

Abstract
This study assessed whether anti-neutrophil cytoplasmic antibodies (ANCAs) interfere with the safe deletion of neutrophils by apoptosis and phagocytic clearance. Tumor necrosis factor (TNF)-primed neutrophils were incubated with normal IgG (N IgG) or ANCA IgG for up to 36 hours. Compared with N IgG, ANCAs accelerated constitutive apoptosis of TNF-α primed neutrophils, as assessed by morphology and confirmed by DNA laddering pattern on gel electrophoresis, and accelerated progression to secondary necrosis. The accelerated apoptosis induced by ANCA was dependent on reactive oxygen species generation, as primed neutrophils from patients with chronic granulomatous disease failed to show an effect of ANCAs on apoptosis. However, there was no change in the rate at which neutrophils exhibited annexin V binding, indicating that externalization of phosphatidylserine was not accelerated by ANCAs. Furthermore, when ANCA-treated primed neutrophils were interacted with human or murine peritoneal macrophages after 12 hours there was significantly less phagocytosis by human macrophages and no difference in phagocytosis by murine peritoneal-derived macrophages when compared with N IgG-treated controls. In conclusion, ANCAs accelerate apoptosis and secondary necrosis in TNF-primed neutrophils by a mechanism dependent on the generation of reactive oxygen species, with uncoupling of nuclear and surface membrane changes, resulting in a “reduced window of opportunity” for phagocytic recognition and engulfment before disintegration.
The elimination of unwanted cells is regarded as an essential component of many normal biological program, including the resolution of inflammation. Programmed cell death, or apoptosis, is a well-recognized process that is accompanied by characteristic phenomena: cell shrinkage, chromatin condensation, DNA fragmentation, membrane blebbing, and finally, in some cell types, fragmentation into membrane-bound apoptotic bodies. 1 During the apoptotic process, the plasma membrane of apoptotic polymorphonuclear leukocytes (PMNs) remains intact, thus preventing release of histiotoxic contents from these cells, which could provoke inflammation and tissue damage. In human PMNs, apoptosis is associated with an impairment of functional activity, including degranulation, respiratory burst, and phagocytosis. 2
There is strong evidence that PMNs migrating to inflamed sites are constitutively deleted by apoptosis, leading to safe phagocytic clearance of the intact dying cell, with resolution of inflammation. The major clearance route is via macrophages (mφ), which have matured from monocytes that lack phagocytic activity. Recognition of apoptotic cells is achieved by displaying surface membrane signals on the apoptotic cell surface. These include phosphatidylserine (PS), a membrane phospholipid usually present on the inner aspect of the membrane. Loss of membrane phospholipid asymmetry and early external expression of PS have been documented as an early marker of apoptosis on many cell types. 3 Recognition of these signals by mφ occurs by uncharacterized but stereospecific receptors for phosphatidylserine. Among other recognition mechanisms, mφ can deploy surface αvβ3 vitronectin receptor integrin in cooperation with CD36 and thrombospondin, which bridges the mφ to uncharacterized sites on the apoptotic PMN. 4 Phagocytosis of apoptotic cells has been documented in vivo in experimental and clinical disease states, supporting the importance of this process in the resolution of inflammation. 5,6 The ingestion of apoptotic leukocytes by phagocytes, unlike the uptake of other biological material, fails to elicit a secretory or proinflammatory response. 7 Some investigators have even detected an immunosuppressive effect of apoptotic leukocyte uptake by mφ, 3,8 which may be important in the successful resolution of inflammation. In contrast, apoptotic cells that fail to be cleared progress to secondary necrosis, with removal of the resulting cell fragments by phagocytes triggering the release of proinflammatory cytokines. 9
It has previously been suggested that failure of the apoptotic process and/or perturbation of clearance of apoptotic PMNs can lead to secondary necrosis in situ, and thence release of toxic leukocyte contents may result in tissue injury and perpetuate the inflammatory response. 10 Such events may be of critical importance in systemic vasculitis. Pathologically, anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis is characterized by a leukocytoclastic picture as shown by light microscopy, and electron microscopy studies provide evidence of leukocytes with apparently degraded nuclear material undergoing disintegration in tissues. 11 This is of interest, as Hebert and colleagues have demonstrated that as PMN apoptosis progresses, cell morphology changes from an early to a late picture with nuclear evanescence 12 ; eventually there is disintegration by secondary necrosis, a highly inflammatory event. Donald speculated that the intravascular cell lysis documented in his study of vasculitic lesions was due to a circulating cytophilic antibody. 11 We were interested in the possibility that this was due to disruption of the constitutive apoptotic process of PMNs by ANCAs, autoantibodies present in the sera of patients with Wegener’s granulomatosis and microscopic polyangiitis. 13-15 The two major antigens recognized by ANCAs are serine proteinase 3 (PR3-ANCA or cANCA), which is found predominantly in Wegener’s granulomatosis, and myeloperoxidase (MPO-ANCA or pANCA), which occurs in 70% of patients with microscopic polyangiitis. 16
In vitro studies have shown that ANCAs can activate tumor necrosis factor-α (TNF-α)-primed PMNs and mononuclear phagocytes to undergo a respiratory burst with the release of reactive oxygen species as well as degranulation of azurophil granules and secretion of proinflammatory cytokines. 17,18 We hypothesized that PMNs incubated with ANCAs would exhibit dysregulation of the apoptotic process. In this study we report ANCA-PMN coculture experiments in which TNF-α-primed PMNs aged for 12 hours showed morphological evidence of accelerated apoptosis that was dependent on the generation of reactive oxygen species. PMNs undergoing accelerated apoptosis failed to exhibit coordinated expression of PS on the cell surface as assessed by annexin V binding, suggesting dysregulated apoptosis. Furthermore, human monocyte-derived mφ showed reduced phagocytosis of apoptotic TNF-α-primed PMNs cultured with ANCAs for 12 hours. These data suggest a novel mechanism for tissue injury in systemic vasculitis whereby ANCA-activated primed PMNs undergo accelerated apoptosis. The potential for progression to deleterious secondary necrosis is promoted by dysregulation of apoptosis such that there is a reduced window of opportunity for safe clearance by macrophages.
Materials and Methods
Isolation of Human PMNs and Culture
Human PMNs were isolated as previously reported. 19 Briefly, freshly isolated blood from six healthy donors was mixed with acid citrate dextrose (ACD) in a ratio of 1:10. This was then mixed with an equal volume of 4:5 Hespan (6% Hetastarch in 0.9% sodium chloride) and with 1:5 diluted ACD (diluted with distilled water to 1:10). After centrifugation (20 × g, 15 minutes) the supernatant was removed and centrifuged (160 × g, 7 minutes). The resulting supernatant was removed and discarded and the cell pellet resuspended in 55% Percoll (Pharmacia, Uppsala, Sweden).
Discontinuous Percoll gradients were prepared. Nine volumes of Percoll was mixed with one volume of 1.5 mol/L sodium chloride. This was then diluted with phosphate-buffered saline (PBS) to concentrations of 81%, 70%, and 55%. Three milliliters of 70% was layered onto 4.5 ml of 81%. Finally, the cell pellet, which was suspended in 3 ml of 55% Percoll, was layered onto the gradients. The band between 81% and 70% was harvested and washed (1200 rpm, 7 minutes). This gave a cell purity of 95% neutrophils with 98% viability, as assessed by trypan blue exclusion. Human monocytes were prepared by retrieving cells at the 70% and 55% interface. These were then washed with PBS and resuspended to 5 × 106cells/ml. One milliliter was added to each well of a 24-well plate (Falcon) and incubated for 1 hour in serum-free Iscove’s Dulbecco’s minimum essential medium (DMEM). Cells were washed with ice-cold saline to remove nonadherent cells and cultured for 4 days in Iscove’s DMEM with added 1% penicillin and streptomycin and 10% autologous serum to mature into mφ (>90% pure).
PMN Incubation
Isolated PMNs were incubated in Iscove’s DMEM (Sigma-Aldrich Company, Dorset, UK) with added antibiotics and supplemented with 10% autologous platelet-poor plasma-derived serum in a humidified atmosphere at 37°C with 4% added CO2. PMNs were cultured in Teflon wells at a concentration of 2 × 10 6 cells/ml. Polymyxin (Sigma) (10 μg/ml) was added to each well, except those with added lipopolysaccharide (LPS), to counteract possible contamination of reagents with endotoxin. Limulus assay showed endotoxin contamination of IgG (<0.06 EU/ml); other reagents were from endotoxin-free accredited sources. Cells were cultured for 36 hours and sampled at various time points. Quiescent PMNs or PMNs primed with TNF-α (2 ng/ml) (NISBC, Potters Bar, UK) were incubated with N IgG or ANCA IgG (either PR3-ANCA or MPO-ANCA) at a concentration of 200 μg/ml. Higher concentrations of IgG produced PMN clumping (data not shown). In some experiments, catalase (5000 U/ml) was added before priming of PMN and the addition of IgG.
Isolation and Culture of Murine mφ
Peritoneal mφ were elicited into the peritoneal cavity of 12–16-week-old BALB/c mice with 3% thioglycollate medium (Difco, Detroit, MI). Peritoneal exudate mφ were harvested 4 days later and cultured in 96-well plates overnight in Dulbecco’s DMEM with 10% fetal calf serum before use in the phagocytosis assay as previously described. 20
IgG Isolation
Serum was collected from patients with vasculitis who fulfilled the Chapel Hill Consensus Conference definitions for Wegener’s granulomatosis or microscopic polyangiitis 21 and patients with antiglomerular basement membrane disease. IgG was prepared from patient and normal human donor sera, using a Hi-trap protein G affinity column (Pharmacia) as previously described. 19 All IgG samples were used individually. ANCA specificity was confirmed by Western blot analysis and enzyme-linked immunosorbent assay (data not shown). 4 MPO-ANCA and 4-PR3 ANCA were isolated, and all ANCAs had a serum titer greater than 1:400 by immunofluorescence.
Assessment of Apoptosis
Fluorescence Microscopy
Cells were sampled at the appropriate times, and the fluorescence staining protocol was used as described previously, with modifications. 22 Briefly, the collected cells were washed in PBS and resuspended at a density of 1 × 10 6 cells/0.5 ml. To this, 20 μl of a 1:1 mixture of ethidium bromide (100 μg/ml; Sigma) and acridine orange (100 μg/ml; Sigma) was added. The cell suspension was incubated in the dark at room temperature for 5 minutes. The stained cells were then centrifuged at 1200 rpm for 5 minutes and resuspended in 20 μl of 10% glycerol. The cell suspension was mounted on a slide and viewed with a fluorescence microscope. At least 500 cells per slide were counted. Normal cell morphology was defined as normal cells appearing large with multilobed yellow-stained nuclei. Apoptotic morphology was observed as yellow fluorescence staining of shrunken nuclei. Cellular ghosts that stain dark green represent late apoptotic PMNs (Figure 1) ▶ .
Figure 1.
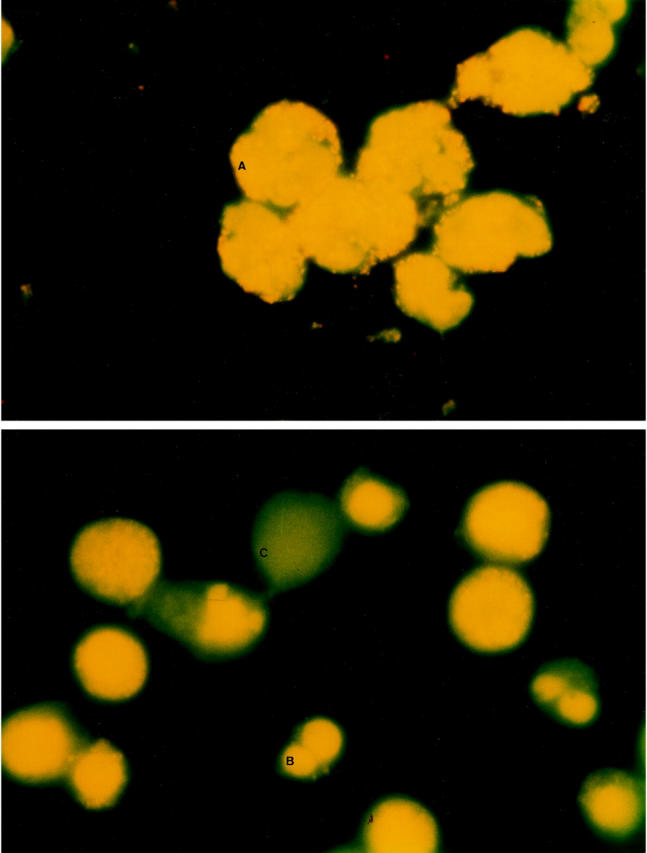
Fluorescence microscopy of aging PMNs, which were stained with a mixture of ethidium bromide and acridine orange. Original magnification, ×100. Top: Freshly isolated cells (A) appear large with multilobed nuclei, staining orange. Bottom: Early apoptotic morphology is observed as yellow staining of shrunken nuclei (B), whereas late apoptotic morphology was seen as cellular ghosts staining dark green with an absence of nuclear material (C).
Flow Cytometric Analysis of Immunofluorescent Stained Cells
Flow cytometry of PMN binding fluorescein isothiocyanate (FITC)-conjugated annexin V (Sigma) was performed as previously described. 23 Isolated PMNs were incubated with saturating amounts of FITC-conjugated annexin V for 10 minutes on ice. Annexin-V-FITC conjugate (Sigma) was diluted by 1:3000 in Hanks’ balanced salt solution with calcium and magnesium. One hundred microliters of PMN mix was added to 400 μl of buffer solution. Trypan blue staining was used to identify dead cells. PMNs were analyzed by fluorescence-activated cell sorter (FACS) within 30 minutes of labeling.
After labeling, PMNs were enumerated in a flow cytometer (Becton Dickinson FACS 440) equipped with an argon laser (wavelength 488 nm). Appropriate settings of forward and side scatter were used to examine PMNs. Cell debris was excluded from analysis by appropriately raising the forward scatter threshold. More than 10,000 cells were examined at each experiment.
Gel Electrophoresis and DNA Fragmentation
DNA fragmentation in cultured cells by phenol:chloroform protein extraction was assessed by electrophoresis through agarose gels as previously described. 24
Assessment of PMN Activation
Production of Superoxide after Stimulation by ANCAs
Superoxide anion production was determined by the superoxide dismutase (SOD) (Sigma) inhibitable reduction of ferricytochrome c (Sigma), as previously described, with modifications. 25 Fifty microliters of PMN culture medium was added to a reaction mixture containing ferricytochrome c (75 μM) with or without SOD (150 U). The difference in reduction of ferricytochrome c in the presence and absence of SOD, as determined spectrophotometrically at 550 nM, was measured and calculated using a molar extinction coefficient of 21.1 × 10−3 M for ferricytochrome c and a light path of 0.6 cm for a final volume of 250 μl. Replicates were carried out at least in quadruplicate.
Myeloperoxidase Release
PMNs primed with TNF-α, as described above, were cultured with IgG for 90 minutes, and supernatants were isolated by centrifugation at 13,000 rpm for 5 minutes and assayed in triplicate for myeloperoxidase (MPO) production. Ninety-six-well flat-bottomed plates (Becton-Dickinson, Oxford, UK) were used. To each well 50 μl of supernatant was added. Myeloperoxidase (Calbiochem-Novabiochem (UK), Nottingham, UK) was used at concentrations of 79–2500 pg/ml, diluted in Iscove’s DMEM, to produce a standard curve. To this was added 200 μl of substrate solution containing 50 mmol/L K3PO4 at pH 6.0, 0.5% hexadecyl trimethyl ammonium bromide (Sigma), 1 mg/ml O-diansidine dihydrochloride (Sigma), and 0.01% H2O2. Supernatants were incubated in the dark for 2 minutes, and then the absorbance was measured at 492 nm with a Multiskan bichromatic analyzer.
Mφ Interaction Assay
A well-established, microscopically quantified phagocytic assay of mφ ingestion of apoptotic PMNs was used as previously described. 26-29 Briefly, PMNs aged for 12 hours were washed once in Hanks’ balanced salt solution, suspended in Iscove’s DMEM without serum (concentration 5 × 106cells/ml). Cells (5 × 106) were added to each well of human mφ in a 24-well plate, and or 2.5 × 10 5 cells (50 μl) were added to murine peritoneal mφ in 96-well plates. Plates were incubated at 37°C for 30 minutes in 4% CO2. The plates were washed with ice-cold saline to remove noningested PMNs. The plates containing the human mφ layer were fixed in 2% glutaraldehyde for 2 minutes and stained for MPO. Using 40× phase-contrast light microscopy, we counted the portion of human mφ ingesting PMNs. Experiments were performed in triplicate, and 100 mφ were counted per well. The phagocytic index was calculated as the percentage of phagocytosing mφ multiplied by the average number of PMNs ingested per mφ. The mφ themselves were routinely negative for peroxidase staining. For murine peritoneal mφ, after a 30-minute interaction, murine mφ were then trypsinized, and a separate cytocentrifuge preparation was prepared for each well as previously described. 29,30 These were fixed with 2% glutaraldehyde and stained for MPO and finally counterstained with Hemalum (BDH, Poole, UK). The proportion of murine mφ containing brown-staining MPO-positive PMNs was then counted.
Statistical Analysis
Data are presented as the mean ± SEM. Statistical significance was determined using ANOVA for multiple comparisons; the Bonferroni test for pairs was used to compare groups.
Results
ANCAs Do Not Affect Constitutive Apoptosis in Unprimed PMNs
PMNs that were not deliberately primed in culture showed a time-dependent increase in the percentage of apoptotic cells, as assessed by morphology and binding of FITC-conjugated annexin V (results not shown). When these cells were cultured with ANCAs they showed no difference in the percentage of cells judged to be apoptotic by either technique at any time point (from 6 to 36 hours) compared with those PMNs incubated with N IgG (results not shown). LPS was used to confirm that PMN apoptosis could be delayed as previously shown. 31 PMNs cultured with added LPS showed significant inhibition of apoptosis at 18 hours (61.44% ± 11 normal morphology and at 24 hours with 40% ± 17.911 normal morphology) compared with cells cultured with N IgG (18 hours 28.64% ± 17.6 11 normal morphology and 24 hours 17.83% ± 15.7 11 normal morphology), P < 0.05; all data quoted refer to assay of apoptosis by morphology and are the results of four different neutrophil donors.
In TNF-α Primed PMNs, ANCAs Accelerate Apoptosis, as Assessed by Morphology and Internucleasomal Cleavage of DNA but Not by Externalization of Phosphatidylserine
Activation of PMNs by ANCAs typically requires TNF-α priming because this increases the surface expression of PR3 and MPO, 32,33 so that Fc receptors are cross-linked by antibody. 34 These observations prompted us to prime freshly isolated PMNs with TNF-α (2ng/ml) before the addition of ANCAs or N IgG. TNF-α accelerates PMN apoptosis at early time points up to 6 hours and delays apoptosis at later times. 31,35 In preliminary experiments, TNF-α addition to incubated PMN without added IgG resulted, as expected, 31,35 in an initial increase in the percentage of apoptotic PMN at 6 hours, which was followed by a decrease in apoptosis at later time points compared with unprimed PMNs, as assessed by morphology (t = 6 hours TNF-primed PMN 19.4 ± 2.7% apoptotic cells, unprimed PMN 11.2 ± 2.3% apoptotic cells, P < 0.05; t = 18 hours TNF-primed PMN 46.9 ± 2.68% apoptotic cells, unprimed PMN 57.98 ± 3.29% apoptotic cells, P < 0.05; N = 3). The correlation coefficient between annexin V binding and morphology was r 2 = 0.88 with P < 0.001.
The effects of ANCAs on apoptosis in primed PMNs was assessed by 1) morphology using immunofluorescence microscopy and dual staining of TNF-α-primed PMN with ethidium bromide and acridine orange, 2) externalization of PS as assessed by binding of FITC-conjugated annexin V, and 3) internucleosomal cleavage of DNA demonstrated by gel electrophoresis. Externalization of PS is considered to be an early feature of apoptosis induced by many different stimuli and, in PMN undergoing apoptosis, is tightly linked to nuclear changes. 36
TNF-α-primed PMNs were incubated for up to 36 hours and assayed at various time points. At 12 hours primed PMNs incubated with ANCAs showed accelerated morphological changes of early apoptosis compared with controls, with ANCA-treated cells showing an increased population with typical nuclear chromatin condensation. Furthermore, in keeping with an acceleration of the constitutive processes of PMN apoptosis, by 18 and 24 hours a greater proportion of ANCA-treated cells exhibited typical nuclear evanescence of late apoptosis compared with cells incubated with N IgG (Figure 2) ▶ . There were no significant differences between PR3-ANCA and MPO-ANCA. Indeed, as one would expect with accelerated apoptosis, there were significantly more cells staining positive for trypan blue at all time points when primed PMNs were incubated with ANCAs compared with those cells incubated with N IgG (Figure 3) ▶ .
Figure 2.

Fluorescence microscopy of primed PMNs that were cultured with TNF only, normal IgG, or ANCA (either anti-MPO or anti-PR3) (200 μg/ml) and stained with a mixture of ethidium bromide and acridine orange. There were accelerated changes of early apoptosis in those cells incubated with ANCA at 12 hours (A) and changes in late apoptosis at 18, 24, and 36 hours compared with normal IgG (*P < 0.05) (B). Data are expressed as a percentage of the total number of cells that showed apoptosis. Data are means ± SD of six experiments.
Figure 3.

Cell viability was assessed by trypan blue staining. TNF-primed PMNs alone or TNF-primed PMNs incubated with normal IgG or ANCA (either anti-MPO or anti-PR3) (*P < 0.05) were assessed for cell viability at various time points. Data are expressed as a percentage of the total number of cells. Data are means ± SD of six experiments.
Further experiments using a disease control antibody were performed to ensure that the accelerated apoptosis with TNF-α-primed PMNs was specific to ANCAs. There was no difference in the rate of apoptosis in those primed cells treated with N IgG or anti-glomerular basement membrane (GBM) antibody (t = 18 hours N IgG 40.7 ± 3.9%, anti-GBM 45.9 ± 4.4% apoptotic cells; t = 24 hours N IgG 55.22 ± 4.2%, anti-GBM 63.1 ± 4.4% apoptotic cells, P > 0.05).
However, TNF-α-primed PMNs incubated with ANCAs showed no significant difference in the percentage of apoptotic PMNs binding annexin V compared with N IgG, despite the morphological evidence of accelerated apoptosis (Figure 4) ▶ . To extend these studies, we investigated whether the treatment of primed PMNs with ANCAs had interfered with DNA fragmentation, which occurs late in apoptosis. Cells undergoing apoptosis usually exhibit DNA fragmentation in a characteristic internucleosomal “ladder” pattern. This represents endogenous endonuclease activation, the amount of DNA in each fragment being a multiple of 180–200 bp. Both ANCA- and N IgG-cultured PMNs showed the characteristic DNA laddering pattern, thereby corroborating the morphological findings despite the failure to accelerate externalization of PS. Thus externalization of PS can also be dissociated from DNA fragmentation (Figure 5) ▶ .
Figure 4.

Coculture of TNF-α primed PMN with TNF only, normal IgG, or ANCA (either anti-MPO or anti-PR3) (200 μg/ml). Primed PMNs incubated with ANCA showed no increase in phosphatidylserine externalization, as assessed by FACS analysis of binding of FITC-conjugated annexin V (*P < 0.05). Data are expressed as a percentage of the total number of cells that showed apoptosis. Data are means ± SD of six experiments.
Figure 5.
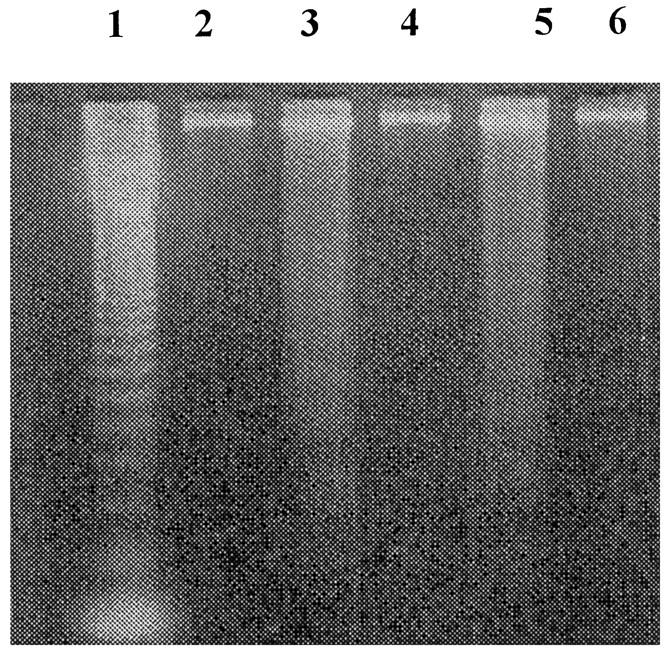
DNA fragmentation of PMNs cultured for 18 hours. Lane 1: markers; lane 2: unprimed neutrophils alone; lane 3: primed PMNs incubated with MPO-ANCA; lane 4: primed PMNs incubated with N IgG; lane 5: primed PMNs incubated with PR3-ANCA; lane 6: primed PMNs alone.
These data demonstrated that in PMNs deliberately primed with TNF-α there was dysregulation of the apoptotic process with uncoupling of plasma membrane changes from nuclear changes, with the result that there was accelerated development of the late apoptotic state and progression to secondary necrosis, but without acceleration of surface changes that lead to safe recognition by phagocytes.
Dysregulated Apoptosis Results in Uncoupling from Safe Phagocytosis
We proceeded to investigate the possibility that uncoupling of plasma membrane changes from nuclear changes would result in failure of mφ recognition by macrophage interaction assays. TNF-α primed PMN incubated with ANCA or N IgG were cultured for 12 hours. Primed PMN were then coincubated with human monocyte-derived mφ, and phagocytosis was assayed. By morphological assessment of PMN apoptosis, primed PMNs incubated with ANCAs showed significantly more apoptosis at 12 hours than those cells incubated with normal IgG (Figure 6A) ▶ . However, despite the increased proportion of PMNs exhibiting morphological evidence of apoptosis, phagocyte uptake for those cells incubated with ANCAs was significantly less than for those cells cultured with N IgG (Figure 6B) ▶ .
Figure 6.
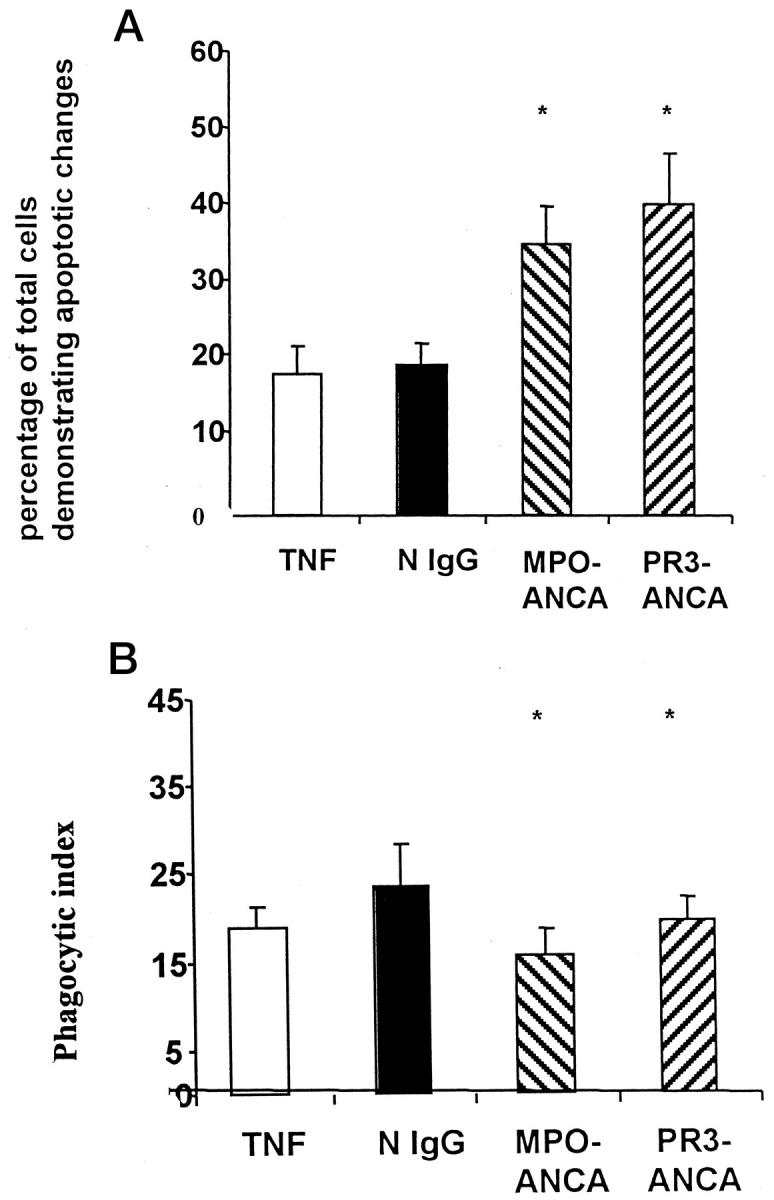
Primed neutrophils were aged in culture for 12 hours with N IgG or ANCA, and apoptosis was assessed by morphology and expressed as a percentage of the total number of cells that showed apoptosis (A). Macrophage interaction experiments were performed using human monocyte-derived macrophages and data expressed by the phagocytic index (B). Data are means ± SD of eight experiments. There were significantly more apoptotic cells when they were incubated with ANCA than when they were incubated with normal IgG (A). However, significantly fewer of those cells incubated with ANCA were phagocytosed compared with those cells incubated with normal IgG (B) (*P < 0.05).
Murine peritoneal-derived mφ phagocytose apoptotic cells via a recognition mechanism dependent on exposure of PS on the target cell. 36-38 To assess whether uncoupling of phagocytosis from morphological changes of apoptosis was directly related to failure of ANCA to accelerate the expression of PS, we used murine peritoneal derived mφ in interaction assays. Despite increased apoptosis as assessed by morphology (Figure 7A) ▶ , there was no significant difference in the phagocytosis of apoptotic PMNs by murine peritoneal-derived mφ in cells incubated with ANCAs compared with cells incubated with N IgG (Figure 7B) ▶ . This suggested that murine peritoneal-derived mφ failed to recognize a population of apoptotic cells because of delayed phosphatidylserine externalization. These studies add weight to our findings that ANCA-accelerated apoptosis is dysregulated with uncoupling of surface membrane changes and nuclear and cytoplasmic changes of apoptosis.
Figure 7.
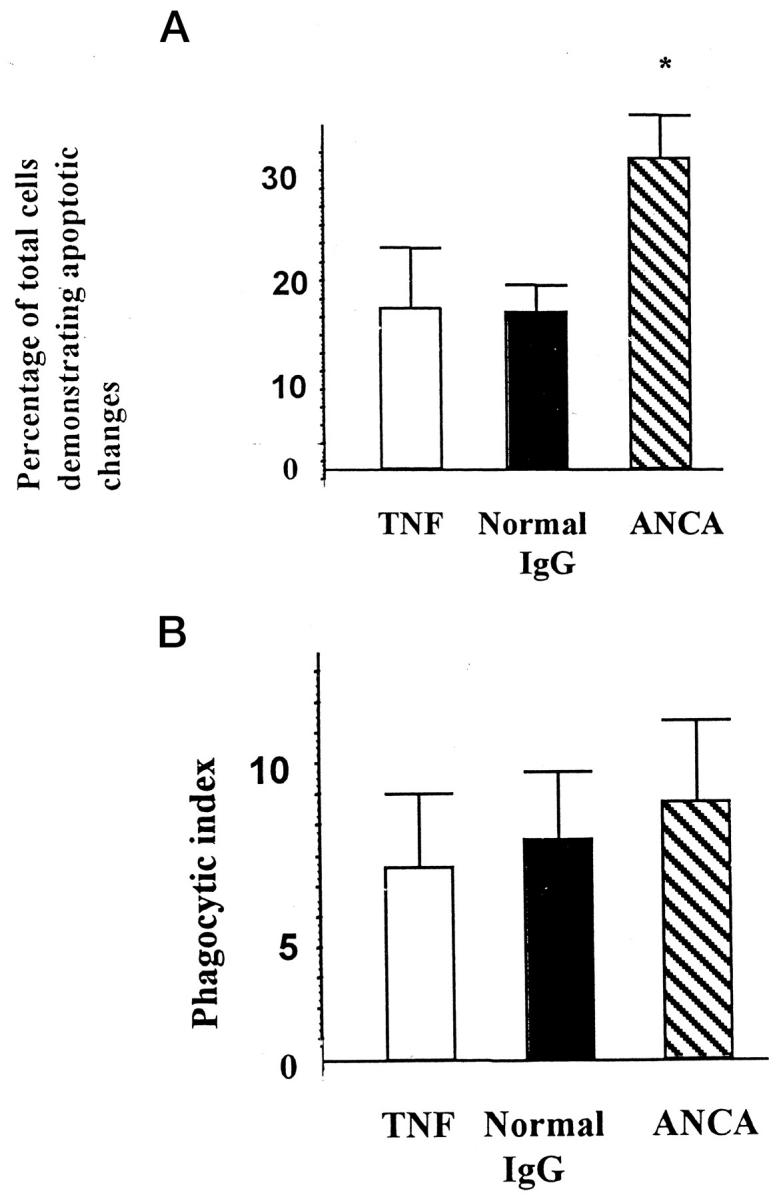
Primed neutrophils aged in culture for 12 hours with N IgG or ANCA. Apoptosis was assessed by morphology and expressed as a percentage of the total number of cells that were apoptotic (A). Macrophage interaction experiments were performed using murine peritoneal-derived macrophages, and data were expressed by the phagocytic index (B). Data are means ± SD of three experiments. There were significantly more apoptotic cells when they were incubated with ANCA than when they were incubated with normal IgG (A). However, there was no difference in the number of cells phagocytosed when they were incubated with ANCA or normal IgG (B) (*P < 0.05).
ANCA-Mediated Dysregulation of PMN Apoptosis Depends on Generation of a Respiratory Burst
Primed PMNs activated by ANCAs undergo a respiratory burst with the release of oxygen products, 17,18 degranulate, and secrete proinflammatory cytokines. Reactive oxygen species have been implicated as an important stimulus to apoptosis. 39-43 As far as we are aware, there have been no previous reports regarding dysregulated apoptosis induced by PMN activation and generation of reactive oxygen species. To investigate further the mechanism of ANCA-dysregulated apoptosis in PMNs, we examined the possible role of superoxide anion production.
We cultured TNF-α-primed PMNs from normal donors and from patients with chronic granulomatous disease (CGD). PMNs from patients with CGD are unable to generate a respiratory burst, although they do degranulate with the release of MPO. 44 TNF-α-primed PMNs cultured with ANCAs showed increased production of superoxide at 100 minutes (PR3-ANCA 11.16 ± 0.86 nmol/L/10 5 cells and MPO-ANCA 12.849 ± 0.81 nmol/L/10 5 cells) compared with those cells incubated with N IgG (8.04 ± 0.89 nmol/L/10 5 cells, P < 0.01). This was also true at 4 hours (PR3-ANCA 4.660 ± 0.97 nmol/L/10 5 cells, MPO-ANCA 3.74 ± 1.99 nmol/L/10 5 cells, and normal IgG 1.01 ± 0.70 nmol/L/10 5 cells, P < 0.05). By 8 hours the production of superoxide had decreased to negligible levels (Table 1) ▶ . TNF-α-primed PMNs isolated from patients with CGD showed no difference in PMN apoptosis between cells incubated with ANCA or N IgG (Figure 8) ▶ . As expected, PMNs isolated from patients with CGD did not undergo a respiratory burst after stimulation with ANCA, although there was activation with the release of MPO (normal PMNs treated with ANCA 426 ± 37.1 pg/ml, N IgG treated 117 ± 29.3 pg/ml, P < 0.01; CGD PMNs treated with ANCA 572 ± 48.5 pg/ml, N IgG 98 treated ± 39.8 pg/ml, P < 0.01).
Table 1.
Production of O2− (nmol/105 cells)
| t = 100 minutes | t = 4 hours | t = 8 hours | t = 12 hours | |
|---|---|---|---|---|
| Normal IgG± SEM | 8.04 | 1.01 | 0.000 | 0.000 |
| 0.89 | 0.70 | 1.202 | 0.181 | |
| PR3-ANCA± SEM | 11.16 | 4.66 | 0.061 | 0.816 |
| 0.86 | 0.697 | 0.840 | 0.197 | |
| MPO-ANCA± SEM | 12.85 | 3.74 | 0.000 | 0.606 |
| 0.81 | 1.441 | 0.505 | 0.849 | |
| Statistics | * | * |
MPO, myeloperoxidase.
*P < 0.05 when compared with those primed cells incubated with normal IgG.
The data are means of six experiments, all performed in at least quadruplicate ± SEM.
Figure 8.

PMNs isolated from patients with chronic granulomatous disease were primed with TNF and cultured alone or with normal IgG or ANCA (anti-MPO or anti-PR3). Apoptosis was assessed by morphology, viewed by fluorescent microscopy, and stained with ethidium bromide and acridine orange. Apoptosis was delayed as expected, and there was no difference between those cells that were primed with TNF and incubated with ANCA compared with TNF-primed PMNs incubated with normal IgG (*P < 0.05). Data are expressed as a percentage of the total number of cells that showed apoptosis. Data are means ± SD of three experiments.
Catalase, an antioxidant that catalyzes the reaction of hydrogen peroxide producing H2O and O2 was added to PMNs form healthy donors before TNF priming and the addition of ANCAs. There was inhibition of the accelerated apoptosis induced by ANCAs (t = 18 hours PR3-ANCA without catalase 85 ± 4% apoptotic cells; PR3-ANCA with catalase 46 ± 3.5% apoptotic cells, P < 0.01; MPO-ANCA without catalase 87 ± 5% apoptotic cells, MPO-ANCA with catalase 56 ± 6% apoptotic cells, P < 0.05). These experiments indicated that in vitro, dysregulation in PMN apoptosis induced by ANCA was critically related to the production of reactive oxygen species.
Discussion
Apoptosis is a physiological form of programmed cell death that is responsible for the deletion of unwanted cells and is essential for the resolution of inflammation. This is the first study to identify an effect of ANCA on TNF-α-primed PMN apoptosis whereby ANCAs accelerate constitutive PMN death by a mechanism dependent on NADPH oxidase and generation of reactive oxygen species. The normal processes of constitutive PMN death in which nuclear changes, cell surface PS exposure, and recognition by macrophages are all closely linked are dysregulated by ANCA. ANCA accelerates the apoptosis of TNF-α-primed PMNs with uncoupling of nuclear and cytoplasmic changes of apoptosis from the plasma membrane changes, when compared with N IgG. This is manifested by the acceleration of sequential morphological stages of apoptosis 12 and DNA fragmentation, without associated acceleration of the externalization of PS.
A key finding of this study was that autoantibodies to PMN components, which are believed to play important roles in the pathogenesis of autoimmune vasculitis, dysregulate PMN apoptosis in a manner that is likely to promote tissue injury at inflamed sites. First, exposure of primed PMNs to ANCA accelerates the progression of constitutive PMN death through apoptosis to secondary necrosis, in which uncontrolled release of injurious PMN contents is inevitable. Because monocytes emigrating into tissues must mature into mφ before they become capable of clearing apoptotic PMN, acceleration of PMN death threatens the synchronicity of monocyte maturation and PMN apoptosis, with undesirable secondary necrosis of PMNs. Second, this threat of PMN disintegration is compounded by ANCA-mediated dysregulation of apoptosis, which results in the acceleration of PMN death without accelerated surface membrane signals such as PS that allow recognition by mφ. Thus there is a shortened time interval during which intact ANCA-activated PMNs are available for phagocyte clearance. Extrapolation of these data to the PMN fate in ANCA-associated vasculitis requires considerable caution. Nevertheless, we submit that ANCA-mediated dysregulation of PMN apoptosis and clearance by phagocytes is a hitherto unrecognized mechanism for leukocytoclasis in vasculitic lesions, the highly characteristic histological picture of PMN disintegration seen in ANCA-associated disorders. 11
There are reported precedents for the uncoupling of nuclear and membrane changes of apoptosis that we have observed in our studies. In a system using the mitochondrial inhibitors antimycin A and oligomycin, Zhuang et al reported dissociation of cell membrane changes from other features of apoptosis in PMNs associated with failure of phagocyte recognition. 45 We have now found that a naturally occurring autoantibody may have functional effects similar to those of these mitochondrial inhibitors.
Our data clearly demonstrate that ANCAs dysregulate the constitutive programmed cell death of TNF-α-primed PMNs in a manner that is dependent on the generation of reactive oxygen species. When we used cells isolated from patients with CGD, there was no acceleration in apoptosis when TNF-α-primed PMNs were incubated with ANCA, despite activation shown by the production of MPO. PMNs from these patients are unable to produce free oxygen radicals because of a defect in one of the components of the NADPH oxidase. 46,47 As a result there is a significant delay in the rate at which these PMNs undergo apoptosis, which can be reversed by the addition of hydrogen peroxide. 41 Other studies have also shown that the production of reactive oxygen species with the activation of PMNs accelerates PMN apoptosis 39-41,43,48,49 . This is exemplified by the accelerated apoptosis seen after Fcγ receptor-mediated phagocytosis of opsonized particles, which is dependent on the generation of intracellular reactive oxygen species. 39,49 Previous studies within our laboratory have suggested that ANCA activation of primed neutrophils generating a respiratory burst is dependent on Fcγ receptor ligation. 34,50 In our studies the addition of catalase to culture media inhibited the accelerated apoptosis of TNF-primed PMNs by ANCA. This provided further evidence of the importance of reactive oxygen intermediaries in the dysregulation of apoptosis of TNF-primed PMNs by ANCA.
The accelerated dysregulated apoptosis induced by ANCA was dependent on priming of PMNs by TNF-α; PMNs incubated with ANCA that were not deliberately primed showed no accelerated apoptosis at any time points up to 36 hours. As PMNs become apoptotic there is an increased expression of PR3 and MPO on the PMN surface; binding of these antigens by ANCA has been suggested as a possible mechanism for PMN activation without the requirement for TNF priming. 51 However, we found this to be unlikely, as stimulating PMNs with ANCA showed a time-dependent decrease in superoxide generation as PMNs aged (manuscript submitted), as previously described with fMLP. 2
In conclusion, we have shown for the first time that TNF-α-primed PMNs incubated with ANCA result in PMN activation, with the generation of reactive oxygen species and acceleration of apoptosis and secondary necrosis. Further studies are required to identify the aberrant pathways dissociating surface changes from other features of apoptosis induced by ANCA. This could reveal important targets for therapeutic manipulation.
Footnotes
Address reprint requests to Prof. Caroline O. S. Savage, Division of Medical Sciences, MRC Centre for Immune Regulation, The Medical School, University of Birmingham, Edgbaston, Birmingham B15 2TT, England. E-mail: c.o.s.savage@bham.ac.uk.
References
- 1.Wyllie A: Glucocorticoid-induced thymocyte apoptosis associated with endogenous endonuclease activation. Nature 1980, 284:555-556 [DOI] [PubMed] [Google Scholar]
- 2.Whyte M, Meagher L, Macdermott J, Haslett C: Impairment of function in ageing neutrophils is associated with apoptosis. J Immunol 1993, 150:5124-5134 [PubMed] [Google Scholar]
- 3.Fadok V, Bratton D, Konowai A, Freed P, Westcott J, Henson P: Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2 and PAF. J Clin Invest 1998, 101:890-898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savill J, Hogg N, Ren Y, Haslett C: Thrombospondin co-operates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J Clin Invest 1992, 90:1513-1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cox G, Crossley J, Xing Z: Macrophage engulfment of apoptotic neutrophils contributes to the resolution of acute pulmonary inflammation in vivo. Am J Respir Cell Mol Biol 1995, 12:232–237 [DOI] [PubMed]
- 6.Lan H, Mitasuhashi H, Ng Y, Nikolic-Pakerson DJ, Yang N, Mu W, Atkins RC: Macrophage apoptosis in rat crescentic glomerulonephritis. Am J Pathol 1997, 151:531-538 [PMC free article] [PubMed] [Google Scholar]
- 7.Meagher L, Savill J, Baker A, Haslett C: Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J Leukoc Biol 1992, 52:260-273 [PubMed] [Google Scholar]
- 8.Voll R, Herrmann M, Roth E, Stach C, Kalden J: Immunosuppressive effects of apoptotic cells. Nature 1997, 390:350-351 [DOI] [PubMed] [Google Scholar]
- 9.Noble P, Henson P, Lucas C, Mora-Worms C, Carre P, Riches D: Transforming growth factor β primes macrophages to express inflammatory gene products in response to particulate by an autocrine/paracrine mechanism. J Immunol 1993, 155:5819-5825 [PubMed] [Google Scholar]
- 10.Savill J, Haslett C: Granulocyte clearance by apoptosis in the resolution of inflammation. Semin Cell Biol 1995, 6:385-393 [DOI] [PubMed] [Google Scholar]
- 11.Donald KJ, Edwards RL, McEvoy JDS: An ultrastructural study of the pathogenesis of tissue injury in limited Wegener’s granulomatosis. Pathology 1976, 8:161- 169 [DOI] [PubMed]
- 12.Hebert M, Takano T, Holthofer H, Brady H: Sequential morphological events during apoptosis of human neutrophils: modulation by lipooxygenase-derived eicosanoids. J Immunol 1996, 157:3105-3115 [PubMed] [Google Scholar]
- 13.Savage COS, Harper L, Adu D: Primary systemic vasculitis. Lancet 1997, 349:553-558 [DOI] [PubMed] [Google Scholar]
- 14.van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, van der Giessen M, van der Hem GK, The TH: Autoantibodies against neutrophils and monocytes: new tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet 1985, ii:425-429 [DOI] [PubMed] [Google Scholar]
- 15.Falk RJ, Jennette JC: Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotising and crescentic glomerulonephritis. N Engl J Med 1988, 318:1651-1657 [DOI] [PubMed] [Google Scholar]
- 16.Kallenberg CGM, Brouwer E, Weening JJ, Cohen Tervaert JW: Anti-neutrophil cytoplasmic antibodies: current diagnostic and pathophysiological potential. Kidney Int 1994, 46:1–15 [DOI] [PubMed]
- 17.Falk RJ, Terrell RS, Charles LA, Jennette JC: Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA 1990, 87:4115-4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Charles L, Falk R, Jennette J: Reactivity of antineutrophil cytoplasmic antibodies with mononuclear phagocytes. J Leukoc Biol 1992, 51:65-68 [DOI] [PubMed] [Google Scholar]
- 19.Brooks CJ, King WJ, Radford DJ, Adu D, McGrath M, Savage COS: IL-1β production by human polymorphonuclear leukocytes stimulated by anti-neutrophil cytoplasmic autoantibodies: relevance to systemic vasculitis. Clin Exp Immunol 1996, 106:273-279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fadok V, Savill J, Haslett C, Bratton DL, Doherty DE, Campbell PA, Henson PM: Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognise and remove apoptotic cells. J Immunol 1992, 149:4029-4035 [PubMed] [Google Scholar]
- 21.Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hager EC, Hoffman GS, Hunder GG, Kallenberg CGM, McCluskey RT, Sinico RA, Rees AJ, van Es LA, Waldherr R, Wick A: Nomenclature of systemic vasculitides: the proposal of an International Consensus Conference. Arthritis Rheum 1994, 37:187-192 [DOI] [PubMed] [Google Scholar]
- 22.McGahon A, Maartin S, Bissonnette R, Mahboubi A, Shi A, Mogil RJ, Nishioka WK, Green DR: The end of the (cell) line: methods for the study of apoptosis in vitro. Methods Cell Biol 1995, 46:153-185 [DOI] [PubMed] [Google Scholar]
- 23.Brown S, Bailey K, Savill J: Actin is cleaved during constitutive apoptosis. Biochem J 1997, 323:233-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith G, Williams G, Kingston R, Jenkinson E, Owen J: Antibodies to CD3/T-cell receptor complex induce death by apoptosis in immature T cells in thymic cultures. Nature 1989, 337:181-183 [DOI] [PubMed] [Google Scholar]
- 25.Mayo LA, Curnutte JT: Kinetic microplate assay for superoxide production by neutrophils and other phagocytic cells. Methods Enzymol 1990, 186:567-575 [DOI] [PubMed] [Google Scholar]
- 26.Savill J, Henson P, Haslett C: Phagocytosis of aged neutrophils by macrophages is mediated by a novel “charge-sensitive” recognition mechanism. J Clin Invest 1989, 84:1518-1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Savill J, Whyllie A, Henson J, Walport M, Henson P, Haslett C: Macrophage phagocytosis of ageing neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest 1989, 83:865–867 [DOI] [PMC free article] [PubMed]
- 28.Savill J, Dransfield I, Hogg N, Haslett C: Vitronectin receptor mediated phagocytosis of cells undergoing apoptosis. Nature 1990, 343:170-173 [DOI] [PubMed] [Google Scholar]
- 29.Ren Y, Savill J: Pro-inflammatory cytokines potentiate thrombospondin-mediated phagocytosis of cells undergoing apoptosis. J Exp Med 1995, 181:1857-1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes J, Liu Y, Van Damme J, Savill J: Human glomerular mesangial cell phagocytosis of apoptotic neutrophils: mediation by a novel CD36-independent vitronectin receptor/thrombospondin recognition mechanism that is uncoupled from chemokine secretion. J Immunol 1997, 158:4389-4397 [PubMed] [Google Scholar]
- 31.Collotta F, Re F, Polentrutti N, Sozzani S, Mantovani A: Modulation of granulocyte survival and programmed cell death by cytokines and bacterial cell products. Blood 1992, 80:2012-2020 [PubMed] [Google Scholar]
- 32.Halbwachs-Mecarelli L, Bessou G, Lesavre P, Lopez S, Witko-Sarsat V: Bimodal distribution of proteinase 3 (PR3) surface expression reflects a constitutive heterogeneity in the polymorphonuclear neutrophil pool. FEBS Lett 1995, 374:29-33 [DOI] [PubMed] [Google Scholar]
- 33.Csernok E, Ernst M, Schmitt W, Bainton DF, Gross WL: Activated neutrophils express PR3 on their plasma membrane in vitro and in vivo. Clin Exp Immunol 1994, 95:244-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Smith A, Dove S, Martin A, Wakelam M, Savage C: Autoantibodies from patients with systemic vasculitis activate primed neutrophils via FCγ receptor dependent pathways. Blood 2000 (in press) [DOI] [PubMed]
- 35.Murray J, Barbara J, Dunkley S, Lopez AF, van Ostade X, Condliffe AM, Dransfield I, Haslett C, Chilvers ER: Regulation of neutrophil apoptosis by tumour necrosis factor-α: requirement for TNFR55 and TNFR75 for induction of apoptosis in vitro. Blood 1997, 90:2772-2783 [PubMed] [Google Scholar]
- 36.Savill J, Fadok V, Henson P, Haslett C: Phagocyte recognition of cells undergoing apoptosis. Immunol Today 1993, 14:131-136 [DOI] [PubMed] [Google Scholar]
- 37.Fadok V, Voelker D, Campbell P, Cohen J, Bratton D, Henson P: Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 1992, 148:2207-2216 [PubMed] [Google Scholar]
- 38.Fadok V, Laszlo D, Noble P, Weinstein L, Riches D, Henson P: Particle digestibility is required for induction of phosphatidylserine recognition mechanism used by murine macrophages to phagocytose apoptotic cells. J Immunol 1992, 151:4274-4285 [PubMed] [Google Scholar]
- 39.Watson R, Redmond H, Huai Wang J, Condron C, Bouchier-Hayes D: Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol 1996, 156:3986–3992 [PubMed]
- 40.Oishi K, Machida K: Inhibition of neutrophil apoptosis by antioxidants in culture medium. Scand J Immunol 1997, 45:21-27 [DOI] [PubMed] [Google Scholar]
- 41.Kasahara Y, Iwai K, Yachie A, Ohta K, Kouou A, Seki H, Miyawaki T, Taniguchi N: Involvement of reactive oxygen intermediaries in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 1997, 89:1748-1753 [PubMed] [Google Scholar]
- 42.McGowan A, Ruiz-Ruiz M, Gorman A, Lopez-Rivas A, Cotter T: Reactive oxygen intermediate(s) (ROI): common mediators of poly(ADP-ribose)polymerase (PARP) cleavage and apoptosis. FEBS Lett 1996, 392:299-303 [DOI] [PubMed] [Google Scholar]
- 43.Jacobson M: Reactive oxygen species and programmed cell death. Trends Biochem Sci 1996, 21:83-87 [PubMed] [Google Scholar]
- 44.Edwards S, Nurcombe H, Hart C: Oxidative inactivation of myeloperoxidase released from human neutrophils. Biochem J 1987, 245:925-928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhuang J, Ren Y, Snowden R, Zhu H, Gogvadez V, Savill J, Cohen G: Dissociation of phagocyte recognition of cells undergoing apoptosis from other features of apoptotic program. J Biol Chem 1998, 273:15628-15632 [DOI] [PubMed] [Google Scholar]
- 46.Tauber A, Borregaard N, Simons E, Wright J: Chronic granulomatous disease: a syndrome of phagocyte oxidase deficiencies. Medicine 1983, 62:286-309 [PubMed] [Google Scholar]
- 47.Curnutte J: Chronic granulomatous disease: the solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol 1993, 67:S2-S15 [DOI] [PubMed] [Google Scholar]
- 48.Gamberale R, Giordano M, Trevani A, Andonegui G, Geffner J: Modulation of human neutrophil apoptosis by immune complexes. J Immunol 1998, 161:3666-3674 [PubMed] [Google Scholar]
- 49.Coxon A, Rieu P, Barkalow F, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN: A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 1996, 5:653-666 [DOI] [PubMed] [Google Scholar]
- 50.Radford D, Lord J, Savage C: The activation of the neutrophil respiratory burst by anti-neutrophil cytoplasm autoantibody (ANCA) from patients with systemic vasculitis requires tyrosine kinases and protein kinase C activation. Clin Exp Immunol 1999, 118:171-179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gilligan H, Bredy B, Brady H, Herberk MJ, Slayter HS, Xu Y, Rauch J, Shia MA, Koh JS, Levine JS: Antineutrophil cytoplasmic autoantibodies interact with primary granule constituents on the surface of apoptotic neutrophils in the absence of neutrophil priming. J Exp Med 1996, 184:2231-2241 [DOI] [PMC free article] [PubMed] [Google Scholar]