

Abstract
Little or no data exist concerning the inactivation of tumor suppressor genes in intraductal lesions surrounding invasive ductal pancreatic carcinomas. Using a novel improved primer extension and preamplification polymerase chain reaction, we analyzed microdissected paraffin-embedded specimens of pancreatic carcinoma (n = 29) and their corresponding pancreatic intraductal lesions (PIL, n = 331) for loss of heterozygosity (LOH) of p16INK4, DPC4, and p53 by microsatellite analysis and for p53 protein by immunohistochemistry. LOH at the p16INK4 locus (9p21) was found in nine of 22 informative tumors (41%), in 15 of 25 tumors (60%) at the DPC4 locus (18q21.1), and in 22 of 27 tumors (81%) at the p53 locus (17p13). Homozygous deletions of p16INK4 and DPC4 were found in eight of 22 (36%) and four of 25 tumors (16%), respectively. Furthermore, 24 of 29 tumors (83%) revealed considerable intratumoral genetic heterogeneity. In 165 of 277 PILs (60%) having suitable DNA for microsatellite analysis, alterations in at least one tumor suppressor gene were found. In individual PILs, up to three alterations were detected, and p53 LOH occurred even in morphologically normal-appearing ductal epithelium near the tumor. Although deletions of all three tumor suppressor genes were found in PILs without nuclear atypia, there was a tendency toward earlier LOH of p16INK4 compared to DPC4 and p53 in these lesions. LOH in tumors accompanied positive p53 immunohistochemistry in 81% but only in 38% in PILs.
Ductal pancreatic carcinoma shows a growing incidence in the Western world, representing the fifth leading cause of cancer death of either sex. 1 Because of its aggressive growth and early metastatic dissemination, only 20% of the patients can be treated by surgery with curative intent at the time of diagnosis. The overall 5-year survival rate of <5% 2 is dismal and emphasizes the need for early diagnosis and/or the identification of susceptible populations. In recent years, considerable insights into the genetic basis of this disease have been generated. In pancreatic carcinoma, tumor suppressor genes controlling critical steps of cell cycle regulation, genomic stability, and growth control, eg, p16INK4, p53, Deleted in Pancreatic Carcinoma 4 (DPC4), as well as oncogenes (eg, K-ras) are commonly altered. Inactivation of p16INK4 and activation of K-ras have been found in >95% of pancreatic carcinomas. 3,4 DPC4 and p53 are deleted in 53% and 76%, respectively, 5 and many more genetic alterations have been described. 6-8
Although much is known about the invasive tumors, little is known about the genetic alterations in progenitor lesions. K-ras mutations have been found in preneoplastic intraductal lesions (PILs) and appear to be early events in pancreatic carcinogenesis. 9-11 To date, analysis of the p53 gene in PILs has only been performed by immunohistochemistry and often without a precise morphological definition of preneoplastic lesions according to the World Health Organization International Histological Classification of Tumors. 12
The available data are controversial and suggest mutational inactivation of p53 at either an early 13 or late stage of pancreatic carcinogenesis. 14-16 In two recent studies that addressed more precisely the different histological forms of preneoplastic lesions in pancreatic carcinoma, p16INK4 has been found inactivated in both low-grade and high-grade ductal lesions. 17,18 Alterations of DPC4 in PILs have not yet been described.
For analysis of loss of heterozygosity (LOH), a homogeneous population of tumor cells or epithelial cells from PILs is required. This can only be achieved by precise microdissection, usually of ∼50 to 200 cells, which allows few specific (nested) polymerase chain reaction (PCR) amplifications. Because LOH studies need to be done with multiple markers, preamplification of DNA by whole genome amplification could be very helpful. For this purpose, we established a protocol which allows multiple DNA analyses of single cells or small cell groups by conducting whole genome amplification followed by locus-specific PCR of multiple specific sites. 19
Microsatellite analysis was performed with multiple markers mapping p16INK4, DPC4, and p53. With this technique, we obtained information about allelic deletion of up to three tumor suppressor genes within a single lesion. In addition, p53 immunohistochemistry was correlated with the molecular data.
Materials and Methods
Tissue Sampling and Morphology
Twenty-nine archival cases of pancreatic adenocarcinoma from the University Clinic of Regensburg were selected for the presence of PILs associated with invasive carcinoma. One case of chronic pancreatitis and one case of nesidioblastosis with PILs were included as controls of nonneoplastic disease. According to the World Health Organization Classification 12 and a recently published study, 17 PILs were classified on the basis of their growth pattern and on the degree of nuclear atypia and were divided into five groups (Figure 1 ▶ , Table 1 ▶ ). Normal appearing epithelia with cuboidal to low columnar cells with round-to-oval nuclei without signs of atypia were graded PIL grade 0. Flat focal lesions with uniformly tall columnar mucin-filled cells without signs of nuclear atypia were graded PIL grade 1. Focal lesions graded PIL grade 2 consisted of mucin-filled cells with basally located nuclei without signs of atypia and a papillary growth pattern, the papillary folds typically showing a fibrovascular stalk. If these lesions exhibited mild nuclear atypia like larger sized round or oval nuclei and a more pronounced chromatin structure with clumped or dense hyperchromatic chromatin pattern, the lesion was graded PIL grade 3. When the epithelial lining showed irregular budding and bridging and signs of severe nuclear atypia like prominent nucleoli, hyperchromasia, loss of polarity, irregular size and contours, the lesion was graded PIL grade 4. In all lesions described, signs of invasion into the surrounding tissue were absent. To avoid examination of infiltrating cancers extending into ducts thereby mimicking a PIL, the first and the last sections were stained with H 38 E to ensure proper alignment of serial sections. If in the last section signs of invasive carcinoma were discovered, the sections were excluded from the study. In addition, only tissue blocks were processed for which a distance of at least 10 mm from the invasive carcinoma was described in the pathology report. For each patient, at least one example of nonneoplastic tissue was microdissected as a normal control.
Figure 1.
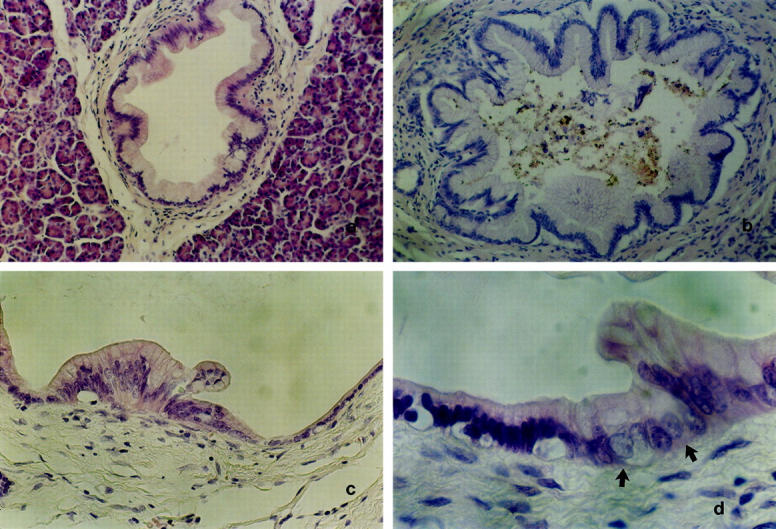
Representative examples of microdissected PILs. a: PIL 1. Mostly flat epithelial lesion composed of uniformly tall columnar mucin-filled cells with mostly basally located nuclei which do not show signs of atypia (H&E; original magnification, ×250). b: PIL 2. Epithelial lesion consisted of mucin-filled cells with basally located nuclei without signs of atypia and a papillary growth pattern, some papillary folds typically showing a fibrovascular stalk (H&E; original magnification, ×250). c: PIL 3. Epithelial lesion exhibiting mucinous epithelium with nuclear crowding and focal loss of nuclear polarity resembling moderate dysplasia falling short of PIL 4 (H&E; original magnification, ×400). d: PIL 4. Epithelial lesion (arrows) showing cytological criteria of severe nuclear abnormalities like prominent nucleoli, hyperchromasia, loss of polarity, irregular size, and contours (H&E; original magnification, ×650).
Table 1.
Histomorphological Classifications of Preneoplastic Lesions
| PanIN* | PIL† | Klöppel et al‡ | ||
|---|---|---|---|---|
| Grade | Description | Grade | Description | Description |
| Normal | Normal | 0 | Normal | — |
| 1A | Mucinous hypertrophy without nuclear atypia | 1 | Flat PIL without nuclear atypia | Tumor-like lesions/mucinous hypertrophy |
| 1B | (Pseudo)papillary PIL without nuclear atypia | 2 | Flat PIL with mild nuclear atypia | Tumor-like lesions/adenomatoid hyperplasia |
| 2 | Papillary PIL with mild nuclear atypia | 3 | Papillary PIL with mild nuclear atypia | Tumor-like lesions/papillary hyperplasia with mild dysplasia |
| 3 | Papillary PIL with severe nuclear atypia/CIS | 4 | Papillary PIL with severe nuclear atypia/CIS | Malignant/severe dysplasia/CIS |
*PanIN grading according to a recently proposed new nomenclature for classification of duct lesions in the pancreas. More information is available at the http://www.pathology.jhu.edu/pancreas_panin website.
†Classification of pancreatic intraductal lesions (PILs) of Moskaluk et al.17 Given is the gradual change (grades 0–4) of morphological appearance of PILs.
‡Classification scheme reported in Ref. 12 .
PanIN, pancreatic intra-epithelial neoplasia; CIS, carcinoma in situ.
Tissue Preparation
Five-micron serial sections of formalin-fixed, paraffin-embedded tissue were deparaffinized by incubating the slides in xylene for 2 × 15 minutes and rehydrating in 99.9% ethanol for 2 × 10 minutes, in 96% ethanol for 2 × 10 minutes, and in 70% ethanol for 2 × 10 minutes.
Microdissection
Methylene blue-stained sections were microdissected (Figure 2) ▶ using a joystick hydraulic micromanipulator (Leitz, Wetzlar, Germany). Between 50 and 200 cells were collected with sterile needles (microlance3R; Becton Dickinson, Franklin Lakes, NJ) and transferred into 10 μl of TL-buffer (1 × Taq PCR buffer, from Life Technologies, Eggenstein, Germany, including 4 mg/ml of Proteinase K and 0.5% Tween 20 from Merck, Darmstadt, Germany). In tumors, the microdissected samples were enriched for a neoplastic cellularity of at least 60% to avoid false-negative results in LOH analysis because of contamination by normal stromal cells present in tumors. Cell lysis was performed by incubation for 16 hours at 50°C and a 10-minute inactivation step at 94°C.
Figure 2.

Microdissection of ductal epithelium. a: Methylene blue staining (original magnification, ×400) of ductal epithelium that shows flat epithelial cells without nuclear atypia (open arrows) that changes into atypical epithelium (closed arrows) with irregular cell shape, high-grade nuclear atypia with hyperdense chromatin, and irregular shaped nuclei. Growth pattern is papillary toward the lumen of the duct without sign of invasion into the surrounding parenchym (PIL grade 4). b: Serial slide of a stained with methylene blue. The microdissected area (arrows) contains only the atypical cells described in a with the surrounding interstitial parenchym containing normal cells still being visible.
PCR Conditions
As a first step, whole genome amplification was performed by using an improved primer extension preamplification (I-PEP)-PCR as described recently 19 using a MJR PTC200 thermocycler (Biozym, Oldenburg, Germany). Briefly, I-PEP PCR was set up by adding 50 μl I-PEP mix (final concentration: 0.05 mg/ml gelatin, 16 μmol/L (N)15 random primer, 0.1 mmol/L dNTP, 3.6 U Taq Expand High Fidelity polymerase, 2.5 mmol/L MgCl2, in 1× PCR buffer No. 3 from Boehringer Mannheim, Mannheim, Germany) to 10 μl of lysed cells. PCR was run for 50 cycles. Step 1: 92°C for 90 seconds; step 2: 92°C for 40 seconds; step 3: 37°C for 2 minutes; step 4: ramp 0.1°C per 1 second to 55°C; step 5: 55°C for 4 minutes; step 6: 68°C for 30 seconds; step 7: go to step 2, 49 times; step 8: 68°C for 15 minutes; step 9: 4°C. The presence and relative quantity of PCR product was ascertained by resolution on a 2% agarose gel. Specific single-round PCR (0.2 mmol/L dNTP, 0.3 μmol/L primers, 0.5 U Taq Expand High Fidelity polymerase) was done using 3-μl aliquots of the preamplified DNA in a final volume of 20 μl in a MJ Research Thermocycler (PTC100, MJ Research, Watertown, MA) for 50 cycles: 94°C for 1 minute, 50 to 60°C for 1 minute, 72°C for 1 minute, followed by a final extension at 72°C for 8 minutes as described previously. 20,21 Primers used are given in Table 2 ▶ . Amplified microsatellites (3 μl) were analyzed by 6.7% polyacrylamide/50% urea gel electrophoresis (1 hour, 1500V, 50°C) in a SequiGen sequencing gel chamber (BioRad, Hercules, CA) and by silver nitrate staining as described previously. 22
Table 2.
Microsatellite Primer
| Gene | Microsatellite primer | Rate of PCR amplification* | % Informative* |
|---|---|---|---|
| p16INK4 | D9S1751 (PKY11) | 30/31 (97%) | 67 |
| D9S942 | 19/31 (61%) | 58 | |
| D9S1748 (PKY3) | 26/31 (84%) | 58 | |
| D9S171 (XC3) | 23/26 (88%) | 83 | |
| DPC4 | D18S46 (CU18007) | 30/31 (97%) | 67 |
| D18S63 | 30/31 (97%) | 70 | |
| p53 | TP53alk | 22/23 (97%) | 77 |
| D17Sp53 | 28/29 (97%) | 72 |
*Data were obtained by amplification of tumor DNA.
Criteria for LOH
LOH was diagnosed when a significantly lower ratio in the signal intensity (<50%) was observed in one of the two alleles in the PIL/tumor sample compared to the matched normal sample after amplification with an informative microsatellite marker. All results of LOH or homozygous deletion were confirmed by at least one repetition of PCR on the same DNA sample or on DNA obtained from serial sections.
Immunohistochemistry
Five-micron sections of formalin-fixed, paraffin-embedded tissue blocks were stained with p53-antibody (clone Bp53–12; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) according to the manufacturer’s instruction. Appropriate positive tissue controls were included for p53 staining in every experiment. p53 immunoreactivity was evaluated according to Baas et al 23 with tumors showing high labeling index (>30%), low labeling index (>1%, <30%), and no positivity (<1%).
Statistical Analysis
The Fisher’s exact test 24 was used to calculate P values using Stat Xact software (Cytel Corporation, Cambridge, MA). A P value of <0.05 was considered statistically significant.
Results
Microsatellite Markers
Table 2 ▶ gives the informative rate and the rate of amplification that could be achieved by the microsatellite markers. These markers have been found suitable for chromosomal mapping for detection of LOH and homozygous deletions of p16INK4 25 and DPC4. 26 In our hands, the microsatellite markers D9S1751 and D9S1748 (p16INK4) and CU18007 and D18S63 (DPC4) gave reliable results. For multiplex PCR, D9S1751 was found to be most suitable. p53 markers D17Sp53 and TP53alk showed a 97% rate of amplification and were informative in 77% and 72% of cases, respectively.
Microsatellite Analysis of Pancreatic Carcinoma
At least two and up to seven tumor loci were microdissected per case (Table 3) ▶ . LOH of p16INK4 was found in nine of 22 cases (41%), and eight of 22 tumors (36%) showed homozygous deletion in multiplex PCR (Figure 3) ▶ . LOH of DPC4 was present in 15 out of 25 cases (60%), and homozygous deletion was detected in four out of 25 cases (16%). p53 showed LOH in 22 out of 27 cases (81%).
Table 3.
Microsatellite Alterations in Tumors and p53 Immunohistochemistry
| Study No. | Staging (acc. to WHO 1978) | p16* | DPC4* | p53* | p53 protein† |
|---|---|---|---|---|---|
| PC 1 | pT1a,pN0,pMX,R0,G2 | HD(3/7) | LOH(5/5) | LOH(5/7) | (+)/(+++) |
| PC 2 | pT1a,pNX,pMX,R0,G3 | HD(2/3), LOH(1/3) | n(2/2) | n(2/2) | (−) |
| PC 3 | pT3,pN1,pMX,G3 | HD(2/3) | HD(2/3) | LOH(1/3) | n.d. |
| PC 4 | pT1b,pN0,pMX,R0,G3 | HD(4/4) | LOH(5/5) | LOH(2/5) | (−) |
| PC 5 | pT2,pN1,pMX,R1,G1 | LOH(2/4) | n(5/5) | n(5/5) | (+++) |
| PC 6 | pT1,pN0,pMX,R0,G1 | n.i. | LOH(2/4) | n(3/3) | n.d. |
| PC 7 | pT2,pN0,pMX,R0,G2 | LOH(3/3) | LOH(3/3) | LOH(2/2) | (++) |
| PC 8 | pT1b,pN1,pMX,R0,G2 | n.d. | HD(1/2), LOH(1/2) | LOH(2/2) | n.d. |
| PC 9 | pT2,pN1,pMX,R0,G2 | n.d. | LOH(2/4) | n(3/3) | n.d. |
| PC 10 | pT3,pN0,pMX,R1,G2 | n.i. | LOH(1/2) | LOH(1/2) | n.d. |
| PC 11 | pT2,pN1,pMX,R0,G3 | n(2/2) | LOH(3/5) | n.i. | (+++) |
| PC 12 | pT2,pN0,pMX,R0,G2 | n(2/2) | n(7/7) | LOH(4/7) | (−) |
| PC 13 | pT2,pN1,pMX,R1,G2 | LOH(1/2) | n(2/2) | LOH(1/1) | (+++) |
| PC 14 | pT2,pN1,pMX,G2 | LOH(1/1) | n.i. | LOH(3/4) | (−)/(+++) |
| PC 15 | pT2,pN1,pM0,R0,G3 | n.i. | n.i. | LOH(1/2) | (+++) |
| PC 16 | pT2,pN1,pMX,R0,G3 | LOH(1/2) | n.i. | LOH(3/3) | (++) |
| PC 17 | pT2,pN0,pMX,R0,G3 | HD(3/3) | LOH(1/3) | LOH(2/3) | (+)/(+++) |
| PC 18 | pT1,pN0,pMX,R0,G1 | HD(1/1) | n(2/2) | n(3/3) | (−) |
| PC 19 | pT2,pN1,pMX,R0,G2 | n.d. | HD(1/2) | LOH(1/2) | n.d. |
| PC 20 | pT2,pN1,pMX,R0,G3 | n(2/2) | n.i. | LOH(2/2) | (−)/(+++) |
| PC 21 | pT2,pN1,pMX,R0,G2 | n(3/3) | n(3/3) | LOH(1/3) | (++) |
| PC 22 | pT2,pN1,pMX,R0,G2 | HD(3/4), LOH(1/4) | HD(3/4), LOH(1/4) | LOH(4/4) | n.d. |
| PC 23 | pT2,pN1,pMX,R0,G2 | n(2/2) | LOH(2/3) | LOH(4/4) | (+++) |
| PC 24 | pT2,pN1,pMX,R0,G2 | HD(2/3), LOH(1/3) | LOH(2/3) | LOH(3/3) | n.d. |
| PC 25 | pT2,pN0,pMX,R0,G2 | n.d. | LOH(4/5) | LOH(3/5) | (+++) |
| PC 26 | pT2,pN1,pMX,R0,G3 | n(3/3) | LOH(2/4) | LOH(3/4) | (−)/(++) |
| PC 27 | pT1a,pN0,pMX,R0,G3 | n(2/2) | LOH(1/3) | LOH(1/3) | (−) |
| PC 28 | pT3,pN1,pMX,R1,G3 | n(2/2) | n(2/2) | n.i. | (+++) |
| PC 29 | pT2,pN1,pMX,R1,G2 | LOH(2/2) | n(2/2) | LOH(2/2) | (++) |
*For tumor suppressor genes, numbers in parentheses show the number of LOHs detected/number of tumor foci microdissected. n, normal; LOH, loss of heterozygosity; HD, homozygous deletion; n.d., not done; n.i., not informative.
†Immunohistochemical staining of p53 protein was classified from no positivity (−), low labeling (+/++), and high labeling (+++).
Figure 3.
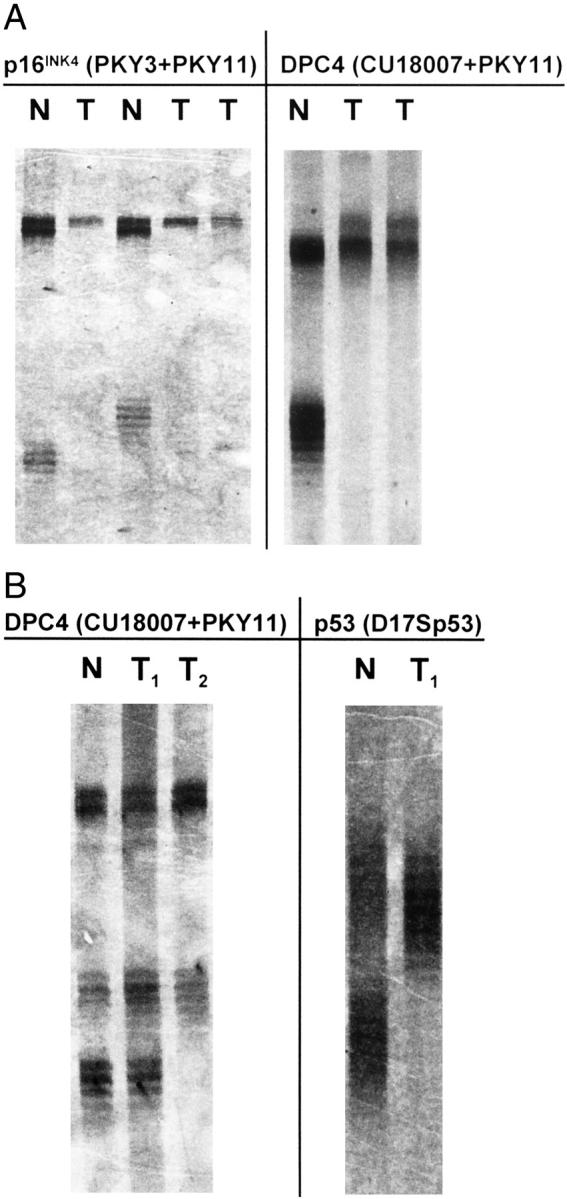
Examples for tumor heterogeneity and homozygous deletions of p16INK4 and DPC4 detected by multiplex PCR and visualized by silver nitrate staining. Microdissected cell clusters of up to 200 cells were preamplified by I-PEP and a 10th aliquot was used for microsatellite PCR. A: Detection of homozygous deletion of the p16INK4 and the DPC4 gene by multiplex PCR. PCR product of PKY11 (larger product, upper bands) that marks a region toward the telomeric end of the chromosome 9p and PCR product of PKY3 (smaller product, lower bands, lanes 1 and 3) that marks a region between the p16 and the p15 gene. The tumor samples (T) show loss of one allele at the PKY11 locus (lanes 2, 4, and 5) and a complete loss of the PKY3 locus in these lanes. At the DPC4 locus, microsatellite marker PKY11 (upper band) shows an additional band in the tumor samples (1 in 29 cases with microsatellite instability). At the DPC4 locus (microsatellite marker CU18007, smaller product, lower bands, lane 6) tumor samples (T) in lanes 7 and 8 show complete loss of the microsatellite marker. B: Detection of intratumoral genetic heterogeneity, multiplex PCR. At the DPC4 locus, upper bands (larger product of microsatellite marker PKY11) show the presence of both alleles in the tumor samples (T1, T2; lanes 2 and 3). The smaller PCR product of microsatellite marker CU18007 (lower bands) shows the presence of both alleles in tumor sample T1 (lane 2), T2 (lane 3) shows LOH. Lanes 4 and 5: LOH at the p53 locus (lane 5, D17Sp53) using the same template DNA (T1, lane 2). N, normal (lanes 1 and 4).
Microdissection of distinct tumor loci uncovered the presence of p16INK4 LOH in three out of eight cases (38%) with homozygous deletion of p16INK4. LOH of DPC4 was coincidentally found in two out of four cases (50%) with homozygous deletion of DPC4. In 13 out of 27 cases (48%) with LOH of p53, at least one microdissected sample showed the presence of both alleles. In these cases, contamination of normal cell-derived DNA masking the detection of LOH was excluded by demonstrating LOH at a different locus, eg, p53, DPC4, p16INK4 or Deleted in Colon Carcinoma (DCC), by using the same template DNA (Figure 3) ▶ .
Overall, the loss of all three tumor suppressor genes was found in 7 out of 29 cases (24%), and two of three tumor suppressor genes were lost in 11 out of 29 cases (38%). Only one of 29 carcinomas (4%) showed no alteration by microsatellite analysis at the three loci investigated but this case did show strong staining for the p53 protein in immunohistochemistry, which is in favor of the presence of a mutated p53 gene in this case. Furthermore, in 24 out of 29 tumors (83%) microdissection revealed the presence of genetic heterogeneity in at least one tumor suppressor gene indicating the presence of different tumor cell subclones. No correlation could be observed between marker loss and either malignancy grade or tumor stage.
Microsatellite Alterations in PIL
From a total of 331 microdissected PILs, the DNA of 277 PILs was suitable for microsatellite analysis (Figures 4 and 5) ▶ ▶ . In total, 163 of the 277 PILs (59%) showed LOH. Two PILs grade 4 (0.7%) exhibited a homozygous deletion at the p16INK4 locus. In two of 22 PILs (9%) having histologically normal epithelium (grade 0), LOH of p53 was detected in two different patients; neither stained positively for p53 protein.
Figure 4.

Examples of genetic alterations of the p53 and DPC4 gene in PILs (P1, P2, P3 = PIL grades 1, 2, 3). Loss of the upper alleles in PILs P2 (lane 4) and P2 (lane 5) at the DPC4 locus. Loss of different alleles of the p53 gene in PILs P3 (lane 5, lower allele; lane 6, upper allele). N, normal; T, tumor.
Figure 5.

Spectrum of deletions in PILs (n = 277). Microdissected cells were preamplified by I-PEP and one-tenth aliquot was used for microsatellite analysis. Given are the deletions of single tumor suppressor genes in percentage. Normal epithelium (PIL grade 0) shows LOH (9%) only at the p53 locus. The frequency of LOH at the p16INK4 locus is not statistically different (P = 0.38) in PILs without nuclear atypia (PIL grade 1 and 2) compared to PILs showing nuclear atypia (PIL grade 3 and 4). At the p53 and DPC4 locus, LOH is more frequent in PILs with nuclear atypia (PIL grade 3 and 4) compared to PILs without nuclear atypia (PIL grade 0 to 2; p53 P < 0.001; DPC4 P = 0.043).
The majority of the PILs investigated contained LOH of at least one single gene (Figure 6) ▶ . Deletions of two of the three tumor suppressor genes was first observed in histologically altered epithelium (grade 1), and triple lesions were first observed in PILs having moderate (grade 3) to severe (grade 4) nuclear atypia. Single lesions were found in 15 of 81 PILs (19%) without nuclear atypia compared to 75 of 196 PILs (38%) showing nuclear atypia. Multiple deletions (double or triple) were found in five of 81 PILs (6%) without nuclear atypia and in 17 of 196 PILs (9%) showing nuclear atypia (Figure 6) ▶ . Of the total 22 PILs with multiple lesions, 17 (77%) were found in PILs showing nuclear atypia. The trend of multiple lesions associated with a PIL with nuclear atypia was found to be not statistically significant (P = 0.538).
Figure 6.

Single- and multiple-genetic lesions in PILs. Compared are PILs without nuclear atypia (PIL grade 0 to 2) and PILs with nuclear atypia (PIL grade 3 to 4). The elevated frequency of multiple lesions in PILs grade 3 and 4 compared with PILs grade 0 to 2 is not statistically significant (P = 0.538).
When the genetic combinations of double lesions in PILs were analyzed, the most frequent combination of LOH was p53 with DPC4 (13 of 20 PILs, 65%) followed by p53 with p16INK4 (6 of 20 PILs, 30%) and p16INK4 with DPC4 (1 of 20 PILs, 5%). Furthermore, we analyzed the presence of mutations in single tumor suppressor genes in relationship to the histological grade (PILs without nuclear atypia versus PILs with low to severe nuclear atypia). In general, LOH of p16INK4, DPC4, or p53 was found in PILs even without nuclear atypia and the number of alterations of these tumor suppressor genes in one individual PIL grows with the grade of nuclear atypia. For p53 and DPC4, we found LOH more often in PILs showing nuclear atypia (P < 0.001 and P = 0.043, respectively) compared to PILs without nuclear atypia. For p16INK4, we found an early accumulation of mutations in PILs without nuclear atypia that was not statistically different from the accumulation of mutations in PILs showing nuclear atypia (P = 0.38) (Figure 5) ▶ .
In the two cases with chronic pancreatitis and nesidioblastosis, a total of 21 PILs were microdissected. Seven of these PILs were grade 3. LOH was not detected in a single lesion.
p53 Immunohistochemistry in Tumors and in PILs
Twenty-one of 29 carcinomas were available for p53 immunostaining. Sixteen of the 21 carcinomas (76%) showed moderate to strong expression of p53 protein, 13 (81%) of these had LOH at the p53 locus. In five carcinomas that stained negative for p53, LOH of p53 was detected in three cases (60%). Three of the 16 tumors (19%) with p53 protein stabilization showed no LOH of p53.
A total of 151 PILs were stained for p53 protein stabilization (Table 4) ▶ . 33 PILs (22%) revealed weak to strong p53 protein staining (Figure 7) ▶ . In comparison, 42 of 151 PILs (28%) showed LOH of p53. Sixteen (38%) of these PILs accompanied positive p53 immunohistochemistry. Of the PILs without nuclear atypia, only two (both grade 2) of 52 PILs (4%) showed a weak staining for p53 protein. All other PILs with p53 protein stabilization had low to severe signs of nuclear atypia. Five of 52 PILs (10%) without nuclear atypia and absence of p53 protein expression showed p53 LOH. Twenty-one of the 99 PILs (21%) showing nuclear atypia and negative staining for p53 protein revealed LOH of p53. On the other hand, 15 of 99 PILs (15%) having nuclear atypia showed p53 protein expression in the absence of LOH. Two of 35 PILs grade 3 (6%) showed both positive staining for p53 and LOH, whereas 14 of 64 PILs grade 4 (22%) exhibited positive staining for p53 in addition to LOH. Of the 33 PILs that stained positive for p53, 16 (48%) revealed LOH at the p53 locus.
Table 4.
Immunohistochemical Staining of p53 Protein in PILs*
| PIL grade | Total | p53 IH(+), LOH | p53 IH(+), n | p53 IH(−), LOH | p53 IH(−), n |
|---|---|---|---|---|---|
| 0 | 13 | 2 (15%) | 11 (85%) | ||
| 1 | 16 | 2 (13%) | 14 (87%) | ||
| 2 | 23 | 2 (9%) | 1 (4%) | 20 (87%) | |
| 3 | 35 | 2 (6%) | 3 (9%) | 10 (28%) | 20 (57%) |
| 4 | 64 | 14 (22%) | 12 (19%) | 11 (17%) | 27 (42%) |
*Compared are results of immunohistochemical staining of PILs (p53IH) with p53 microsatellite analysis of PILs from a corresponding serial tissue section. (+), positive staining of p53 protein ranging from low to high labeling index; (−), negative staining result (<1%) of p53 immunohistochemistry; LOH, loss of heterozygosity; n, normal allele status without detectable LOH.
Figure 7.

Example of histological growth pattern and immunohistochemical staining for p53 protein in PILs. Ductal epithelium showing a beginning papillary growth pattern (right side of image) with no to mild nuclear atypia that changes into irregular growth pattern (middle) with severe nuclear atypia and irregular cell shape (PIL grade 4). Notice the sudden appearance of nuclear staining for p53 protein (arrow) that continues to the left side of the image, indicating a clonal outgrowth of ductal epithelium that harbors a mutation of the p53 gene. There are no visible signs of invasion into the underlying parenchyme. Original magnification, ×400.
Of the two cases with chronic pancreatitis and nesidioblastosis, none showed p53 protein stabilization.
Discussion
The stepwise accumulation of genetic mutations involving proto-oncogenes, tumor suppressor genes, and DNA repair genes during carcinogenesis is now widely accepted for many sporadic neoplasms. 27-29 The development and improvement of molecular genetic analysis has given insight into the genetic alterations in precancerous lesions that are found in the vicinity of many solid tumors. These lesions show well-known histological features of cellular and nuclear atypia of various degrees, but lack signs of invasion into the surrounding tissue. In pancreatic carcinoma, previous studies reported mutations of oncogenes (eg, K-ras) and tumor suppressor genes (p53, p16INK4, DPC4), 5,30 however, the precise molecular mechanisms that give rise to pancreatic carcinoma are still poorly understood. 17,18
We demonstrated multiple genetic alterations within a single tumor in one to three tumor suppressor genes, revealing widespread genetic heterogeneity in pancreatic carcinoma. Considerable intratumoral genetic heterogeneity in primary pancreatic cancers has been described before by using cytogenetic techniques. 6,31 In these studies, up to 76% of tumors were found to harbor up to 39 related and 54 unrelated clones. However, intratumoral genetic heterogeneity was not found in a study where xenografted pancreatic carcinomas were microdissected. 32 The reason for this discrepancy remains obscure, but xenografting of primary tumors may introduce some bias in the results obtained. For example, a selection for subclones within a cancer cell population may be the reason for a variation of mutational profile or expression patterns of various genes in primary tumors versus xenografts. 33-37
In our study, the presence of LOH in one tumor focus and the simultaneous presence of both alleles in a different tumor focus was shown not to be because of sample contamination by normal cells. A point of concern during amplification of single or few cells is the occurrence of allele drop out, especially if tissue sections are microdissected because chromosomes or parts of chromosomes could get lost during specimen cutting. 38-40 The I-PEP PCR technique used in our study has been extensively evaluated to demonstrate the reliability, reproducibility, and limitations of this method for mutation analysis of single or several cells obtained in routine tumor pathology. 19 In this study we have demonstrated accurate biallelic amplification of various microsatellite markers by I-PEP even in 30 cells microdissected from formalin-fixed, paraffin-embedded normal tissues. In light of the data presented we believe that our LOH data reflect true mutational events, as we always amplified 50 or more cells thereby minimizing chances for differential allelic amplification or loss.
Our detected frequency of deletions at the p16INK4 and DPC4 locus was lower than those reported by others previously using different techniques such as xenografting of tumors with subsequent tumor microdissection 5,30,32 and higher for the p16INK4 locus than the frequency of deletions obtained by conventional microdissecting of primary tumors. 41,42 A major advantage of our microdissection technique is that the frequency of deletions may resemble more closely the in vivo situation as compared to tumor xenografts, where (eg, p53) mutations are over-represented (up to 100%). 33 Single foci of LOH or simultaneous LOH and homozygous deletion within one tumor area would have been overlooked in conventional studies using less precise microdissection techniques or only one tumor focus for mutation analysis, as it has been shown previously for prostate cancer. 43 Consequently, differences in the methodological approaches used may account, in part, for the findings obtained.
This is the first study to systematically investigate LOH in three tumor suppressor genes in a large number of preneoplastic lesions in pancreatic carcinoma. For tumor suppressor genes, the only data that are available so far have reported p16INK4 mutations together with K-ras mutations in a small number of cases. 17 An immunohistochemical analysis of p16 protein expression in a wide variety of preneoplastic lesions described loss of p16 protein expression in both low-grade and high-grade duct lesions. 18 For p53, only immunohistochemical analyses have been performed so far. 13,16,44,45 For DPC4, no data have been reported yet.
Molecular analysis of precursor lesions are of significant interest, because of the need 1) to understand of the genetic alterations that occur during the multistep carcinogenic process; 2) to develop a pancreatic cancer-specific expression profile; and 3) to create a screening method that could be developed to detect preneoplastic lesions as early as possible to identify patients with a high risk of developing cancer.
The analysis of K-ras mutations as a screening method for patients at risk of developing pancreatic cancer has been questioned, because K-ras mutations are often detected in PILs from healthy individuals not at risk for developing cancer. 46-48 Clear evidence has accumulated that PILs are true neoplastic precursor lesions in pancreatic carcinoma. 17,18,49 Our molecular data strongly support these findings, demonstrating genetic alterations of p16INK4, DPC4, and p53 genes occurring early in carcinogenesis of pancreatic carcinoma. Our findings show that more than one genetic alteration of the carcinoma can be found in adjacent PILs and that a rough correlation between the grade of histological atypia and the number of accumulated mutations exists.
Most interestingly, we found LOH of p53 in two independent PILs that had histological normal epithelium, whereas LOH of p16INK4 and DPC4 could not be detected within truly normal epithelium in our series. Very recently, Gansauge et al 50 found p53 mutations by sequence analysis in normal appearing epithelium in eight of 80 cases of chronic pancreatitis, highlighting the role of p53 mutations in pancreatic carcinogenesis.
Although deletion of all three tumor suppressor genes could be detected at the earliest stages of histological change, there was a statistically significant tendency for p53 and DPC4 mutations to accumulate in PILs with nuclear atypia, whereas no statistically significant difference could be found for p16INK4. This suggests an earlier accumulation of p16INK4 mutations in pancreatic tumorigenesis compared to DPC4 and p53 mutations. This observation is of importance in regard to molecular screening of patients being at risk of developing pancreatic adenocarcinoma. The significance of our observations in regard to the development of pancreatic carcinoma has yet to be established, and further studies of larger series of tissues including chronic pancreatitis may be informative.
Because we found in PILs only a low number of double and triple lesions compared to a high number of single lesions, the sensitivity of our methods for detecting double lesions is poor. But the detection of a PIL harboring multiple lesions seems to be associated with high-grade atypia, although this trend is not statistically significant. When double lesions were analyzed for combinations of tumor suppressor genes, we found the most frequent combination to be p53 LOH and DPC4 LOH, followed by the combination of p53 with p16INK4 and then by p16INK4 and DPC4. These findings, along with previous data, suggest an important role for mutations of p53 during carcinogenesis in pancreatic carcinoma, as is true in many other malignancies. 51 Rozenblum et al 5 reported a high concordance of DPC4 and p16INK4 inactivation in pancreatic cancer, suggesting that the inactivation of p16INK4 increases the selective advantage of subsequent mutation of DPC4. It is unclear why we did not find a concordance of p16INK4 and DPC4 mutations in PILs, although a clonal outgrowth of tumor cells having p16INK4 and DPC4 mutations might occur at later stages of tumorigenesis among lesions with p53 mutations. In addition, the number of PILs with inactivation of the p16INK4 gene is probably underestimated in this study, because methylation analysis was not performed.
A question that is not addressed in our study is the mutational status of the allele that is not affected by LOH and the mutational status of the epithelium that had no detectable LOH. As has been shown for preneoplastic lesions in Barrett’s esophagus, 52 LOH of one allele may not be necessarily accompanied by a mutation of the other allele to produce loss of functional protein. To determine more precisely the time point of complete mutational inactivation of tumor suppressor genes during tumorigenesis of the pancreas, sequence analysis and methylation analysis need to be performed in precursor lesions. In most tumors, p53 protein stabilization indicates loss of function of the protein on the basis of mutational inactivation of one allele in conjunction with the loss of the wild-type allele. Because we found only 22% of the PILs investigated had positive p53 immunohistochemistry compared to 28% of PILs with LOH of p53, a possible explanation might be the alteration of one allele and the presence of a functionally intact second allele, which may not lead to p53 protein stabilization. In contrast to our tumor samples, positive p53 immunohistochemistry in PILs was less often accompanied with LOH at the p53 locus. One explanation for this might be the observation that PILs with positive p53 immunohistochemistry often showed a mixed staining pattern (Figure 7) ▶ , indicating the presence of cells with at least one functional allele in close approximation with epithelial cells that have lost function of both alleles. Such a mixed cell population may have masked the detection of LOH in some cases.
Our data may have impact on the screening for true precancerous lesions in chronic pancreatitis, which is considered to be a common condition predisposing to pancreatic carcinoma. 53 The predictive value of K-ras mutations is still highly controversial, and immunohistochemical detection of p53 protein stabilization seems not to be useful because p53 protein stabilization in chronic pancreatitis appears to be because of the accumulation of wild-type p53 protein. 54
In conclusion, by using precise microdissection techniques and a novel PCR protocol, we show 1) that genetic heterogeneity is a common feature of carcinomas of the exocrine pancreas; 2) LOH of the p16INK4, DPC4, and p53 genes can be found in ductal lesions with low-grade dysplasia and even rarely in normal epithelium surrounding invasive carcinoma, suggesting that the latter represents a true preneoplastic lesion that can be screened for by simple microsatellite analysis; and 3) immunohistochemical expression of p53 protein accompanied LOH at the p53 locus in 81% of invasive carcinoma but only in up to 22% in PILs.
Acknowledgments
We thank William P. Bennett and Lawrence M. Weiss for helpful discussions and comments on the manuscript and Astrid Schwend for skillful technical assistance.
Footnotes
Address reprint requests to Ernst Heinmöller, M.D., c/o Josef Rüschoff, M.D., Institute of Pathology Klinikum Kassel, Mönchebergstrasse 41–43, 34125 Kassel, Germany. E-mail: ruesch@klinikum-kassel.de.
References
- 1.Parker SL, Tong T, Bolden S, Wingo PA: Cancer statistics. CA Cancer J Clin 1996, 46:5-27 [DOI] [PubMed] [Google Scholar]
- 2.Warshaw AL, Castillo CFD: Pancreatic carcinoma. N Engl J Med 1992, 326:455-465 [DOI] [PubMed] [Google Scholar]
- 3.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, Baylin SB, Kern SE, Herman JG: Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997, 57:3126-3130 [PubMed] [Google Scholar]
- 4.Redston MS, Caldas C, Seymour AB, Hruban RH, Da Costa L, Yeo CJ, Kern SE: p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994, 54:3025-3033 [PubMed] [Google Scholar]
- 5.Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE: Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 1997, 57:1731-1734 [PubMed] [Google Scholar]
- 6.Gorunova L, Höglund M, Andren-Sandberg A, Dawiskiba S, Jin Y, Mitelman F, Johansson B: Cytogenetic analysis of pancreatic carcinomas: intratumor heterogeneity and nonrandom pattern of chromosome aberrations. Genes Chromosom Cancer 1998, 23:81-99 [DOI] [PubMed] [Google Scholar]
- 7.Gress TM, Müller-Pillasch F, Geng M, Zimmerhackl F, Zehetner G, Friess H, Büchler M, Adler G, Lehrach H: A pancreatic cancer-specific expression profile. Oncogene 1997, 13:1819-1830 [PubMed] [Google Scholar]
- 8.Solinas-Toldo S, Wallrap C, Muller-Pillasch F, Bentz M, Gress T, Lichter P: Mapping of chromosomal imbalances in pancreatic carcinoma by comparative genomic hybridization. Cancer Res 1997, 56:3803-3807 [PubMed] [Google Scholar]
- 9.Lemoine NR, Jain S, Hughes CM, Staddon SL, Maillet B, Hall PA, Klöppel G: Ki-ras oncogene activation in preinvasive pancreatic cancer. Gastroenterology 1992, 102:230-236 [DOI] [PubMed] [Google Scholar]
- 10.Caldas C, Hahn SA, Hruban RH, Redston MS, Yeo CJ, Kern SE: Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res 1994, 54:3568-3573 [PubMed] [Google Scholar]
- 11.Sugio K, Molberg K, Albores-Saavedra J, Virmani A, Kishimoto Y, Gazdar AF: K-ras mutations and allelic loss at 5q and 18q in the development of human pancreatic cancers. Int J Pancreatol 1997, 1:205-217 [DOI] [PubMed] [Google Scholar]
- 12.Klöppel G, Solcia E, Longnecker DS, Capela C, Sobin LH: Histological typing of tumours of the exocrine pancreas. ed 2 WHO International Histological Classification of Tumours, 1996, Heidelberg, New York, Springer, Berlin
- 13.Boschman CR, Stryker S, Reddy JK, Rao MS: Expression of p53 protein in precursor lesions and adenocarcinoma of human pancreas. Am J Pathol 1994, 145:1291-1295 [PMC free article] [PubMed] [Google Scholar]
- 14.van den Berg FM, Polak M, Baas IO, Offerhaus GJA: Detection of p53 overexpression in routinely paraffin embedded tissue of human carcinomas using a novel target unmasking fluid. Am J Pathol 1993, 142:381-385 [PMC free article] [PubMed] [Google Scholar]
- 15.di Guiseppe JA, Hruban RH, Goodman SN, Polak M, van den Berg FM, Allison DC, Cameron JL, Offerhaus JA: Overexpression of p53 protein in adenocarcinoma of the pancreas. Am J Clin Pathol 1994, 101:684-688 [DOI] [PubMed] [Google Scholar]
- 16.Zhang SY, Ruggeri B, Agarwal P, Sorling AF, Obara T, Ura H, Namiki M, Klein-Szanto AJP: Immunohistochemical analysis of p53 expression in human pancreatic carcinomas. Arch Pathol Lab Med 1994, 118:150-154 [PubMed] [Google Scholar]
- 17.Moskaluk CA, Hruban RH, Kern SE: p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res 1997, 57:2140-2143 [PubMed] [Google Scholar]
- 18.Wilentz RE, Geradts J, Maynard R, Offerhaus GJA, Kang M, Goggins M, Yeo CJ, Kern SE, Hruban RH: Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res 1998, 58:4740-4744 [PubMed] [Google Scholar]
- 19.Dietmaier W, Hartmann A, Wallinger S, Heinmöller E, Kerner T, Endl E, Jauch KW, Hofstädter F, Rüschoff J: Multiple mutation analyses in single tumor cells enabled by improved whole genome amplification. Am J Pathol 1999, 154:83-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Rüschoff J: Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997, 57:4749-4756 [PubMed] [Google Scholar]
- 21.Rüschoff J, Dietmaier W, Lüttges J, Seitz G, Bocker T, Zirngibl H, Schlegel J, Schackert HK, Jauch KW, Hofstädter F: Poorly differentiated colonic adenocarcinoma, medullary type. Am J Pathol 1997, 150:1815-1825 [PMC free article] [PubMed] [Google Scholar]
- 22.Schlegel J, Bocker T, Zirngibl H, Hofstädter F, Rüschoff J: Detection of microsatellite instability in human colorectal carcinomas using a non-radioactive PCR-based screening method. Virchows Arch 1995, 426:223-227 [DOI] [PubMed] [Google Scholar]
- 23.Baas IO, Mulder JWR, Offerhaus GJA, Vogelstein B, Hamilton SR: An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994, 172:5-12 [DOI] [PubMed] [Google Scholar]
- 24.Mehta CR, Pantel NR: A network algorithm for performing Fisher’s exact test in r x c contingency tables. J Am Stat Assoc 1983, 78:427-434 [Google Scholar]
- 25.Cairns P, Polascik TJ, Eby Y, Tokino K, Califano J, Merlo A, Mao L, Herath J, Jenkins R, Westra W, Rutter JL, Buckler A, Gabrielson E, Tockman M, Cho KR, Hedrick L, Bova GS, Isaacs W, Koch W, Schwab D, Sidranski D: Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet 1995, 11:210-212 [DOI] [PubMed] [Google Scholar]
- 26.Hahn SA, Shamsul Hoque ATM, Moskaluk CA, Da Costa LT, Schutte M, Rozenblum E, Seymour AB, Weinstein CL, Yeo CJ, Hruban RH, Kern SE: Homozygous deletions map at 18q21.1 in pancreatic cancer. Cancer Res 1996, 56:490–494 [PubMed]
- 27.Fearon ER, Vogelstein B: A genetic model for colorectal carcinogenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 28.Mao L, Lee JS, Kurie JM, Fan YH, Lippman SM, Lee JJ, Ro JY, Broxson A, Yu R, Morice RC, Kemp BL, Khuri FR, Walsh GL, Hittelman WN, Hong WK: Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst 1997, 89:857-862 [DOI] [PubMed] [Google Scholar]
- 29.O’Connel P, Pekkel V, Fuqua SAW, Osborne CK, Clark GM, Allred DC: Analysis of loss of heterozygosity in 399 premalignant breast lesions at 15 genetic loci. J Natl Cancer Inst 1998, 90:697-703 [DOI] [PubMed] [Google Scholar]
- 30.Hahn SA, Schutte M, Shamsul Hoque ATM, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271:350–353 [DOI] [PubMed]
- 31.Gorunova L, Johansson B, Dawiskiba S, Andren-Sandberg A, Yin Y, Mandahl N, Heim S, Mitelman F: Massive cytogenetic heterogeneity in a pancreatic carcinoma: fifty-four karyotypically unrelated clones. Genes Chromosom Cancer 1995, 14:259-266 [DOI] [PubMed] [Google Scholar]
- 32.Hahn SA, Seymour AB, Shamsul Hoque ATM, Schutte M, da Costa LT, Redston MS, Caldas C, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res 1995, 55:4670–4675 [PubMed]
- 33.Reyes G, Villanueva A, Garcia C, Sancho FJ, Piulats J, Lluis F, Capella G: Orthotopic xenografts of human pancreatic carcinomas acquire genetic aberrations during dissemination in nude mice. Cancer Res 1996, 56:5713-5719 [PubMed] [Google Scholar]
- 34.Goike HM, Asplund AC, Pettersson EH, Liu L, Sanoudou D, Collins VP: Acquired rearrangement of an amplified epidermal growth factor receptor (EGFR) gene in a human glioblastoma xenograft. J Neuropathol Exp Neurol 1999, 58:697-701 [DOI] [PubMed] [Google Scholar]
- 35.Schmidt EE, Ichimura K, Goike HM, Moshref A, Liu L, Collins VP: Mutational profile of the PTEN gene in primary human astrocytic tumors and cultivated xenografts. J Neuropathol Exp Neurol 1999, 58:1170-1183 [DOI] [PubMed] [Google Scholar]
- 36.Bocsi J, Zalatnai A: Establishment and long-term xenografting of human pancreatic carcinomas in immunosuppressed mice: changes and stability in morphology, DNA ploidy and proliferation activity. J Cancer Res Clin Oncol 1999, 125:9-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zalatnai A: Epidermal growth factor receptor, somatostatin and bcl-2 in human pancreatic tumor xenografts. An immunohistochemical study. Pathol Oncol Res 1999, 5:146-151 [DOI] [PubMed] [Google Scholar]
- 38.Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N: Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci USA 1992, 89:5847-5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrett MT, Reid BJ, Geoffrey J: Genotypic analysis of multiple loci in somatic cells by whole genome amplification. Nucleic Acids Res 1995, 23:3488-3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duddy SK, Gorospe S, Bleavins MR: Genetic analysis of multiple loci in microsamples of fixed paraffin-embedded tissue. Toxicol Sci 1998, 46:317-323 [DOI] [PubMed] [Google Scholar]
- 41.Bartsch D, Shevlin DW, Tung WS, Kisker O, Wells SA, Goodfellow PJ: Frequent mutations of CDKN1 in primary pancreatic adenocarcinomas. Genes Chromosom Cancer 1995, 14:189-195 [DOI] [PubMed] [Google Scholar]
- 42.Huang L, Goodrow TL, Zhang SY, Klein-Szanto AJP, Chang H, Ruggeri BA: Deletion and mutation analyses of the p16/MTS-1 tumor suppressor gene in human ductal pancreatic cancer reveals a higher frequency of abnormalities in tumor-derived cell lines than in primary ductal adenocarcinomas. Cancer Res 1996, 56:1137-1141 [PubMed] [Google Scholar]
- 43.Macintosh CA, Stower M, Reid N, Maitland NJ: Precise microdissection of human prostate cancers reveals genotypic heterogeneity. Cancer Res 1998, 58:23-28 [PubMed] [Google Scholar]
- 44.Berrozpe G, Schaeffer J, Peinado MA, Real FX, Perucho M: Comparative analysis of mutations of the p53 and K-ras genes in pancreatic cancer. Int J Cancer 1994, 58:185-191 [DOI] [PubMed] [Google Scholar]
- 45.Pellegata NS, Sessa F, Renault B, Bonato M, Leone BE, Solcia E, Ranzani GN: K-ras and p53 gene mutations in pancreatic cancer: ductal and nonductal tumors progress through different genetic lesions. Cancer Res 1994, 54:1556-1560 [PubMed] [Google Scholar]
- 46.Yanagisawa A, Ohtrake K, Ohashi K, Hori M, Kitagawa T, Sugano H, Kato Y: Frequent Ki-ras oncogene activation in mucous cell hyperplasia of pancreas suffering from chronic inflammation. Cancer Res 1993, 53:953-956 [PubMed] [Google Scholar]
- 47.Longnecker DS: The quest for preneoplastic lesions in the pancreas. Arch Pathol Lab Med 1994, 118:226. [PubMed] [Google Scholar]
- 48.Tada M, Ohashi M, Shiratori Y, Okudaira T, Komatsu Y, Kawabe T, Yoshida H, Machinami R, Kishi K, Omata M: Analysis of K-ras gene mutation in hyperplastic duct cells of the pancreas without pancreatic disease. Gastroenterology 1996, 110:227-231 [DOI] [PubMed] [Google Scholar]
- 49.Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH: Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am J Surg Pathol 1998, 22:163-169 [DOI] [PubMed] [Google Scholar]
- 50.Gansauge S, Schmid RM, Muller J, Adler G, Mattfeldt T, Beger HG: Genetic alterations in chronic pancreatitis: evidence for early occurrence of p53 but not K-ras mutations. Br J Surg 1998, 85:337-340 [DOI] [PubMed] [Google Scholar]
- 51.Rodin SN, Rodin AS: Strand asymmetry of CpG transitions as indicator of G1 phase-dependent origin of multiple tumorigenic p53 mutations in stem cells. Proc Natl Acad Sci 1998, 95:11927-11932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrett MT, Sanchez CA, Galipeau PC, Neshat K, Emond M, Reid BJ: Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene 1996, 13:1867-1873 [PubMed] [Google Scholar]
- 53.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellöf L: Pancreatitis and the risk of pancreatic cancer. N Engl J Med 1993, 328:1433-1437 [DOI] [PubMed] [Google Scholar]
- 54.Maacke H, Kessler A, Schmiegel W, Roeder C, Vogel I, Deppert W, Kalthoff H: Overexpression of p53 protein during pancreatitis. Br J Cancer 1997, 75:1501-1504 [DOI] [PMC free article] [PubMed] [Google Scholar]