<0.05 – significant differences relative to wild type mice for similar concentration of tastants (two-way ANOVA, Bonferroni post-hoc analyses).
3.3. Saccharin and Sucrose consumption in limited access procedure
Consumption of water during the 4 hour period was greater in null female mice than in wild type mice (14.9 ± 4.4 g/kg body weight and 9.2 ± 1.8 g/kg body weight, respectively). Null male mice and wild type males consumed similar amount of water (12.5 ± 2.5 g/kg body weight and 11.1 ± 2.7 g/kg body weight, respectively). To attempt to correct for these differences in initial intake and facilitate presentation of the data, intake of saccharin and sucrose was calculated as a percentage of the mean consumption of water by dividing the amount of saccharin and sucrose solution consumed on subsequent 4-hrs sessions by the amount of water consumed during 4-h session. No differences in response to saccharin were found in female mice (F1,63 = 1.3; P> .05, main effect of genotype; F2,63 = 2.5; P> .05, main effect of concentration) (Fig.4 a). One-way ANOVA revealed a trend in increase of saccharin intake compare with water intake only in wild type female mice (F2,27 = 3.1; P= .06) but not in null females (F2,36 = 0.5; P= .6) In contrast, preprodynorphin null female mice consumed significantly smaller amounts of sucrose than wild type females (F1,84 = 4.6; P< .05, main effect of genotype; F3,84 = 11.1; P< .0001, main effect of concentration) (Fig.4 b). In males, no differences in response to saccharin were found and this is similar to results obtained with female mice (F1,66 = 0.8; P> .05, main effect of genotype; F2,66 = 1.6; P> .05, main effect of concentration) (Fig.4 c). No response to saccharin was also found in both wild type (F2,27 = 1.1; P> .05, one-way ANOVA) and null male mice (F2,39 = 0.4; P> .05, one-way ANOVA) (Fig.4 c). Although, both wild type (F3,52 = 15.2; P< .0001, one-way ANOVA) and null male mice (F3,52 = 3.6; P< .05, one-way ANOVA) responded to sucrose, only trend to difference between two genotypes was found (F1,88 = 3.4; P= .06, main effect of genotype; F3,88 = 13.3; P< .0001, main effect of concentration) (Fig.4 d).
FIG.4.
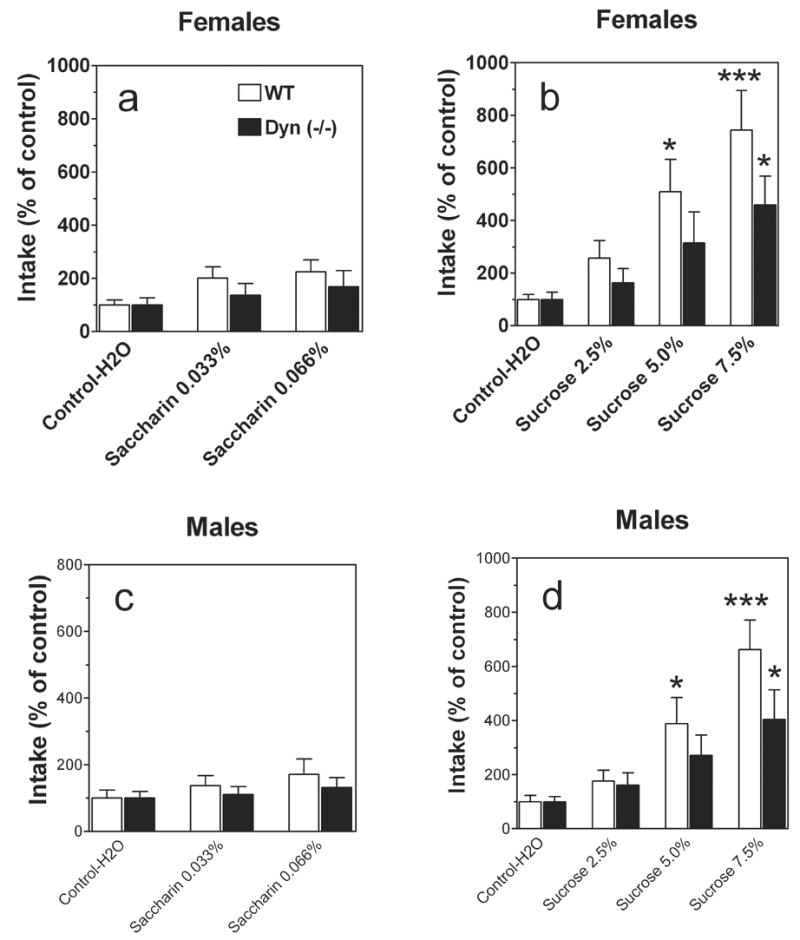
Limited access to saccharin or sucrose solutions in prodynorphin deficient mice.
A. Females. Amount of consumed saccharin solutions. B. Females. Amount of consumed sucrose solutions. C. Males. Amount of consumed saccharin solutions. D. Males. Amount of consumed sucrose solutions. n= 10–14 per genotype and sex. * - p<0.05; *** - p<0.001– significant differences relative to correspondent control (one-way ANOVA, Bonferroni post-hoc analyses).
3.4. Conditioned Taste Aversion
Because null mice showed a reduced preference for saccharin, two variants of conditioned taste aversion with two different conditional stimuli (saccharin and NaCl) were explored. Although there was no difference in consumption of saccharin on trial 0 (before conditioning) (wild type and preprodynorphin null female mice: 79 ± 3 g/kg body weight and 80 ± 7 g/kg body weight, respectively; wild type and preprodynorphin null male mice: 53 ± 4 g/kg body weight and 57 ± 3 g/kg body weight, respectively), consumption of NaCl on trial 0 was slightly greater in wild type female mice than in null females (131 ± 7 g/kg body weight and 119 ± 8 g/kg body weight, respectively) and in null male mice compare with wild type males (105 ± 5 g/kg body weight and 97 ± 7 g/kg body weight, respectively). To attempt to correct for these initial differences in tastant intake and facilitate presentation of the data, intake was calculated as a percentage of the trial 0 consumption for each subject by dividing the amount of saccharin (or NaCl) solution consumed on subsequent conditioning trials by the amount of saccharin (or NaCl) solution consumed on trial 0 (before conditioning). Ethanol-saccharin as well as ethanol-NaCl pairings produced reductions in saccharin (females: F4,90 = 2.6; P< .05 and males: F4,90 = 4.1; P< .01, main effect of trial) or NaCl (females: F4,80 = 4.9; P< .01 and males: F4,80 = 7.6; P< .0001, main effect of trial) intake across trials, indicating the development of CTA in both genotypes (Fig.5). However, there were no differences in development of CTA between wild type and preprodynorphin null mice (females-saccharin: F1,90 = 0.001; P> .05 and females-NaCl: F1,80 = 0.3; P> .05; males-saccharin: F1,90 = 0.4; P> .05 and males-NaCl: F1,80 = 0.3; P> .05, main effect of genotype). In saline-treated groups of mice only differences between wild type and null male mice were found in experiment with saccharin as conditioned stimulus (F1,34 = 14.7; P< .0001, main effect of genotype).
FIG.5.
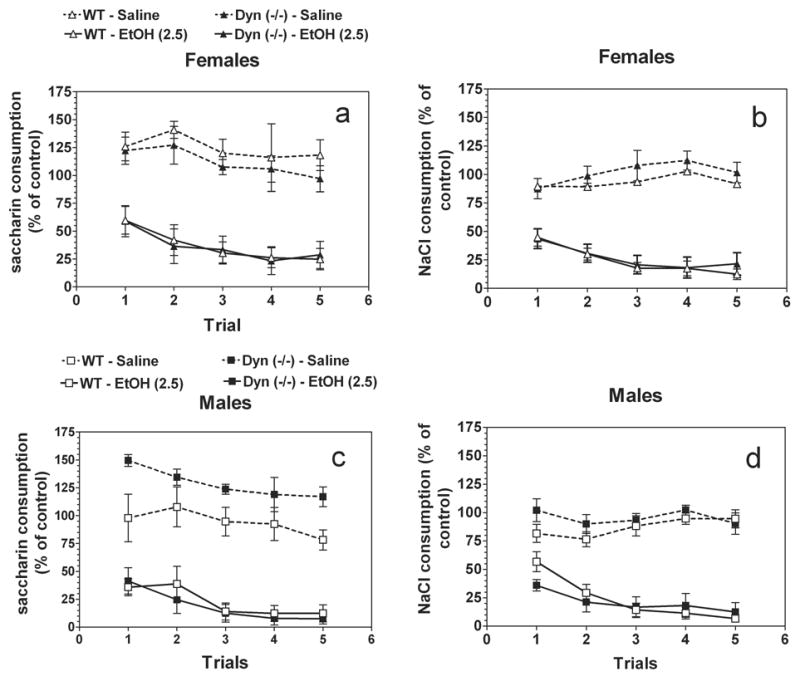
Ethanol induced conditioned taste aversion in prodynorphin deficient mice.
Females. Saccharin is the conditioned stimulus. B. Females. NaCl is the conditioned stimulus. C. Males. Saccharin is the conditioned stimulus. B. Males. NaCl is the conditioned stimulus. n = 5–6 for saline injection for all genotypes. n = 9–10 for groups with ethanol injection.
3.5. Place Conditioning
Following control saline injections, female mice of both genotypes spent substantially less time on the bar floor than on the wire mesh floor (F1,40 = 12.2; < .01, main effect of floor) (Fig. 6a). No main effect of genotype was found (F1,40 = 0.01; P> .05). In male mice only trend to reduction of time spent on bar floor was found in both genotypes (F1,40 = 3.6; P= .06, main effect of floor; F1,40 = 0.01; P> .05, main effect of genotype) (Fig.6 e). Taking into account floor preference found in female mice, we calculated place conditioning for both sexes only for the group of mice injected with ethanol paired with their less favorite floor (the bar floor, GRID). The percent of time spent on the bar floor by saline- and ethanol-injected female mice of each genotype is shown in figure Fig. 6 b. Female mice of both genotypes spent more time on the bar floor when it was paired with ethanol than when paired with saline, reflecting development of conditioned place preference (F1,38 = 22.1; P< .0001, main effect of treatment). However, there was no difference in development of place conditioning between the two genotypes (F1,38 = 0.3; P> .05, main effect of genotype). Like female mice males of both genotypes developed similar conditioned place preference and preferred the bar floor when it was paired with ethanol than when paired with saline (F1,37 = 12.5; P< .01, main effect of treatment; F1,37 = 0.8; P> .05, main effect of genotype) (Fig.6 f).
FIG.6.
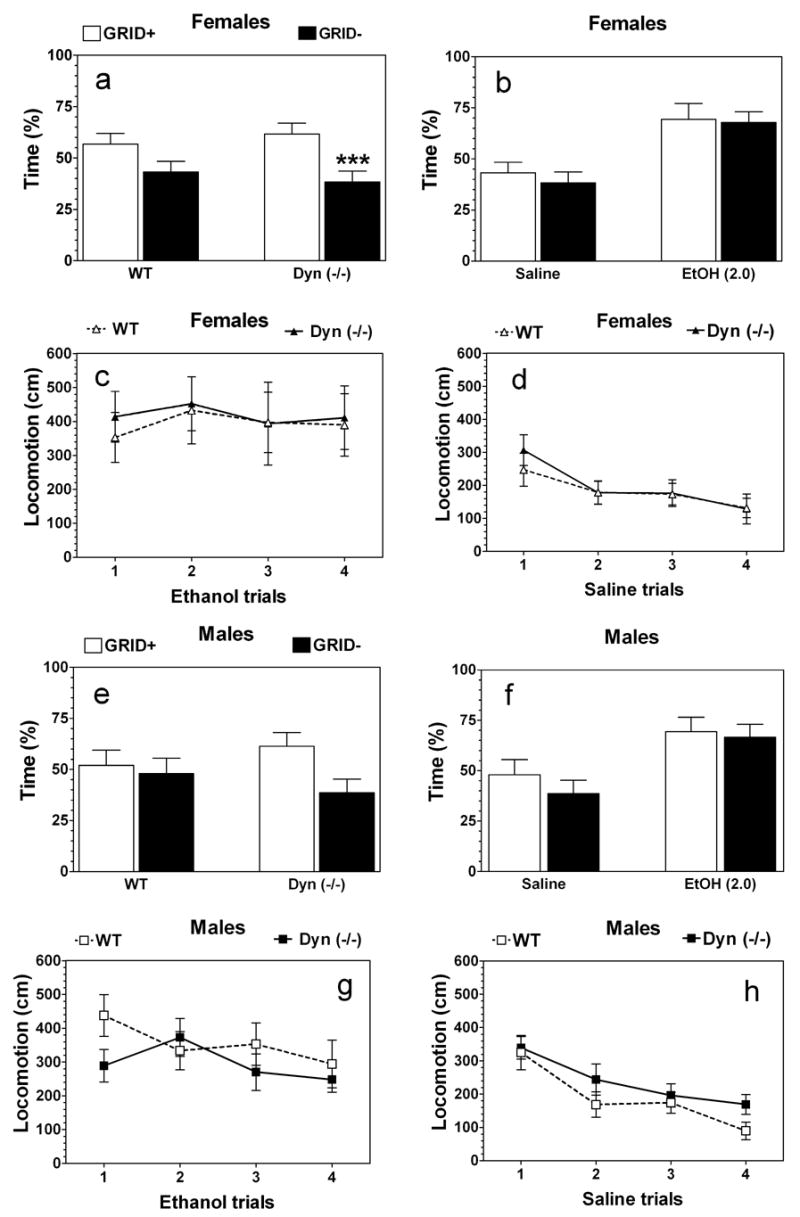
Ethanol-induced conditioned place preference in prodynorphin deficient mice.
Females. Percent of time spent on different floors during 30-min test session in control saline-treated groups. B. Females. Percent of time spent on the bars by saline-treated groups and GRID- conditioned subgroups (ethanol injection was paired with bars). C. Females. Motor activity during each 5-min ethanol conditioned trials (CS+). D. Females. Motor activity during each 5-min saline conditioned trials (CS-). E. Males. Percent of time spent on different floors during 30-min test session in control saline-treated groups. F. Males. Percent of time spent on the bars by saline-treated groups and GRID- conditioned subgroups (ethanol injection was paired with bars). G. Males. Motor activity during each 5-min ethanol conditioned trials (CS+). H. Males. Motor activity during each 5-min saline conditioned trials (CS-). n = 11 for saline-injected groups of each genotype, n = 9–11 for ethanol-injected groups of each genotype and sex. *** - p<0.001– significant differences between different types of floor for the same genotype (two-way ANOVA, Bonferroni post-hoc analyses).
Mean activity of female mice during each 5-min ethanol (CS+) and saline (CS-) conditioning trial are depicted in Fig. 6 c,d. The first injection of ethanol (Trial 1) produced slight increase in activity relative to saline in both preprodynorphin knockout and wild type female mice. However, this increase did not reach statistical significance (F1,36 = 2.9; P= .09, main effect of treatment). In female mice of both genotypes, injections of ethanol in the following trials (2, 3 and 4) did not induce further motor activation (F1,72 = 0.1; P> .05, main effect of genotype and F3,72 = 0.2; P> .05, main effect of trial) (Fig.6 c). Activity on saline trials slightly decreased across trials in preprodynorphin null female mice (F3,40 = 3.7; P< .05, one-way ANOVA) but not in wild type female mice (F3,40 = 1.8; P> .05, one-way ANOVA) (Fig.6 d). There was no effect of the first injection of ethanol (Trial 1) in both wild type and null male mice (F1,32 = 0.4; P> .05, main effect of treatment and F1,32 = 1.9; P> .05, main effect of genotype) (Fig.6 g,h). Multiple injections of ethanol (Trials 1, 2, 3 and 4) did not change the motor activity in both wild type (F3,32 = 1.0; P> .05, one-way ANOVA) and null male mice (F1,36 = 1.2; P> .05, one-way ANOVA) (Fig. 6 g). No genotype dependent differences between wild type and null male mice were found (F1,66 = 2.2; P> .05, main effect of genotype and F3,66 = 1.1; P> .05, main effect of trial) (Fig. 6 g). Saline injections reduced the motor activity in both wild type (F3,32 = 6.7; P< .01, one-way ANOVA) and null male mice(F3,36 = 4.2; P< .05, one-way ANOVA) (Fig. 6 h). However, only effect of treatment (F3,67 = 10.3; P< .0001) but not genotype (F1,67 = 3.2; P>.05) was found (Fig.6 h).
3.6. Ethanol Acute Withdrawal Severity
A single 4 g/kg ethanol dose suppressed basal HIC in both the knockout and wild type mice of both sexes for about 5 hours, followed by increased HIC (Fig. 7 a,c). Preprodynorphin null and wild type mice of both sexes did not differ in levels of basal HIC (Fig. 7 a,c). Animals of both genotypes demonstrated signs of withdrawal (HIC scores higher than the basal level) (Fig. 7 a,c). However, there were no significant genotype differences in the area under the curves for HIC during withdrawal for female mice (P> .05, t-Student test) as well as for male mice (P> .05, -Student test) (Fig. 7 b,d).
FIG.7.
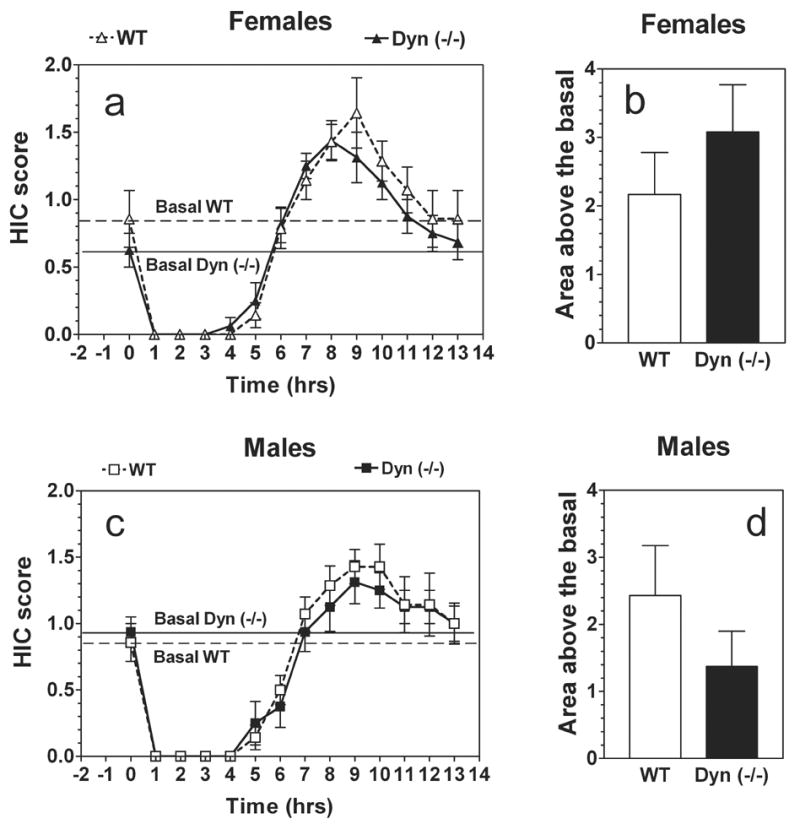
Severity of ethanol-induced withdrawal in prodynorphin deficient mice.
A. Females. HIC scores. B. Females. Area under HIC curve and above the basal HIC level. C. Males. HIC scores. D. Males. Area under HIC curve and above the basal HIC level. n = 7–8 for both genotypes and sex.
3.7. Depressant Effects of Ethanol
Only effect of dose (F1,44 = 5.1; P< .05 for female mice and F1,24 = 14.1; P< .01 for male mice) but not genotype (F1,44 = 0.5; P> .05 for female mice and F1,24 = 0.1; P> .05 for male mice) were found in the anesthetic (depressant) effects of ethanol (Fig.8 a,b).
FIG.8.
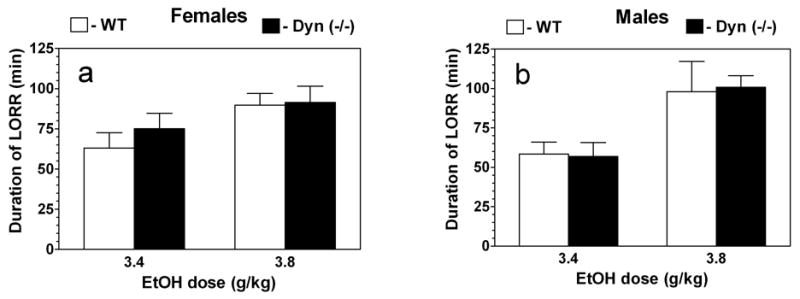
Ethanol-induced loss of righting reflex in prodynorphin deficient mice.
A. Females. Duration of LORR. B. Males. Duration of LORR. n = 9–16 for females of both genotypes per dose of ethanol. n = 6–9 for males of both genotypes per dose of ethanol.
3.8. Ethanol metabolism
There were no differences in metabolism of ethanol between control and any of the knockout mice (data not shown).
4. Discussion
A summary of the behavioral results obtained from male and female of preprodynorphin knockout mice are presented in Table 1. It is clear that deletion of the preprodynorphin gene from this genetic background leads to substantial reduction of alcohol consumption and preference in female, but not in male, mice. A possible explanation for this sex-dependent reduction could be decreased preference for sweet taste observed in both sexes of null mice which was however accompanied by reduced avoidance of bitter solutions of quinine observed only in male null mice. On the other hand, it should be noted that effects of mutations on voluntary ethanol consumption strongly depends on genetic background (Blednov et al., 2006; Crabbe et al., 2006). Therefore, it is quite possible that a different genetic background could reveal a reduction in ethanol intake in male mice.
Table 1.
Summary of effects of deletion of preprodynorphin gene on ethanol-related behaviors in male and female mice.
| Behavior | Males | Females |
|---|---|---|
| 2 bottle choice: Ethanol intake (preference) | 0 | ↓ |
| 2 bottle choice: Ethanol intake (g/kg) | 0 | ↓ |
| 2 bottle choice: Total intake (g/kg) | 0 | ↑ |
| 2 bottle choice: Saccharin intake (preference) | ↓ | ↓ |
| 2 bottle choice: Quinine intake (avoidance) | ↓ | 0 |
| Limited access: Saccharin intake (g/kg) | 0 | 0 |
| Limited access: Sucrose intake (g/kg) | 0 | ↓ |
| Conditioned taste aversion | 0 | 0 |
| Conditioned place preference | 0 | 0 |
| Loss of righting reflex | 0 | 0 |
| Acute withdrawal | 0 | 0 |
↓ Decreased behavior in comparison with wild type. ↑- Increased behavior in comparison with wild type. 0 – No change in comparison with wild type.
Endorphin and enkephalin interactions with μ- and δ-opioid receptors increase dopamine release in the nucleus accumbens and may initiate the processes associated with reward and reinforcement, whereas binding of dynorphin to κ-opioid receptors produce aversive states and decrease dopamine release, and reduce reinforcement (Koob, 1992). If this view is correct, we might expect that the removal of tonic inhibition mediated by κ-opioid receptors and dynorphins will facilitate reinforcing properties of alcohol and will increase alcohol consumption. However, mice lacking preprodynorphin (present study) or κ-opioid receptors (Kovacs et al., 2005) showed reduced consumption of alcohol and of sucrose. Ethanol self-administration might also be reduced by increased aversive effects of ethanol (Broadbent et al., 2002), but preprodynorphin knockout mice did not differ from wild type in development of conditioned taste aversion to alcohol. Alternatively, mutation could increase rewarding properties of alcohol and mice could reduce alcohol consumption because less alcohol is needed to produce reinforcement. However, deletion of preprodynorphin did not change the rewarding properties of alcohol measured in the conditioned place preference test. A negative correlation between hypnotic (loss of righting reflex) effects of ethanol and voluntary ethanol consumption has been shown (Hodge et al., 1999; Thiele et al., 1998). However, there was no difference between preprodynorphin knockout and wild type mice in the hypnotic effect of ethanol. Alcohol withdrawal severity is inversely correlated with alcohol consumption in mice (Metten at al. (1998) raising the possibility that increased alcohol withdrawal could account for the differences in alcohol consumption. However, preprodynorphin knockout mice did not differ from wild type mice in severity of acute ethanol-induced withdrawal. Together, these data showed that deletion of preprodynorphin does not change several pharmacological effects of alcohol which can be linked to ethanol consumption.
In addition to the pharmacological effects of alcohol, alcohol consumption depends on caloric value, taste, olfaction and palatability (Bachmanov et al., 2003; Belknap et al., 1993; Kiefer et al., 1998; McMillen & Williams, 1998). It is well known, that opioid systems are involved in food intake regulation (see Glass et al., 1999; Yamamoto, 2003 for rev.), but the precise role opioids have in feeding behavior is unclear. One possibility is that opioids mediate the rewarding or palatable aspects of feeding. Thus, the ability of opioid antagonists to suppress intake is greater when palatable food items (lard or diets sweetened with sucrose) or drink are offered (Giraudo et al., 1993; Levine et al., 1982, 1995; Lynch, 1986). Hypothalamic dynorphin A1-17 and prodynorphin mRNA levels are elevated by feeding a palatable diet rich in fat and sucrose whereas caloric restriction of the same diet decreases mRNA levels of prodynorphin, proenkephalin and POMC, as well as levels of dynorphin A1-17 (Welch et al., 1996). Thus, the activation of the prodynorphin system may be required to signal palatability and to link consumption and palatability. This is in agreement with our data showing that deletion of preprodynorphin leads to reduction of response to palatable solutions of sucrose. However, this conclusion is not consistent with reports about aversive effects of κ-opioid agonists in both humans and rodents (Kumor et al., 1986; Mucha & Herz, 1985). On the other hand, it is known that manipulations that block the development of satiation may also increase the intake of concentrated sucrose solutions (Sclafani, 1991). Thus, the facilitatory effect of activation of κ-opioid neurotransmission on the intake of concentrated sucrose solution may be due, at least in part, to a reduction in satiation (Badiani et al., 2001). It is also important to note that κ-opioid agonists do not increase the intake of palatable but non-caloric saccharin solutions (Gosnell & Majchrzak, 1989) and activation of κ-opioid receptors has opposite effects on feeding and drinking in the rat, enhancing food-intake while suppressing water-intake (Badiani & Stewart, 1992, 1993; Nencini & Graziani, 1990). These results are in agreement with our data showing that mice lacking preprodynorphin do not demonstrate reduced response to saccharin solution under conditions of non-active consummatory behavior but they increase total fluid consumption under conditions of 24 hours continuous drinking.
Of course, one of important question is whether compensatory changes in expression of other genes occur as a result of deletion of preprodynorphin. Several studies show that lack of preprodynorphin leads to blockade of dysphoric effects of cannabinoids. Thus, preprodynorphin-deficient mice show specific changes in the behavioral effects of delta-9-tetrahydrocannabinol (THC), including a reduction of spinal THC analgesia and the absence of THC-induced conditioned place aversion (Zimmer et al., 2001). On the other hand, Mendizabal et al. (2006) demonstrated a shift to the left in the dose-intake curve for self-administration of the cannabinoid agonist WIN 55,212–2 in preprodynorphin knockout mice. Both cannabinoids and opioids are the active players in regulation of palatability (see Cooper, 2004 for rev.), but changes in cannabinoid neurotransmission observed in preprodynorphin knockout mice should increase rather than reduce palatability.
Recent findings also suggest that dynorphin-A may play a role in the function of the orexin neurons and orexin stimulates both food intake and energy expenditure (Nakamachi et al., 2006; Wang et al., 2003). Chou et al. (2001) showed that nearly all (94%) neurons of the rat lateral hypothalamic area (LHA) expressing prepro-orexin mRNA also expressed prodynorphin mRNA. Although, orexin knockout mice do not show decreased expression of neuronal dynorphin mRNA in LHA (Chou et al., 2001), the possibility that within the orexin neurons dynorphin may play a role in coordination with orexin can not be ruled out.
It should be also noted that deletion of preprodynorphin leads to compensatory up-regulation of κ-opioid receptors mainly in limbic brain area (Clarke et al., 2003). Chefer and Shippenberg (2006) showed that this up-regulation is functional because the inhibitory effects of the selective κ-opioid receptor agonist U-69593 on nucleus accumbens dialysate dopamine (DA) levels are enhanced in preprodynorphin knockout mice. Furthermore, they showed that constitutive deletion of preprodynorphin is associated with decreased basal extracellular DA levels. In vitro studies have shown that not only dynorphin but met-enkephalin and β-endorphin can also bind with relatively high affinity to κ-opioid receptors (Kosterlitz et al., 1989). Therefore, a more likely explanation for the attenuation of basal DA release in preprodynorphin knockout mice is that deletion of dynorphin leads to a compensatory increase in κ-opioid receptor number and that, in the absence of dynorphin, endogenous met-enkephalin and/or β-endorphin activate κ-opioid receptors. Interestingly, the increase of intake of concentrated sucrose solution observed after administration of κ-opioid agonist U-50,488H is also accompanied by reduction of extracellular DA concentration in the nucleus accumbens (Badiani et al., 2001). These data are consistent with the notion that the activation of mesolimbic DA transmission plays a critical role in food reward (Wise, 1994). However, it is unclear what this up-regulation of κ-opioid receptors in preprodynorphin knockout mice has in common with behavioral responses to ethanol, because behavioral phenotype of these mice is completely identical with behavioral ethanol phenotype described in mice lacking κ-opioid receptors (Kovacs et al., 2005).
Our results showing decreased alcohol consumption, lower saccharin preference, and lower sucrose intake in preprodynorphin knockout mice are consistent with previous observations of opioid involvement in regulation of food intake. Together with similar ethanol behavioral phenotypes demonstrated in mice lacking κ-opioid receptors (Kovacs et al., 2005), these findings indicate the important role of dynorphin/κ-opioid receptors system in regulation of orosensory reward.
Acknowledgments
This work was supported by NIAAA INIA Consortium and NIH grants AA13520 and AA06399
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature
- Amalric M, Cline EJ, Marinez JL, Bloom FE, Koob GF. Rewarding properties of β-endorphin as measured by conditioned place preference. Psychopharmacology. 1987;91:14–19. doi: 10.1007/BF00690919. [DOI] [PubMed] [Google Scholar]
- Bachmanov AA, Kiefer SW, Molina JC, Tordoff MG, Duffy VB, Bartoshuk LM, Mennella JA. Chemosensory factors influencing alcohol perception, preferences, and consumption. Alcohol Clin Exp Res. 2003;27:220–231. doi: 10.1097/01.ALC.0000051021.99641.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani A, Rajabi H, Nencini P, Stewart J. Modulation of food intake by the kappa opioid U-50,488H: evidence for an effect on satiation. Behav Brain Res. 2001;118:179–186. doi: 10.1016/s0166-4328(00)00325-9. [DOI] [PubMed] [Google Scholar]
- Badiani A, Stewart J. The kappa-opioid U-50,488H suppresses the initiation of nocturnal spontaneous drinking in normally hydrated rats. Psychopharmacology. 1992;106:463–473. doi: 10.1007/BF02244816. [DOI] [PubMed] [Google Scholar]
- Badiani A, Stewart J. Enhancement of the prophagic but not of the antidipsogenic effect of U-50, 488H after chronic amphetamine. Pharmacol Biochem Behav. 1993;44:77–86. doi: 10.1016/0091-3057(93)90283-y. [DOI] [PubMed] [Google Scholar]
- Becker A, Grecksch G, Kraus J, Loh HH, Schroeder H, Hollt V. Rewarding effects of ethanol and cocaine in mu opioid receptor-deficient mice. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:296–302. doi: 10.1007/s00210-002-0533-2. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Crabbe JC, Young ER. Voluntary consumption of ethanol in 15 inbred mouse strains. Psychopharmacology. 1993;112:503–510. doi: 10.1007/BF02244901. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Stoffel M, Chang SR, Harris RA. Potassium channels as targets for ethanol: studies of G-protein-coupled inwardly rectifying potassium channel 2 (GIRK2) null mutant mice. J Pharmacol Exp Ther. 2001;298:521–530. [PubMed] [Google Scholar]
- Blednov YA, Walker D, Alva H, Creech K, Findlay G, Harris RA. GABAA receptor alpha 1 and beta 2 subunit null mutant mice: behavioral responses to ethanol. J Pharmacol Exp Ther. 2003;305:854–863. doi: 10.1124/jpet.103.049478. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Walker D, Alva H, Harris RA. Reduced ethanol consumption and increased ethanol-induced conditioned taste aversion in mice lacking preproenkephalin. Alcohol Clin Exp Res. 2004;28:10.a.3. Suppl. [Google Scholar]
- Blednov YA, Walker D, Ramirez M, Harris RA. Increased ethanol consumption and palatability in mice lacking glutamic acid decarboxylase (GAD65) Alcohol Clin Exp Res. 2006;30:240. Suppl. [Google Scholar]
- Broadbent J, Muccino KJ, Cunningham CL. Ethanol-induced conditioned taste aversion in 15 inbred mouse strains. Behav Neurosci. 2002;116:138–148. [PubMed] [Google Scholar]
- Chefer VI, Shippenberg TS. Paradoxical effects of prodynorphin gene deletion on basal and cocaine-evoked dopaminergic neurotransmission in the nucleus accumbens. Eur J Neurosci. 2006;23:229–238. doi: 10.1111/j.1460-9568.2005.04525.x. [DOI] [PubMed] [Google Scholar]
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE. Orexin (hypocretin) neurons contain dynorphin. J Neurosci. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke S, Zimmer A, Zimmer AM, Hill RG, Kitchen I. Region selective up-regulation of micro-, delta- and kappa-opioid receptors but not opioid receptor-like 1 receptors in the brains of enkephalin and dynorphin knockout mice. Neuroscience. 2003;122:479–489. doi: 10.1016/j.neuroscience.2003.07.011. [DOI] [PubMed] [Google Scholar]
- Coonfield DL, Hill KG, Kaczmarek HJ, Ferraro FM, 3rd, Kiefer SW. Low doses of naltrexone reduce palatability and consumption of ethanol in outbred rats. Alcohol. 2002;26:43–47. doi: 10.1016/s0741-8329(01)00180-x. [DOI] [PubMed] [Google Scholar]
- Cooper SJ. Endocannabinoids and food consumption: comparisons with benzodiazepine and opioid palatability-dependent appetite. Eur J Pharmacol. 2004;500:37–49. doi: 10.1016/j.ejphar.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Crabbe JC, Merrill C, Belknap JK. Acute dependence on depressant drugs is determined by common genes in mice. J Pharmacol Exp Ther. 1991;257:663–667. [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Harris RA, Arends MA, Koob GF. Alcohol-related genes: Contributions from studies with genetically engineered mice. Addiction Biology. 2006;11:195–270. doi: 10.1111/j.1369-1600.2006.00038.x. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Niehus JS, Noble D. Species difference in sensitivity to ethanol's hedonic effects. Alcohol. 1993;10:97–102. doi: 10.1016/0741-8329(93)90087-5. [DOI] [PubMed] [Google Scholar]
- Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–418. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- Froehlich JC, Harts J, Lumeng L, Li TK. Naloxone attenuates voluntary ethanol intake in rats selectively bred for high ethanol preference. Pharmacol Biochem Behav. 1990;35:385–390. doi: 10.1016/0091-3057(90)90174-g. [DOI] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology. 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- Herz A, Spanagel R. Endogenous opioids and addiction. In: Tseng LF, editor. The pharmacology of opioids. Germany: Harwood; 1995. pp. 445–462. [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, Wang D, Sanchez-Perez AM, Messing RO. Supersensitivity to allosteric GABA(A) receptor modulators and alcohol in mice lacking PKCepsilon. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- Hyytia P, Kiianmaa K. Suppression of ethanol responding by centrally administered CTOP and naltrindole in AA and Wistar rats. Alcohol Clin Exp Res. 2001;25:25–33. doi: 10.1111/j.1530-0277.2001.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Gianoulakis C. Endogenous opioids and addiction to alcohol and other drugs of abuse. Curr Top Med Chem. 2004;4:39–50. doi: 10.2174/1568026043451573. [DOI] [PubMed] [Google Scholar]
- Giraudo SQ, Grace MK, Welch CC, Billington CJ, Levine AS. Naloxone's anorectic effect is dependent upon the relative palatability of food. Pharmacol Biochem Behav. 1993;46:917–921. doi: 10.1016/0091-3057(93)90222-f. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Billington CJ, Levine AS. Opioids and food intake: distributed functional neural pathways? Neuropeptides. 1999;33:360–368. doi: 10.1054/npep.1999.0050. [DOI] [PubMed] [Google Scholar]
- Gosnell BA, Majchrzak MJ. Centrally administered opioid peptides stimulate saccharin intake in nondeprived rats. Pharmacol Biochem Behav. 1989;33:805–810. doi: 10.1016/0091-3057(89)90474-7. [DOI] [PubMed] [Google Scholar]
- Grahame NJ, Low MJ, Cunningham CL. Intravenous self-administration of ethanol in β-endorphin-deficient mice. Alcohol Clin Exp Res. 1998;22:1093–1098. [PubMed] [Google Scholar]
- Green PK, Wilkinson CW, Woods SC. Intraventricular corticosterone increases the rate of body weight gain in underweight adrenalectomized rats. Endocrinology. 1992;130:269–275. doi: 10.1210/endo.130.1.1727703. [DOI] [PubMed] [Google Scholar]
- Grisel JE, Mogil JS, Grahame NJ, Rubinstein M, Belknap JK, Crabbe JC, Low MJ. Ethanol oral self-administration is increased in mutant mice with decreased beta-endorphin expression. Brain Res. 1999;835:62–67. doi: 10.1016/s0006-8993(99)01384-0. [DOI] [PubMed] [Google Scholar]
- Kakidani K, Furutani Y, Takahashi H, Noda M, Morimoto Y, Hiroshi T, Numa S. Cloning and sequence analysis of cDNA for porcine β-neoendorphin/dynorphin precursors. Nature. 1982;298:245–249. doi: 10.1038/298245a0. [DOI] [PubMed] [Google Scholar]
- Kiefer SW, Hill KG, Kaczmarek HJ. Taste reactivity to alcohol and basic tastes in outbred mice. Alcohol Clin Exp Res. 1998;22:1146–1151. [PubMed] [Google Scholar]
- Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- Koob GF, Roberts AJ, Kieffer BL, Heyser CJ, Katner SN, Ciccocioppo R, Weiss F. Animal models of motivation for drinking in rodents with a focus on opioid receptor neuropharmacology. Recent Dev Alcohol. 2003;16:263–281. doi: 10.1007/0-306-47939-7_19. [DOI] [PubMed] [Google Scholar]
- Kosterlitz HW, Corbett AD, Paterson SJ. Opioid receptors and ligands. NIDA Res Monogr. 1989;95:159–166. [PubMed] [Google Scholar]
- Kovacs KM, Szakall I, O'Brien D, Wang R, Vinod KY, Saito M, Simonin F, Kieffer BL, Vadasz C. Decreased oral self-administration of alcohol in kappa-opioid receptor knock-out mice. Alcohol Clin Exp Res. 2005;29:730–738. doi: 10.1097/01.alc.0000164361.62346.d6. [DOI] [PubMed] [Google Scholar]
- Kumor KM, Haertzen CA, Johnson RE, Kocher T, Jasinski D. Human psychopharmacology of ketocyclazocine as compared with cyclazocine, morphine and placebo. J Pharmacol Exp Ther. 1986;238:960–968. [PubMed] [Google Scholar]
- Levine AS, Murray SS, Kneip J, Grace M, Morley JE. Flavor enhances the antidipsogenic effect of naloxone. Physiol Behav. 1982;28:23–25. doi: 10.1016/0031-9384(82)90095-6. [DOI] [PubMed] [Google Scholar]
- Levine AS, Weldon DT, Grace M, Cleary JP, Billington CJ. Naloxone blocks that portion of feeding driven by sweet taste in food-restricted rats. Am J Physiol. 1995;268:R248–252. doi: 10.1152/ajpregu.1995.268.1.R248. [DOI] [PubMed] [Google Scholar]
- Lindholm S, Werme M, Brene S, Franck J. The selective kappa-opioid receptor agonist U50,488H attenuates voluntary ethanol intake in the rat. Behav Brain Res. 2001;120:137–146. doi: 10.1016/s0166-4328(00)00368-5. [DOI] [PubMed] [Google Scholar]
- Lundquist F. The determination of ethyl alcohol in blood and tissue. Methods Biochem Anal. 1959;7:217–251. [Google Scholar]
- Lynch WC. Opiate blockade inhibits saccharin intake and blocks normal preference acquisition. Pharmacol Biochem Behav. 1986;24:833–836. doi: 10.1016/0091-3057(86)90420-x. [DOI] [PubMed] [Google Scholar]
- McMillen BA, Williams HL. Role of taste and calories in the selection of ethanol by C57BL/6NHsd and Hsd:ICR mice. Alcohol. 1998;15:193–198. doi: 10.1016/s0741-8329(97)00111-0. [DOI] [PubMed] [Google Scholar]
- Mendizabal V, Zimmer A, Maldonado R. Involvement of kappa/dynorphin system in WIN 55,212–2 self-administration in mice. Neuropsychopharmacology. 2006;31:1957–1966. doi: 10.1038/sj.npp.1300957. [DOI] [PubMed] [Google Scholar]
- Metten P, Phillips TJ, Crabbe JC, Tarantino LM, McClearn GE, Plomin R, Erwin VG, Belknap JK. High genetic susceptibility to ethanol withdrawal predicts low ethanol consumption. Mamm Genome. 1998;9:983–990. doi: 10.1007/s003359900911. [DOI] [PubMed] [Google Scholar]
- Mezey E, Kiss J, Mueller GP, Eskay R, O'Donohue TL, Palkovits M. Distribution of the pro-opiomelanocortin derived peptides (ACTH, alpha-MSH, beta-endorphin) in the rat hypothalamus. Brain Res. 1985;328:341–347. doi: 10.1016/0006-8993(85)91046-7. [DOI] [PubMed] [Google Scholar]
- Middaugh LD, Kelley BM, Cuison ER, Jr, Groseclose CH. Naltrexone effects on ethanol reward and discrimination in C57BL/6 mice. Alcohol Clin Exp Res. 1999;23:456–464. [PubMed] [Google Scholar]
- Mucha RF, Herz A. Motivational properties of kappa and mu opioid receptor agonists studied with place and taste preference conditioning. Psychopharmacology. 1985;86:274–280. doi: 10.1007/BF00432213. [DOI] [PubMed] [Google Scholar]
- Myers RD, Robinson DE. Mu and D2 receptor antisense oligonucleotides injected in nucleus accumbens suppress high alcohol intake in genetic drinking HEP rats. Alcohol. 1999;18:225–233. doi: 10.1016/s0741-8329(99)00015-4. [DOI] [PubMed] [Google Scholar]
- Nakamachi T, Matsuda K, Maruyama K, Miura T, Uchiyama M, Funahashi H, Sakurai T, Shioda S. Regulation by orexin of feeding behaviour and locomotor activity in the goldfish. J Neuroendocrinol. 2006;18:290–297. doi: 10.1111/j.1365-2826.2006.01415.x. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Inoue A, Kita T, Nakamura M, Chang AC, Cohen SN, Numa S. Nucleotide sequence of cloned cDNA for bovine corticotropin-b-lipotropin precursor. Nature. 1979;278:423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- Nencini P, Graziani M. Opiatergic modulation of preparatory and consummatory components of feeding and drinking. Pharmacol Biochem Behav. 1990;37:531–537. doi: 10.1016/0091-3057(90)90024-c. [DOI] [PubMed] [Google Scholar]
- Noda M, Teranishi Y, Takahashi H, Toyosato M, Notake M, Nakanishi S, Numa S. Isolation and structural organization of the human preproenkephalin gene. Nature. 1982;297:431–434. doi: 10.1038/297431a0. [DOI] [PubMed] [Google Scholar]
- Oswald LM, Wand GS. Opioids and alcoholism. Physiol Behav. 2004;81:339–358. doi: 10.1016/j.physbeh.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Reisine T, Bell GI. Molecular biology of opioid receptors. Trends Neurol Sci. 1993;16:506–510. doi: 10.1016/0166-2236(93)90194-q. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, Koob GF. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res. 2001;25:1249–1256. [PubMed] [Google Scholar]
- Roberts AJ, McDonald JC, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, Gold LH. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- Sclafani A. The hedonics of sugar and starch. In: Bolles RC, editor. The hedonics of taste. Hillsdale, NJ: Lawrence Erlbaum; 1991. pp. 59–87. [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U. Generation of dynorphin knockout mice. Brain Res Mol Brain Res. 2001;86:70–75. doi: 10.1016/s0169-328x(00)00264-3. [DOI] [PubMed] [Google Scholar]
- Sindelar DK, Palmiter RD, Woods SC, Schwartz MW. Attenuated feeding responses to circadian and palatability cues in mice lacking neuropeptide Y. Peptides. 2005;26:2597–2602. doi: 10.1016/j.peptides.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Bals-Kubik R, Shippenberg TS. β-Endorphin-induced locomotor stimulation and reinforcement are associated with an increase in dopamine release in the nucleus accumbens. Psychopharmacology. 1991;104:51–56. doi: 10.1007/BF02244553. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Marsh DJ, Ste Marie L, Bernstein IL, Palmiter RD. Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature. 1998;396:366–369. doi: 10.1038/24614. [DOI] [PubMed] [Google Scholar]
- Wang J, Osaka T, Inoue S. Orexin-A-sensitive site for energy expenditure localized in the arcuate nucleus of the hypothalamus. Brain Res. 2003;971:128–134. doi: 10.1016/s0006-8993(03)02437-5. [DOI] [PubMed] [Google Scholar]
- Welch CC, Kim EM, Grace MK, Billington CJ, Levine AS. Palatability-induced hyperphagia increases hypothalamic Dynorphin peptide and mRNA levels. Brain Res. 1996;721:126–131. doi: 10.1016/0006-8993(96)00151-5. [DOI] [PubMed] [Google Scholar]
- Wilcox JN, Roberts JL, Chronwall BM, Bishop JF, O'Donohue T. Localization of proopiomelanocortin mRNA in functional subsets of neurons defined by their axonal projections. J Neurosci Res. 1986;16:89–96. doi: 10.1002/jnr.490160109. [DOI] [PubMed] [Google Scholar]
- Wise RA. A brief history of the anhedonia hypothesis. In: Legg CR, Booth D, editors. Appetite: neural and behavioural bases. Oxford, England: Oxford University Press; 1994. pp. 243–263. [Google Scholar]
- Yamamoto T. Brain mechanisms of sweetness and palatability of sugars. Nutr Rev. 2003;61:S5–9. doi: 10.1301/nr.2003.may.S5-S9. [DOI] [PubMed] [Google Scholar]
- Zimmer A, Valjent E, Konig M, Zimmer AM, Robledo P, Hahn H, Valverde O, Maldonado R. Absence of delta -9-tetrahydrocannabinol dysphoric effects in dynorphin-deficient mice. J Neurosci. 2001;21:9499–9505. doi: 10.1523/JNEUROSCI.21-23-09499.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]