

Abstract
Recent magnetic resonance (MR) studies of multiple sclerosis lesions indicate that axonal injury is a major correlate of permanent clinical deficit. In the present study we systematically quantified acute axonal injury, defined by immunoreactivity for beta-amyloid-precursor-protein in dystrophic neurites, in the central nervous system of 22 multiple sclerosis patients and 18 rats with myelin-oligodendrocyte glycoprotein (MOG)-induced chronic autoimmune encephalomyelitis (EAE). The highest incidence of acute axonal injury was found during active demyelination, which was associated with axonal damage in periplaque and in the normal appearing white matter of actively demyelinating cases. In addition, low but significant axonal injury was also observed in inactive demyelinated plaques. In contrast, no significant axonal damage was found in remyelinated shadow plaques. The patterns of axonal pathology in chronic active EAE were qualitatively and quantitatively similar to those found in multiple sclerosis. Our studies confirm previous observations of axonal destruction in multiple sclerosis lesions during active demyelination, but also indicate that ongoing axonal damage in inactive lesions may significantly contribute to the clinical progression of the disease. The results further emphasize that MOG-induced EAE may serve as a suitable model for testing axon-protective therapies in inflammatory demyelinating conditions.
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system characterized by widespread inflammation, focal demyelination, and a variable degree of axonal loss. 1 Axonal damage in MS lesions has recently attracted significant attention, because neuroimaging studies suggest that it may be the major pathological correlate of permanent functional deficit. 2-13 The presence of axonal degeneration in multiple sclerosis has long been recognized, 1,14-26 but only recently have efforts been made toward a quantitative assessment of axonal injury with particular emphasis on early, actively demyelinating lesions. 27,28
Similarly, the presence of axonal degeneration has been shown in animal models of the human demyelinating disease, such as experimental autoimmune encephalomyelitis (EAE). 29-31 Detailed, quantitative results which allow a comparison with MS lesions, are so far lacking, however.
In the present study we give a detailed account of acute axonal injury, as revealed by the axonal immunoreactivity for the beta-amyloid-precursor protein (βAPP) in myelinated and demyelinated areas of the central nervous system of 22 MS patients. Immunocytochemistry for βAPP has been shown to be an early and sensitive marker for axonal damage in various disorders of the central nervous system, 32,33 including MS. 27 Our data show that acute axonal injury occurs at a very high incidence during the phase of active demyelination, at a very early time point of lesion formation. In addition, however, there is a low incidence of axonal damage in inactive demyelinated plaques, which may be responsible for disease progression in the chronic inflammatory inactive stage of the disease. In contrast, remyelination seems to protect axons within MS plaques from further degeneration. The acute phase of axonal destruction in actively demyelinating lesions is closely reflected in chronic autoimmune encephalomyelitis in rats. EAE induced by active sensitization with myelin-oligo-dendrocyte glycoprotein (MOG) thus may serve as a good model for testing axon-protective therapeutic strategies in inflammatory demyelination.
Material and Methods
MS Patients and Controls
The study was performed on autopsy tissue from 20 and biopsy tissue from 2 MS patients. Biopsy material was included in this study to determine the reliability of the immunocytochemical techniques in the autopsy cases. In addition, 9 control brains were included in this series from patients (age, 63.56 ± 12.4 years; male/female, 5/4) without evidence of neurological disease or neuropathological alterations. Clinical background data as well as the number and characteristics of lesional areas studied in each patient are summarized in Table 1 ▶ . All material was fixed in 4% paraformaldehyde and embedded in paraffin wax.
Table 1.
Number and Characteristics of Patients Included in the Study
| Patient no. | Age/sex | Disease duration | Disease course |
|---|---|---|---|
| 1 | 47 /F | 3.5 months | acute |
| 2 | 46 /F | 12 days | acute |
| 3 | 29 /M | 1.5 months | acute |
| 4 | 51 /F | 7 months | acute |
| 5 | 46 /M | 3 months | acute |
| 6 | 28 /F | 8 months | acute |
| 7 | 68 /F | 1.5 months | acute |
| 8 | 45 /M | 3 weeks | acute |
| 9 | 20 /F | 4 years | RR/acute |
| 10 | 35 /M | 1.5 months | acute |
| 11 | 35 /F | 4 years | RR/acute |
| 12 | 53 /M | 1 month | RR/acute |
| 13 | 39 /F | 3 years | SP |
| 14 | 43 /F | 20 years | SP |
| 15 | 53 /F | 21 years | SP |
| 16 | 42 /F | 11 years | RR |
| 17 | 45 /F | 16 years | SP |
| 18 | 37 /F | 2 years | PP |
| 19 | 34 /F | 13 years | SP |
| 20 | 33 /F | 10 years | SP |
| 21 | 30 /M | 6 years | SP |
| 22 | 40 /F | 10 years | SP |
RR, relapsing-remitting MS; SP, secondary progressive MS; PP, primary progressive MS.
EAE Material
Lewis 1N rats were kindly provided by Prof. H. Hedrich. 34 EAE was induced in 18 rats by active sensitization with 20 to 100 micrograms of recombinant MOG as described in detail before. 35 Three healthy rats of the same strain served as controls. Animals were weighed and examined daily for clinical signs of EAE. At various time points after sensitization (days 11–61), animals were sacrificed and perfused via the aorta with 4% paraformaldehyde. Brains and spinal cords were dissected and routinely embedded in paraffin wax.
Neuropathology and Immunocytochemistry
Serial sections 2 to 4 μm thick were stained with hematoxylin/eosin (HE), Luxol fast blue (LFB)/Periodic acid-Schiff (PAS), and Bielschowsky silver impregnation to assess inflammation, demyelination, and axonal pathology, respectively. Immunohistochemistry was performed on adjacent serial sections using an avidin-biotin or an alkaline phosphatase/anti-alkaline phosphatase technique. Primary antibodies were used against the following targets: Myelin-oligodendrocyte glycoprotein (anti-MOG; Department of Biochemistry, Cardiff, UK), proteolipid protein (anti-PLP; Serotec, Oxford, UK), 2′-3′-cyclic nucleotide phosphodiesterase (anti-CNPase, Affinity Research Products, Ilkeston, UK), myelin basic protein (anti-MBP, BioGenex, San Ramon, CA), human T cells (anti-CD3, Serotec), human macrophages (anti-CD68, Dako, Glostrup, Denmark), human common leukocyte antigen (anti-CD 45, Pharmingen, San Diego, CA), rat T cells (anti-CD43-equivalent, clone W3/13; Harlan Sera-Lab, Loughborough, UK), macrophages/activated microglia in rats (anti-ED 1; Serotec), early activated human hematopoietic macrophages (anti-MRP 14, BMA Biomedicals, Augst, Switzerland; anti-27E10; BMA Biomedicals), beta-amyloid precursor protein (anti-APP, Boehringer Mannheim, Mannheim, Germany). Control sections were incubated in the absence of primary antibody. In situ hybridization for PLP mRNA was performed according to Breitschopf et al. 36
Selection of Demyelinated Plaques and Definition of Lesional Staging
In both MS and EAE, areas of normal white matter, periplaque white matter, actively demyelinating plaques, inactive demyelinated plaques, and remyelinated shadow plaques were selected for further analysis of axonal pathology. Normal white matter (NWM) was defined as an area that showed no evidence of demyelination by macroscopic inspection and histology within the area and the surrounding tissue. Periplaque white matter (PP) represented a strip of tissue of 5 mm adjacent to the border of active or inactive plaques.
The following categories for demyelinated plaques were defined:
Early active lesions (EA): These lesions were heavily infiltrated by T cells and macrophages. Myelin sheaths were in the process of disintegration and macrophages contained degradation products, which were stained by Luxol fast blue and were immunoreactive for all myelin proteins, including MOG or CNPase.
Late active lesions (LA): In these lesions myelin was already destroyed and removed from axons. Macrophages contained degradation products reactive for major myelin proteins, such as PLP, but were negative for MOG or CNPase.
Inactive lesions with active border (IA + A): The inactive center of radially expanding lesions still showed pronounced inflammation and macrophage infiltration. The macrophages revealed empty vacuoles and showed no immunoreactivity for myelin proteins.
Inactive lesions (IA): These lesions showed no evidence for ongoing myelin destruction at their borders. Although some of these lesions too contained T cells and macrophages, their number was much lower compared to active ones.
Remyelinated shadow plaques (RM): These lesions were characterized by myelin pallor, due to abnormally thin myelin sheaths and a pronounced expression of PLP mRNA in oligodendrocytes. Similar to inactive lesions, residual inflammation was present.
Patient Groups
Patients (n = 22) were divided into groups according to the following criteria (see Results and Tables 3 and 4 ▶ ▶ ). Those classified as active MS (n = 17) besides inactive lesions, had at least one actively demyelinating lesion present in the CNS; those classified inactive MS (n = 5) had only inactive and/or remyelinated lesions and no evidence for ongoing demyelinating activity in the whole CNS.
Table 3.
Acute Axonal Injury in Normal and Periplaque White Matter of Multiple Sclerosis
| Control white matter | Periplaque white matter (active MS) | Normal white matter (active MS) | Periplaque white matter (inactive MS) | Normal white matter (inactive MS) | |
|---|---|---|---|---|---|
| βAPP-positive axons/0.01 mm2 | 0.05 ± 0.02 | 1.07 ± 0.17 | 0.27 ± 0.09 | 0.07 ± 0.02 | 0.05 ± 0.02 |
| -fold increase compared to control WM | 21.4 | 5.4 | 1.4 | 1 | |
| P values | CO/PP: <0.0001 | CO/NWM: < 0.01 | CO/PP: n.s. | CO/NWM: n.s. |
Table 4.
Acute Axonal Injury in Acute versus Chronic Multiple Sclerosis
| Active (early + late active) | Inactive + active edge | Inactive | Periplaque white matter | Normal white matter | ||
|---|---|---|---|---|---|---|
| βAPP-positive axons/0.01 mm2 | Acute MS | 47.27 ± 6.02 | 7.6 ± 1.43 | 1.48 ± 0.17 | 1.3 ± 0.21 | 0.41 ± 0.14 |
| Chronic MS | 26.6 ± 4.29 | 3 ± 0.72 | 0.23 ± 0.05 | 0.19 ± 0.04 | 0.06 ± 0.01 | |
| P | <0.05 | <0.05 | <0.01 | <0.0001 | <0.001 |
Patients were considered to have either acute or chronic MS (Table 4) ▶ as follows:
Acute MS (malignant MS; n = 12) patients exhibited a rapidly progressing disease course, leading to significant disability in multiple neurological systems and often to death. 37
Classical or chronic MS (n = 10) patients exhibited relapsing-remitting, secondary progressive, primary progressive disease, with duration ranging from 3 to 21 years.
Quantitative Determination of Acute Axonal Injury and Inflammation
Camera lucida drawings of demyelinating lesions were made in order to define precisely the pattern of myelin destruction for each lesional area. In the selected area, the demyelinating activity was determined by the presence or absence of myelin degradation products within macrophages immunoreactive for MOG, proteolipid protein (PLP), and CNPase as described above. On adjacent serial sections the number of βAPP- and common leukocyte antigen (CLA)-positive elements stained per square unit of tissue was counted. A 0.01-mm 2 field, defined by an ocular morphometric grid, taken throughout the middle of each lesional area was selected. In this field βAPP-positive fibers were counted under a 100× objective. In inactive demyelinated lesions, shadow plaques as well as in periplaque and normal white matter for each lesional area the average number of βAPP-positive axons in 10 adjacent fields of 0.01 mm 2 were taken for quantification because of the much lower density of injured axons.
To assess the extent of inflammation, CLA-positive elements were counted in a 0.1 mm 2 field in the respective lesions. Total number of MS lesional areas analyzed: n = 240; among those: n = 36 early active (EA), n = 13 late active (LA), n = 29 inactive center with active border (IA + A), n = 21 inactive (IA), n = 26 remyelination (RM), n = 36 normal white matter (NWM) of cases with active lesions (active MS), n = 41 periplaque white matter (PP) of cases with active lesions (active MS), n = 15 normal white matter (NWM) of cases with only inactive and/or remyelinating lesions (inactive MS), n = 23 periplaque white matter (PP) of cases with only inactive and/or remyelinating lesions (inactive MS); n = 27 white matter of controls (CO). Because there were no regional or interindividual differences in βAPP reactivity in normal white matter of controls, the same controls were used for all comparisons.
The total number of EAE lesional areas analyzed: n = 55; among those, n = 16 early active (EA), n = 12 late active (LA), n = 11 inactive plus active border (IA + A), n = 4 remyelination with superimposed demyelinating activity (RM-A), n = 12 white matter of control animals (CO).
Mann-Whitney U test and chi-square test were used for statistical analysis.
Results
Acute Axonal Injury in MS and EAE
Axonal pathology was qualitatively assessed by Bielschowsky’s silver impregnation method. This staining technique revealed many swollen and distorted axons as well as axonal spheroids in actively demyelinating lesions of MS and EAE (Figures 1 and 2) ▶ ▶ . In inactive lesions and in shadow plaques without concomitant active demyelination silver stained axonal spheroids were found only occasionally. For the quantitative assessment of acute axonal injury in MS and EAE, immunohistochemistry for βAPP protein was performed. βAPP serves as a marker for disturbance of axonal transport. 32,38-40 As described previously, 27 βAPP reactivity was found in some axons of normal axonal caliber, being most prominent in focal axonal swellings and in terminal ovoids (Figures 1 and 2) ▶ ▶ .
Figure 1.
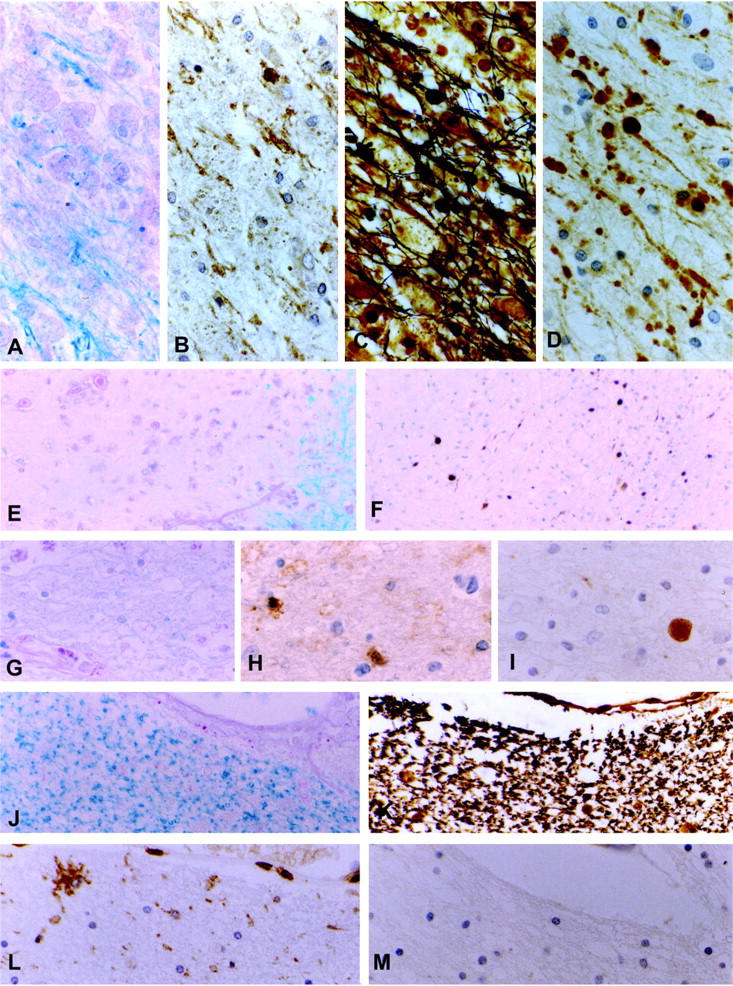
Axonal pathology in multiple sclerosis. A–D: Early active MS lesion. Macrophages containing myelin degradation products (A, B), immunoreactive for all myelin proteins including MOG (A: Luxol fast blue (LFB), B: immunocytochemistry for MOG). C. Bielschowsky silver impregnation for axons reveals apparently normal axons as well as axonal swellings and spheroids. D: Immunocytochemistry for βAPP shows numerous injured axons and axonal end-bulbs. E–F: Inactive demyelinated plaque with ongoing demyelinating activity at the border. Injured axons are found at the plaque edge as well as in the demyelinated plaque center (E: LFB, F: immunocytochemistry for βAPP). G–I: Inactive demyelinated plaque without any evidence for active demyelination, but residual inflammation reveals a singular axonal spheroid by immunocytochemistry for βAPP (G: LFB, H: CLA, I: βAPP) J–M: Remyelinated shadow plaque as revealed by the presence of thin myelin pallor (J: LFB). K: Bielschowsky silver impregnation shows the absence of axonal swellings and spheroids. Residual inflammation, in particular activated microglia is still found (L: CLA), but no evidence for acute axonal injury (M: βAPP). Original magnifications, ×484 (A–D, G–M) and ×123 (E and F).
Figure 2.
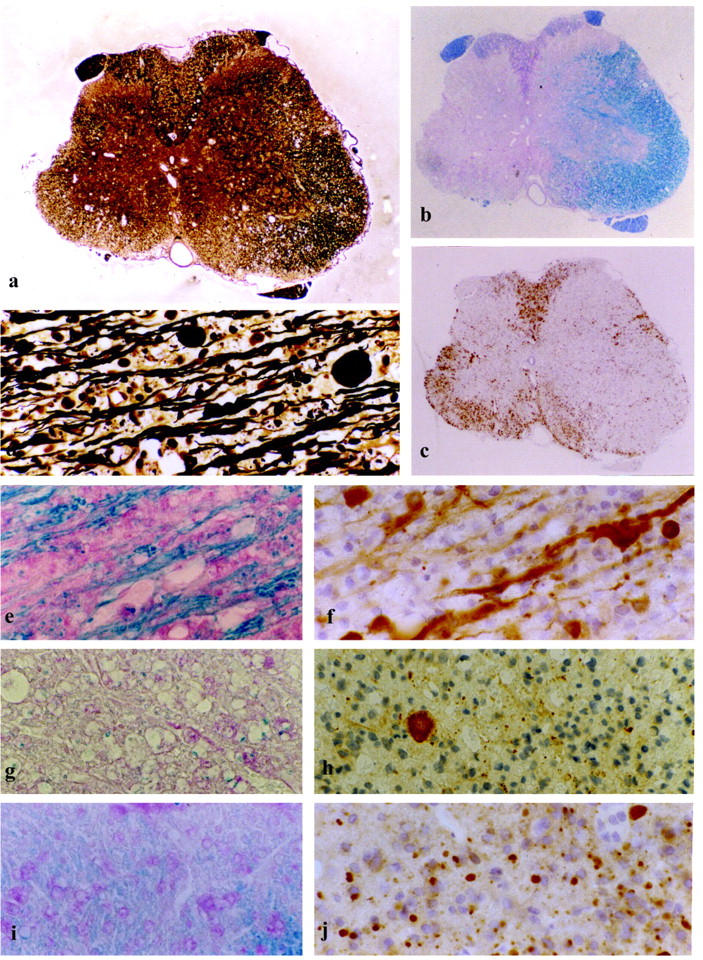
Axonal pathology in chronic experimental autoimmune encephalomyelitis. a–c: Spinal cord atrophy in an animal sacrificed on day 48 post-immunization. a: Bielschowsky silver impregnation for axons showing reduction of axonal density on one side of the spinal cord in comparison to the opposite side, where damage to myelin is only minor. b: LFB myelin stain reveals demyelination of the atrophic side of the spinal cord A. c: Massive macrophage infiltration within the demyelinated area. (ED1) d: Axonal swellings and spheroids in an actively demyelinating EAE lesion (Bielschowsky). e–j: Acute axonal injury in lesions of different stages of myelin degradation as revealed by immunocytochemistry for βAPP. e and f: Same area of active demyelination as shown in d infiltrated by macrophages containing myelin debris (e, LFB) and numerous axonal swellings and spheroids immunoreactive for βAPP (f, βAPP). g and h: Demyelinated plaque with residual activity at the border (g, LFB) showing only one βAPP-positive axonal spheroid beside some glial reactivity for βAPP (h, βAPP). i and j: Shadow plaque with thin myelin pallor indicating remyelination, infiltrated by myelin degrading macrophages (i, LFB) and many βAPP-reactive axons (j, βAPP). Original magnifications, ×40 (a), ×31 (b and c), ×389 (d–j).
Incidence of βAPP-Positive Axons in Human Control Brain Tissue
Brain tissue of nine individuals without macroscopic or microscopic evidence for CNS disease served as control tissue. In each individual three distinct areas of 10 × 0.01 mm 2 were selected. The areas studied were located on brain regions where MS plaques are frequently found, such as the frontal, parietal, temporal, or occipital periventricular white matter or the subcortical white matter. βAPP-positive axons were observed only exceptionally (0.05 ±0.02 mean ± SE) and there were no differences noted between different brain areas. Therefore the data were pooled and used as controls for all comparisons.
Incidence of βAPP-Positive Axons in Actively Demyelinating Lesions of MS
The highest incidence of βAPP-positive axons was observed in areas of early and late active demyelination. Although in total βAPP levels in early active lesions were slightly higher compared to late active lesions, some early active lesions showed only minor axonal injury (EA: 41.26 ± 5.59 range: 1–136 βAPP-positive axons/0.01 mm2, LA: 39.85 ± 7.72 range: 13–101 βAPP-positive axons/0.01 mm2). The difference to late active lesions was not statistically significant (CO/EA: p < 0.0001; CO/LA: p < 0.0001, EA/LA: n.s.; Table 2 ▶ ).
Table 2.
Acute Axonal Injury in Multiple Sclerosis Lesions
| Control white matter | Early active | Late active | Inactive + active edge | Inactive | Remyelination | |
|---|---|---|---|---|---|---|
| βAPP-positive axons/ 0.01 mm2 | 0.05 ± 0.02 | 41.26 ± 5.59 | 39.85 ± 7.72 | 5.38 ± 0.91 | 0.47 ± 0.12 | 0.07 ± 0.01 |
| -fold increase compared to control WM | 825.2 | 797 | 107.6 | 9.4 | 1.4 | |
| P values | CO/EA: < 0.0001 | CO/LA: < 0.0001 | CO/IA+ A: < 0.0001 | CO/IA: <0.01 | CO/RM: n.s. |
Incidence of βAPP-Positive Axons in Inactive Demyelinated Plaques of MS
βAPP-positive axons were also abundant in the inactive center of lesions with ongoing demyelinating activity at their border (CO/IA + A, P < 0.0001). Their number, though, was significantly lower compared to early and to late active lesions (EA/IA + A, p < 0.0001; LA/IA + A, P < 0.0001). In addition, 15 completely inactive lesions of five patients were analyzed where no area of ongoing demyelination was found in the whole CNS. Even in these chronic inactive lesions a significant number of βAPP-positive axons was found, when compared to the control areas (CO/IA, P < 0.01), but it was significantly lower compared to inactive lesions with an active edge (IA/IA + A, P < 0.0001; Table 2 ▶ ).
Incidence of βAPP-Positive Axons in Remyelinated Shadow Plaques
There was no evidence for active demyelination in all shadow plaques analyzed, though inflammation of some T cells, macrophages, and, in particular, activated microglia, was still present and comparable to that in inactive demyelinated lesions (Figure 1) ▶ . However, inactive demyelinated and remyelinated plaques differed in βAPP reactivity; whereas significant axonal injury was found in inactive demyelinated lesions, axonal damage was minor in shadow plaques (CO/RM, n.s.; IA/RM, P < 0.05; Table 2 ▶ ).
Incidence of βAPP-Positive Axons in Periplaque and Normal White Matter
In comparison to controls, there was also a low, but significant number of injured axons found in periplaque white matter of actively demyelinating lesions (active MS; CO/PP, P < 0.0001). In these “active” cases significant axonal damage was even found in normal white matter, far distant from established plaques (active MS, CO/NWM, P < 0.01, Table 3 ▶ ).
In order to determine whether acute axonal injury in the normal and periplaque white matter was restricted to cases with active demyelination, we also analyzed axonal βAPP reactivity in cases with only inactive and/or remyelinated lesions (inactive MS; Table 3 ▶ ). In such inactive cases some increase in the number of βAPP-positive axons was also found in the periplaque white matter, but it did not reach statistical significance. βAPP immunoreactivity in normal white matter of inactive MS cases was comparable to that in control white matter (inactive MS, CO/PP, n.s.; CO/NWM, n.s., Table 3 ▶ ).
Incidence of βAPP in Acute versus Chronic MS
Acute MS is characterized by a fulminant onset and rapid clinical deterioration leading to significant disability or even death within a short time period. 37 In all lesions of patients who presented with acute MS, the number of βAPP-positive axons was significantly higher compared to the respective lesions of chronic MS cases. A significant difference was also noted in normal and periplaque white matter (Table 4) ▶ .
Relation between Acute Axonal Injury and Inflammation
Inflammation was assessed by the presence of CLA-positive cells within and around the MS lesions. As expected, inflammation was most prominent in MS plaques with evidence for active demyelination, but still present in inactive demyelinated and remyelinated lesions (Table 5) ▶ . Similar to a previous study, 27 a significant correlation between CLA-positive cells and βAPP-positive axons was found in the lesions of MS (P < 0.0001). When the different types of lesions were analyzed separately, a highly significant correlation was found for late active and inactive lesions, but only a weak correlation was found for remyelinated lesions and none for early active lesions. The latter finding may be due to the fact that in some very early lesions inflammation was pronounced, whereas axonal injury was not yet fully developed (CLA/APP; EA: n.s.; LA: P < 0.01; IA + A: P < 0.01; IA: P < 0.0001; RM: P < 0.05).
Table 5.
Inflammation in Multiple Sclerosis Lesions
| Early active | Late active | Inactive + active edge | Inactive | Remyelination | |
|---|---|---|---|---|---|
| CLA-positive cells/mm2 | 1582.86 ± 117.79 | 1097 ± 166.33 | 740 ± 171.68 | 380.5 ± 44.81 | 274.29 ± 53.02 |
Inactive demyelinated and remyelinated plaques revealed similar values for inflammation (Table 5) ▶ , but differed in βAPP reactivity. Whereas significant axonal injury was found in inactive demyelinated lesions, axonal damage was minor in shadow plaques (Table 2) ▶ .
Incidence of βAPP in Chronic EAE
Acute axonal injury in chronic active EAE lesions was qualitatively and quantitatively similar to that in active MS plaques (Figure 2 ▶ and Table 6 ▶ ). Also, axonal damage occurred most prominently during a short time window when myelin was in the process of disruption (CO/EA, P < 0.0001; CO/LA, P < 0.0001). Although βAPP levels were generally higher in early active lesions, there was no statistically significant difference to late active lesions (EA/LA: n.s.).
Table 6.
Acute Axonal Injury in EAE Lesions
| Control white matter | Early active | Late active | Inactive + active edge | Remyelination + active demyelination | |
|---|---|---|---|---|---|
| βAPP-positive axons/0.01 mm2 | 0.2 ± 0.07 | 87.38 ± 16.59 | 67.08 ± 16.15 | 7 ± 2.09 | 30.25 ± 15.59 |
| β-fold increase compared to control WM | 436.9 | 335.4 | 35 | 151.25 | |
| P values | CO/EA: <0.0001 | CO/LA: <0.0001 | CO/IA+ A: <0.0001 | CO/RM-A: <0.01 |
In contrast to MS, no completely inactive lesions were present in the sample of EAE material. Such as in multiple sclerosis lesions the number of βAPP-positive axons in inactive lesions with an active edge was significantly increased compared to control white matter, but also significantly lower compared to early and late active lesions (CO/IA + A, P < 0.0001; EA/IA + A, P < 0.0001; LA/IA + A, P < 0.0001). In addition, most remyelinated shadow plaques in EAE showed evidence of recurrent demyelinating activity (Figure 2) ▶ . This may explain the high incidence of axonal βAPP reactivity in remyelinated EAE plaques (CO/RM-A, P < 0.01).
Discussion
The present study confirms some aspects of axonal damage reported in previous quantitative studies. As reported before, a high incidence of axonal damage apparently occurs in actively demyelinating lesions. 27,28 There are, however, several aspects of our present study that shed new light on the patterns of axonal damage in inflammatory demyelinating lesions.
For the detection of acute axonal injury, immunohistochemistry for βAPP was performed. βAPP is a membrane-spanning glycoprotein and a normal constituent of neuronal cells. 32,38 It is transported by fast axonal transport. Normal levels of βAPP in axons are not detectable by standard immunohistochemistry in formalin-fixed tissue. Its accumulation at sites of injury is probably due to the disturbance of axonal transport. 32,38 Therefore, βAPP has been suggested to be the immunoctyochemical marker of choice for the detection of injured axons. 39 As has been demonstrated in fatal head injury, βAPP stains damaged axons within 2 hours after injury 32 and remains detectable in axons and bulbs for 10 to 14 days. 40 With a survival time longer than 2 weeks, βAPP reactivity disappears from the injured axons as well as from bulbs. 40 βAPP, therefore, may serve as a marker for early axonal damage, while more advanced stages of axonal degeneration may not be detected by this method.
By correlating acute axonal injury with the presence of myelin degradation products in our study, we can more precisely define the time window during which axonal damage occurs in MS lesions. Because myelin proteins are rapidly degraded when taken up by macrophages, the high incidence of axonal APP reactivity in lesional areas, which contain macrophages with degradation products immunoreactive for MOG, CNPase, and PLP, suggests that the most pronounced acute axonal injury occurs immediately within this short time window of plaque formation, eg, during early and late active demyelination. 41,42 According to the time course of myelin degradation by macrophages in vitro and in vivo in EAE, 43 (Lassmann H, unpublished data), our data suggest that the time window of active demyelination lasts for approximately 1 to 2 weeks after initiation of the demyelinating process. This observation may be important for the design of axon-protective therapeutic strategies in MS. In some early active lesions only minor axonal injury was found. This may be due to the fact that very early, incipient lesions, where myelin disruption and possibly also axonal damage were incomplete, were included in the study. On the other hand, an interindividual variability in terms of tissue destruction may account for this phenomenon.
As reported before, 28 axonal injury in MS is not only restricted to demyelinated lesions, but also affects the periplaque area as well as the normal white matter, far distant from established demyelinated plaques. This diffuse axonal injury may be reflected in abnormalities of N-acetyl aspartate levels in the NWM in magnetic resonance spectroscopy. 6-8 Since significant acute axonal injury in the periplaque and normal white matter in our study was only found in cases with ongoing active demyelination, and since the changes were more pronounced in the periplaque, compared to the normal white matter, it is likely that these alterations occur secondary to axonal destruction in actively demyelinating plaques. To what extent the inflammatory process, which is much more widely distributed in the CNS compared to demyelination, 44 contributes to axonal injury in the so-called normal white matter remains to be determined.
Recent work of axonal changes after trauma suggests that some axonal damage may be repaired. 45 This may also be reflected in the normalization of NAA levels within MS lesions and NAWM six months after the acute phase of lesion formation in early MS. 46 Therefore, reactive axonal changes as seen by immunocytochemistry for βAPP may be reversible unless they have not led to the formation of fully developed terminal ovoids. Intervention at an early time point of plaque formation, therefore, might not only reduce acute axonal injury, but also reduce the number of injured axons that undergo definite degeneration.
Besides acute axonal injury during active demyelination, we also found significant low-burning axonal damage even in plaques, completely devoid of active myelin destruction in cases where no active lesion was detected (Table 2 ▶ and Figure 1 ▶ ). This finding may explain, at least in part, clinical progression in cases where no MRI-activity can be detected. 47 The mechanisms of axonal degeneration, however, remain speculative. It has to be noted that ongoing axonal destruction in inactive MS lesions occurs on the background of a residual inflammatory process, reflected by infiltration of the lesions by some T cells, macrophages, and activated microglia. Thus, the inflammatory process may play an important role in the ongoing axonal destruction. Alternatively, ongoing axonal destruction may be due to a lack of trophic support provided by myelin and oligodendrocytes.
In remyelinated lesions inflammation was still found, but acute axonal injury was significantly less pronounced in remyelinated shadow plaques compared to that in inactive demyelinated lesions. This observation further supports the view that myelination exerts a protective effect against axonal damage. 48 This could be due to the protective effect of the myelin sheath against the inflammatory environment, as has been shown for nitric oxide. 49 On the other hand, trophic supply by oligodendrocytes may protect axons against gradual degeneration. Such a mechanism of axonal damage has been suggested in experimental models deficient for myelin proteins such as myelin-associated glycoprotein (MAG−/− animals) 50 or proteolipid protein (PLP-DM20−/− mice). 51
In conclusion, our data suggest that extensive axonal damage occurs during plaque formation in a very short time window after onset of demyelination. During active demyelination, significant axonal injury is also found in the periplaque and normal white matter. Second, there is ongoing low-burning axonal destruction in inactive demyelinated lesions. This process, in contrast to acute axonal injury in active lesions, may be continued for prolonged time periods and may explain the observed profound reduction of axonal density in established MS plaques. 20,52,53 Finally, our results show that axonal pathology in inflammatory demyelinating lesions of chronic MOG-induced autoimmune encephalomyelitis closely reflects that found in MS. This particular model of MOG-induced EAE in Lewis N rats was selected because it is pathologically characterized by focal, very large plaques of demyelination. 35 In contrast to MS, however, the development of lesions in this model is condensed to a time period of 1 to 2 months. Thus, most of the lesions studied here still present demyelinating activity at least at their borders to the adjacent periplaque white matter. Furthermore, in contrast to most shadow plaques in MS, remyelinating lesions in this model generally show signs of recurrent active demyelination. Thus, this model closely reflects axonal pathology in actively demyelinating MS cases, but does not, so far, allow study of the slow axonal degeneration in chronic MS cases. Despite these differences in disease activity, MOG-induced EAE may serve as an ideal model for testing the effectiveness of axon-protective therapeutic strategies for chronic inflammatory demyelinating diseases.
Acknowledgments
We thank Helene Breitschopf, Angela Kury, Marianne Leisser, Jutta Wakley-Neuninger, and Petra Tassotti for expert technical assistance and Drs. Christine Stadelmann and Helmut Rauschka for carefully reading the manuscript and for discussion.
Footnotes
Address reprint requests to Prof. Dr. Hans Lassmann, Division of Neuroimmunology, Brain Research Institute, University of Vienna, Spitalgasse 4, A-1090 Wien, Austria. E-mail: hans.lassmann@univie.ac.at.
Supported by the Austrian Science Foundation Project P 12658-MED, by the Austrian Ministry for Science (GZ 650.223/2-III/2a/99) and the EC Biomed 2-Project BMH 4-97-2027.
References
- 1.Charcot JM: Histologie de la sclérose en plaques. Gazette des Hopitaux civils et militaires 1868, 140:554–555 and 141:557–558 and 143:566
- 2.Arnold DL, Riess GT, Matthews PM, Francis GS, Collins DL, Wolfson C, Antel JP: Use of proton magnetic resonance spectroscopy for monitoring disease progression in multiple sclerosis. Ann Neurol 1994, 36:76-82 [DOI] [PubMed] [Google Scholar]
- 3.Barnes D, Munro PMG, Youl BD: The longstanding MS lesion: a quantitative MRI and electron microscopic study. Brain 1991, 114:1271-1280 [DOI] [PubMed] [Google Scholar]
- 4.Davie CA, Hawkins CP, Barker GJ, Brennan A, Tofts PS, Miller DH, McDonald WI: Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain 1994, 117:49-58 [DOI] [PubMed] [Google Scholar]
- 5.Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, Harding AE, McDonald WI, Miller DH: Persistent functional deficit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axonal loss. Brain 1995, 118:1583-1592 [DOI] [PubMed] [Google Scholar]
- 6.Davie CA, Barker GJ, Thompson AJ, Tofts PS, McDonald WI, Miller DH: 1H magnetic resonance spectroscopy of chronic cerebral white matter lesions and normal appearing white matter in multiple sclerosis. J Neurol Neurosurg Psychiat 1997, 63:736-742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Stephano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS, Antel JP, Arnold DL: Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis: results of a longitudinal magnetic resonance spectroscopy study. Brain 1998, 121:1469-1477 [DOI] [PubMed] [Google Scholar]
- 8.Fu L, Matthews PM, De Stefano N, Worsley KJ, Narayanan S, Francis GS, Antel JP, Wolfson C, Arnold DL: Imaging axonal damage of normal appearing white matter in multiple sclerosis. Brain 1998, 121:103-113 [DOI] [PubMed] [Google Scholar]
- 9.Losseff NA, Wang L, Lai HM, Yoo DS, Gawne-Cain ML, McDonald WI, Miller DH, Thompson AJ: Progressive cerebral atrophy in multiple sclerosis: a serial MRI study. Brain 1996, 119:2009-2019 [DOI] [PubMed] [Google Scholar]
- 10.Losseff NA, Webb SL, O’Riordan JI, Page R, Wang L, Barker GJ, Tofts PS, McDonald WI, Miller DH, Thompson AJ: Spinal cord atrophy and disability in multiple sclerosis: a new reproducible and sensitive MRI method with potential to monitor disease progression. Brain 1996, 119:701-708 [DOI] [PubMed] [Google Scholar]
- 11.van Walderveen MAA, Barkhof F, Hommes OR, Polman CH, Tobi H, Frequin STFM, Valk J: Correlating MRI and clinical disease activity in multiple sclerosis: relevance of hypointense lesions on short-TR/short-TE (T1-weighted) spin echo images. Neurology 1995, 45:1684-1690 [DOI] [PubMed] [Google Scholar]
- 12.van Walderveen MAA, Kamphorst W, Scheltens P, van Waesberghe JHTM, Ravid R, Polman CH, Barkhof F: Histopathologic correlate of hypointense lesions on T1-weighted spin-echo MRI in multiple sclerosis. Neurology 1998, 50:1282-1288 [DOI] [PubMed] [Google Scholar]
- 13.van Walderveen MAA, Barkhof F, Pouwels PJW, van Schijndel RA, Polman CH, Castelijns JA: Neuronal damage in T1-hypointense multiple sclerosis lesions demonstrated in vivo using proton magnetic resonance spectroscopy. Ann Neurol 1999, 46:79-87 [DOI] [PubMed] [Google Scholar]
- 14.Bielschowsky M: Zur Histologie der multiplen sklerose. Neurol Centralblatt 1903, 22:770-777 [Google Scholar]
- 15.Bielschowsky M: Die marklosen Nervenfasern in den Herden der multiplen Sklerose: eine Antwort an Herrn Strähuber. Neurol Centralblatt 1904, 16:59-62 [Google Scholar]
- 16.Marburg O: Die sogenannte “akute multiple Sklerose” (Encephalomyelitis peraxialis scleroticans). Jahrb Neurol Psychiatr 1906, 27:211-312 [Google Scholar]
- 17.Doinikow B: Über De- und Regenerationserscheinungen an Achsenzylindern bei der multiplen Sklerose. Z Gesamt Neurol Psychiatr 1915, 27:151-178 [Google Scholar]
- 18.Fraenkel M, Jakob A: Zur Pathologie der multiplen Sklerose mit besonderer Berücksichtigung der akuten Formen. Z Gesamt Neurol Psychiatr 1913, 14:565-603 [Google Scholar]
- 19.Siemerling E, Raecke J: Beiträge zur Klinik und Pathologie der multiplen Sklerose mit besonderer Berücksichtigung ihrer Pathogenese. Archiv für Psychiatrie und Nervenkrankheiten 1914, 53 (Heft II): 385–564
- 20.Putnam TJ: Studies in multiple sclerosis. Arch Neurol Psychiatry 1936, 35:1289-1308 [Google Scholar]
- 21.Greenfield JG, King LS: Observations on the histopathology of the cerebral lesions in disseminated sclerosis. Brain 1936, 59:445-458 [Google Scholar]
- 22.Lumsden CE: The neuropathology of multiple sclerosis. Handbook of Clinical Neurology 1970, Vol. 9: Multiple Sclerosis and Other Demyelinating Diseases. Edited by PJ Vinken, GW Bruyn. New York, Elsevier, pp 217–309
- 23.Prineas JW, Connel F: The fine structure of chronically active multiple sclerosis plaques. Neurology 1978, 28 (suppl.):68-75 [DOI] [PubMed] [Google Scholar]
- 24.Dahl D, Perides G, Bignami A: Axonal regeneration in old multiple sclerosis plaques. Acta Neuropathol (Berl) 1989, 79:154-159 [DOI] [PubMed] [Google Scholar]
- 25.Shintaku M, Hirano A, Llena JF: Increased diameter of demyelinated axons in chronic multiple sclerosis of the spinal cord. Neuropathol Appl Neurobiol 1988, 14:505-510 [DOI] [PubMed] [Google Scholar]
- 26.Kornek B, Lassmann H: Axonal pathology in multiple sclerosis: a historical note. Brain Pathol 1999, 9:651-656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferguson B, Matyszak MK, Esiri MM, Perry VH: Axonal damage in acute multiple sclerosis. Brain 1997, 120:393-399 [DOI] [PubMed] [Google Scholar]
- 28.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L: Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998, 338:278-285 [DOI] [PubMed] [Google Scholar]
- 29.Madrid RE, Wisniewski HM: Axonal degeneration in demyelinating disorders. J Neurocytol 1977, 6:103-117 [DOI] [PubMed] [Google Scholar]
- 30.Raine CS, Mokhtarian F, MacFarlin DE: Adoptively transferred chronic relapsing experimental autoimmune encephalomyelitis in the mouse: neuropathologic analysis. Lab Invest 1984, 51:534-546 [PubMed] [Google Scholar]
- 31.Raine CS, Cross AH: Axonal dystrophy as a consequence of long-term demyelination. Lab Invest 1989, 60:714-725 [PubMed] [Google Scholar]
- 32.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW: Beta-amyloid precursor protein as a marker for axonal injury after head injury. Neurosci Lett 1993, 160:139-144 [DOI] [PubMed] [Google Scholar]
- 33.An SF, Giometto B, Groves M, Miller RF, Beckett AAJ, Gray F, Tavolato B, Scaravilli F: Axonal damage revealed by accumulation of β-APP in HIV-positive individuals without AIDS. J Neuropathol Exp Neurol 1997, 56:1262-1268 [DOI] [PubMed] [Google Scholar]
- 34.Hedrich HJ, ed: Genetic Monitoring of Inbred Strains of Rats. Stuttgart, New York, Gustav Fischer, 1990
- 35.Storch MK, Stefferl A, Brehm U, Weissert R, Wallstroem E, Kerschensteiner M, Olsson T, Linington C, Lassmann H: Autoimmunity to myelin oligodendrocyte glycoprotein in rats mimics the spectrum of multiple sclerosis pathology. Brain Pathol 1998, 8:681-694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Breitschopf H, Suchanek G, Gould RM, Colman DR, Lassmann H: In situ hybridization with digoxigenin-labeled probes: sensitive and reliable detection method applied to myelinating rat brain. Acta Neuropathol (Berl) 1992, 84:581-587 [DOI] [PubMed] [Google Scholar]
- 37.Lublin FD, Reingold SC: Defining the clinical course of multiple sclerosis: results of an international survey. Neurology 1996, 46:907-911 [DOI] [PubMed] [Google Scholar]
- 38.Sheriff FE, Bridges LR, Sivaloganathan S: Early detection of axonal injury after human head trauma using immunocytochemistry for β-amyloid precursor protein. Acta Neuropathol (Berlin) 1994, 87:55-62 [DOI] [PubMed] [Google Scholar]
- 39.Sheriff FE, Bridges LR, Gentleman SM, Sivaloganathan S, Wilson S: Markers of axonal injury in post mortem human brain. Acta Neuropathol (Berlin) 1994, 88:433-439 [DOI] [PubMed] [Google Scholar]
- 40.Geddes JF, Vowles GH, Beer TW, Ellison DW: The diagnosis of diffuse axonal injury: implications for forensic practice. Neuropathol Appl Neurobiol 1997, 23:339-347 [PubMed] [Google Scholar]
- 41.Brück W, Schmied M, Suchanek G, Brück Y, Breitschopf H, Poser S, Piddlesden S, Lassmann H: Oligodendrocytes in the early course of multiple sclerosis. Ann Neurol 1994, 35:65-73 [DOI] [PubMed] [Google Scholar]
- 42.Brück W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretschmar HA, Lassmann H: Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol 1995, 38:788-796 [DOI] [PubMed] [Google Scholar]
- 43.Lassmann H, Wisniewski HM: Chronic relapsing experimental allergic encephalomyelitis: morphological sequence of myelin degradation. Brain Res 1979, 169:357-368 [DOI] [PubMed] [Google Scholar]
- 44.Lassmann H: The pathology of multiple sclerosis and its evolution. Phil Trans Roy Soc London (B) 1999, 354:1635-1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gennarelli TA: The spectrum of traumatic axonal injury. Neuropathol Appl Neurobiol 1996, 22:509-513 [DOI] [PubMed] [Google Scholar]
- 46.DeStefano N, Narayanan S, Matthews PM, Francis GS, Antel JP, Arnold DL: In vivo evidence for axonal dysfunction remote from focal cerebral demyelination of the type seen in multiple sclerosis. Brain 1999, 122:1933-1939 [DOI] [PubMed] [Google Scholar]
- 47.Truyen L, van Waesberghe JHTM, van Walderveen MAA, van Oosten BW, Polman CH, Hommes OR, Ader HJA, Barkhof F. Accumulation of hypointense lesions (“black holes”) on T1 spin-echo MRI correlates with disease progression in multiple sclerosis. Neurology 1996, 47:1469–1476 [DOI] [PubMed]
- 48.Duncan ID: Glial cell transplantation and remyelination of the central nervous system (review). Neuropathol Appl Neurobiol 1996, 22:87-100 [DOI] [PubMed] [Google Scholar]
- 49.Redford EJ, Kapoor R, Smith KJ: Nitric oxide donors reversibly block axonal function: demyelinated axons are especially susceptible. Brain 1997, 120:2149-2157 [DOI] [PubMed] [Google Scholar]
- 50.Yin X, Crawford TO, Griffin JW, Tu PH, Lee VMY, Li C, Roder J, Trapp BD: Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci 1998, 18:1953-1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffiths I, Klugmann M, Anderson T, Yool D, Thompson C, Schwab HM, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA: Axonal swellings and degeneration in mice lacking the major proteolipid protein of myelin. Science 1998, 280:1610-1613 [DOI] [PubMed] [Google Scholar]
- 52.Mews I, Bergmann M, Bunkowski S, Gullotta F, Brück W: Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult Scler 1998, 4:55-62 [DOI] [PubMed] [Google Scholar]
- 53.Lassmann H: The pathology of multiple sclerosis. McAlpine’s Multiple Sclerosis, 3rd edition. Edited by A Compston, G Ebers, H Lassmann, I McDonald, B. Matthews, H Wekerle. London, Churchill Livingstone, 1998