

Abstract
Acute and chronic pulmonary diseases are characterized by impaired fibrinolytic activity within the lung. To determine the role of the fibrinolytic system in regulating the pathologies associated with lung injury, we examined the effect of bleomycin, an agent that induces the development of pulmonary fibrosis, in mice deficient for plasminogen (Pg−/−), urokinase (u-PA−/−), urokinase receptor (u-PAR−/−), or tissue plasminogen activator (t-PA−/−), and in control wild-type (WT) mice. Pg−/− and t-PA−/− mice demonstrated an enhanced increase in lung collagen content relative to that observed in WT mice. Levels in u-PA−/− and u-PAR−/− mice were similar to those in WT mice. Histological analysis 14 days after lung injury confirmed enhanced interstitial fibrosis in Pg−/−, u-PA−/−, and t-PA−/− mice relative to WT and u-PAR−/− mice. Areas of pulmonary hemorrhage were observed in bleomycin-treated WT mice and not in Pg−/−, u-PA−/−, and u-PAR−/− mice or saline controls. Instead, extensive areas of fibrosis were present throughout the lungs of bleomycin-treated Pg−/− and u-PA−/− mice. A mixed phenotype (hemorrhage and fibrosis) was observed in t-PA−/− and Pg+/− mice. Hemosiderin-laden macrophages were abundant in the lungs of mice exhibiting hemorrhage and these mice were prone to an early death. Enhanced macrophage levels in the lungs and activation of matrix metalloelastase (MMP-12) were found in mice with a hemorrhage phenotype. The results of these studies indicate a role for the fibrinolytic system in acute lung injury and suggests that intra-alveolar hemorrhage is the result of basement membrane degradation through cell-mediated u-PA activation of Pg with possible involvement of matrix metalloproteinases. Absence of these two components of the fibrinolytic system, either urokinase or plasminogen, results in accelerated fibrosis.
Lung disease is one of the leading causes of death in the United States, claiming more than 300,000 lives every year. 1 Among pulmonary diseases, those caused by acute lung injury have a variety of clinical pathologies, some of which eventually develop into pulmonary fibrosis. Lung injury triggers an inflammatory response that includes migration and activation of both resident and circulating inflammatory cells and production of cytokines and growth factors. This inflammatory cascade results in cell proliferation, extracellular matrix remodeling, and changes in vascular tone. In addition, macrophages produce enzymes that modulate thrombosis and coagulation. Acute and chronic pulmonary diseases are characterized by severe inflammation, impaired fibrinolytic activity, and increased deposition of collagen within the lung. 2-9
The primary physiological fibrinolytic system is the plasminogen activation pathway. 10 Components of this system include the zymogen, plasminogen (Pg); plasminogen activators, urokinase (u-PA), and tissue plasminogen activator (t-PA); plasminogen activator inhibitors, PAI-1 and PAI-2; plasmin (Pm), the enzymatically active form of Pg responsible for the degradation of fibrin; and the Pm inhibitor, α2 anti-plasmin. The development of mice deficient for components of the fibrinolytic system has provided the means to examine the role of these components in various physiological processes. These include hemostasis and cell migratory events associated with inflammation, reproduction, angiogenesis, and tumor metastasis. 11-19 Indeed, studies in these mice have confirmed the importance of Pg in fibrinolysis and maintenance of vascular patency. 20 Other indirect studies have shown that Pm-generated fibrin(ogen) degradation products can serve as chemoattractants for leukocytes, hence magnifying the inflammatory response. 21,22 Pm itself has been shown to act as a chemoattractant agent specifically for macrophages. 23 In addition, Pm is thought to activate matrix metalloproteinases, 24 which are capable of degrading many extracellular matrix components including collagen, elastin, fibronectin, proteoglycan, and laminin, 25 and hence can indirectly facilitate cell migration. Therefore, Pm could be involved directly or indirectly with several aspects of repair mechanisms after acute lung injury.
It has been reported that overexpression of PAI-1 results in increased deposition of collagen and fibrin in bleomycin-treated mice. 26 On the other hand, deficiency of PAI-1 attenuated the development of pulmonary fibrosis. In the former case, inhibition of Pg activation led to an increase in fibrin clots in the interstitium and alveolar spaces. 26 Additionally, u-PA treatment has been shown to diminish the development of bleomycin-induced pulmonary fibrosis in rats 27 and mice. 28 This study showed decreased lung collagen levels after treatment with u-PA, demonstrating the potential use of Pg activators as therapeutic agents for pulmonary fibrotic disorders. The current investigation was designed to evaluate the mechanism by which Pg and its activators, u-PA and t-PA, are involved in the development of pulmonary fibrosis, using a model of inflammatory injury and fibrosis. Lack of one or more of the components of the fibrinolytic system resulted in alterations in the response to acute lung injury and development of pulmonary fibrosis. Based on these findings, the current investigation is aimed at further delineation of the roles of genes involved in the fibrinolytic system in repair of lung injury induced by bleomycin, which may serve as a model of oxidative lung injury.
Materials and Methods
Animals
WT, Pg−/−, u-PA−/−, u-PAR−/−, t-PA−/−, of mixed (50:50) C57BL6/129 strain background were developed and characterized as previously described. 11,12,19,29,30 Mice were housed in micro-isolation cages on a constant 12-hour light/12-hour dark cycle in a temperature- (22.2°C) and humidity-controlled room and given ad libitum access to food and water. Sexually mature Pg+/− females were mated with Pg+/− males, and t-PA−/−, u-PA−/−, u-PAR−/− females were mated with their male counterparts. Experimental mice were 8 to 12 weeks of age and included males and females. All animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Research Advisory Committee.
Bleomycin Treatment
Intratracheal administration of a single dose of 0.075 U bleomycin (Bristol-Myers Squibb Co., Princeton, NJ) in 50 μl of sterile saline was performed by infusion through the vocal cords using a fiber optic light source for illuminating the entrance into the trachea while the mice were under intraperitoneal ketamine/xylazine anesthesia. This dose of bleomycin has been previously shown to produce pulmonary fibrosis in mice of similar genetic background. 26 Control animals included nontreated and saline-treated mice. At defined time points after bleomycin infusion, mice were anesthetized with isofluorane and lung vasculature perfused free of blood by slowly injecting 3 ml of phosphate buffered saline (PBS) into the right ventricle.
Hydroxyproline Assay
To estimate the total amount of collagen in the lung, hydroxyproline was measured as described 26 from bleomycin- or saline-treated mice 14 days after treatment. The lung vasculature was perfused as described and the left lung was excised and homogenized in 2 ml cold PBS using a hand homogenizer. A 1-ml aliquot of the homogenate was vacuum-dried, weighed, and hydrolyzed in 6 N HCl at 110°C for 12 hours. Samples were filtered through a 45-μm filter and 50 μl of the filtered sample was used for the assay which was conducted in duplicate. An aliquot of 50 μl of citrate/acetate buffer (5% citric acid, 1.2% glacial acetic acid, 7.24% sodium acetate, 3.4% sodium hydroxide) and 1 ml chloramine T solution (0.564 g chloramine T, 4 ml n-propanol, 32 ml citrate/acetate buffer) were added to the sample and allowed to incubate at room temperature for 20 minutes. A 1-ml aliquot of Ehrlich’s solution (4.5 g ρ-dimethylaminobenzaldehyde in 18.6 ml n-propanol, 7.8 ml of 70% perchloric acid) was added to the sample and incubated at 65°C for 15 minutes. Absorbance was measured at 550 nm and the amount of hydroxyproline was determined against a standard curve generated from known concentrations of reagent hydroxyproline (Sigma Chemical Co., St. Louis, MO). Samples containing known amounts of purified collagen (Vitrogen-100, Celtrix Laboratories, Palo Alto, CA) were used to confirm the ability of the assay to completely hydrolyze and recover hydroxyproline from collagen.
Isolation and Identification of Lung Leukocytes
To study the inflammatory response during the acute phase after lung injury, leukocytes were isolated from whole lungs. 31 Mice were anesthetized at day 0 and at 3, 5, 7, or 9 days after treatment with bleomycin or vehicle and lung vasculature was perfused free of blood. The left lung was excised and the trachea and hilar nodes were removed from the lung sample. Lungs were minced and digested in 15 ml of a digest solution containing collagenase (Type A, 1 mg/ml, Sigma Chemical); DNase I (50 U/ml, Sigma Chemical); Hanks’ balanced salt solution (HBSS; Gibco BRL, Grand Island, NY); antibiotics (Pen-Strep, 1%, Gibco BRL); and supplemented with 5% fetal calf serum (Gibco BRL). After digestion, the tissue was mechanically disrupted by multiple passages through a 10-ml plastic syringe. Contaminating erythrocytes were lysed in 0.075% NH4Cl in Tris-HCl, pH 7.2, and the leukocytes recovered free of cell debris by filtration through a nytex screen (3–100/35 mesh; Tekco, Briarcliff Manor, NY). To ensure a homogeneous leukocyte preparation, a 40% Percoll (Sigma Chemical) gradient was used and the leukocyte pellet resuspended in HBSS media containing antibiotics and serum. Cell viability was determined using trypan blue exclusion and total number of leukocytes per lung were quantitated using a hemocytometer.
To determine the type of leukocytes obtained from the lung digests, 100-μl samples containing 10 6 cells/ml were centrifuged onto a glass slide using a cytocentrifuge (Shandon, Sewickley, PA). Samples were then stained with Wright stain (EM Science, Gibbstown, NJ) and visualized by light microscopy. The number of macrophages was determined for a particular field of view (200 cells) in duplicate and expressed as mean ± SE.
Western Blot Analysis
To determine the protein expression and activation of MMP-12 whole lung extracts from saline- or bleomycin-treated WT and Pg−/− mice were homogenized in 2 ml of PBS containing 1% sodium dodecyl sulfate (SDS) and proteinase inhibitors (1 mmol/L phenylmethylsulfonyl fluoride, 10− 5 mol/L leupeptin, 1 mmol/L EDTA). Protein was extracted for 1 hour at room temperature, centrifuged, and the supernatant was used for total protein determination using the bicinchoninate (BCA, Pierce, Rockford, IL) method. SDS-polyacrylamide gel electrophoresis (PAGE) was performed using 80 μg total protein. Proteins were transferred onto an immobilon-P membrane (Millipore, Bedford, MA) and nonspecific protein binding was blocked in 5% powdered milk Tris-buffered saline (TBS) containing 0.05% Tween-20 (BLOTTO). A polyclonal antibody against recombinant mouse MMP-12 was kindly provided by Dr. Steve Shapiro (Washington University School of Medicine, St. Louis, MO). Membranes were incubated in anti-MMP-12 antibody (1:2000 in BLOTTO) at 4°C overnight. Membranes were washed in TBS-Tween, reblocked, then incubated with HRP-conjugated swine anti-rabbit IgG antibody (1:2000 in BLOTTO) and washed again. Finally, membranes were developed using the ECL Plus Kit (Amersham, Arlington Heights, IL).
Histology and Immunohistochemistry
To study the histological changes and alterations in the deposition of fibrin and collagen in situ during the acute and organizing stages after lung injury, right lungs from bleomycin- and saline-treated mice were examined at 7 and 14 days after treatment. Animals were anesthetized and perfused as described above. After excision of the left lung for other studies, such as collagen content, the right lung was instilled intrabronchially with Histochoice (Amresco, Solon, OH). The airway was then ligated, excised, and embedded in paraffin. Paraffin sections 7 μm thick were placed on slides coated with polyL-lysine (Sigma Chemical). Tissue sections were deparaffinized, rehydrated, and immersed in running water (for hematoxylin and eosin, Masson’s trichrome, and Prussian blue stains) or TBS/bovine serum albumin (BSA) or PBS/BSA (for immunohistochemistry).
Fibrin was identified by immunohistochemistry using a polyclonal antibody against mouse fibrin(ogen) (DAKO Corp., Carpinteria, CA). Histological sections were incubated with peroxidase-labeled rabbit anti-goat IgG (Dakopatts, Copenhagen, Denmark) and peroxidase activity was developed by incubation in 0.05 mol/L Tris-HCl, pH 7.0, containing 0.06% 3,3-diaminobenzidine and 0.1% H2O2. MMP-12 was localized in sections of lung using the same anti-MMP-12 antibody used for Western blots at 1:500 in PBS containing 0.1% BSA. Sections were incubated with biotinylated secondary antibody, followed by the ABC reagent from Vectastain ABC Kit (Vector Laboratories, Burlingame, CA). Peroxidase activity was detected as described above. Counterstaining was performed with hematoxylin.
Statistical Analysis
Values were expressed as means ± SE. Comparisons were made using Student’s t-test and analysis of variance when more than two values were compared. P values <0.05 were considered significant.
Results
Collagen Content of Bleomycin-Treated Lungs
Because collagen content has been used as a measure of the degree of fibrosis in the lung, 32 these measurements were made in the lungs of saline- or bleomycin-treated mice. Eitzman and others 26 have demonstrated that pulmonary fibrosis peaks 14 days after bleomycin treatment in mice of a similar genetic strain. Therefore, lungs were examined 14 days after treatment except for t-PA−/− mice who survived only 11 days post-drug treatment. After intratracheal administration of bleomycin, collagen levels increased significantly above saline controls in Pg−/−, Pg+/−, and t-PA+/− mice (150.06 ± 28.40 vs. 33.53 ± 6.00, P = 0.02; 147.02 ± 25.70 vs. 60.07 ± 1.6, P = 0.04; 152.70 ± 8.50 vs. 55.67 ± 8.20, P = 0.0005 μg hydroxyproline/left lung; Figure 1 ▶ ). In WT, u-PA−/−, and u-PAR−/− mice there was also an increase in collagen with values approaching significance (89.38 ± 19.20 vs. 47.97 ± 25.80, P = 0.24; 100.90 ± 15.14 vs. 63.53 ± 5.30, P = 0.12; 78.76 ± 11.60 vs. 50.70 ± 7.45, P = 0.18 μg hydroxyproline/left lung; Figure 1 ▶ ). Collagen levels observed in Pg−/− and t-PA−/− mice were higher than those observed in WT, u-PA−/−, and u-PAR−/− mice (150.06 ± 28.4 and 152.70 ± 8.50 μg hydroxyproline/left lung, respectively, vs. 89.38 ± 19.20 μg hydroxyproline/left lung for WT mice; Figure 1 ▶ ).
Figure 1.

Hydroxyproline content in lungs 14 days after saline (n = 3 for each genotype) or bleomycin treatment of WT (n = 4), Pg+/− (n = 5), Pg−/− (n = 5), u-PA−/− (n = 5), u-PAR−/− (n = 7), and t-PA−/− (n = 4) mice. Values are means ± SE of 4 to 7 mice, performed in duplicate.
Histology
To better understand the histological changes associated with this acute lung injury model, the right lungs from bleomycin- or saline-treated mice were collected 14 days after treatment. Lung tissue sections were stained with hematoxylin and eosin, Masson’s trichrome (for collagen identification), or Prussian blue (for identifying areas of chronic hemorrhage). The lungs of saline-treated mice appeared normal regardless of genotype or time point after treatment. Collagen was identified in these lungs only in areas surrounding large vessels and airways (Figure 2A) ▶ . At 14 days after bleomycin treatment, focal areas of fibrosis in the interstitium as well as proteinaceous exudate in the intra-alveolar spaces were observed in some areas of the lungs of WT mice (Figure 2B) ▶ . However, an unexpected hemorrhage phenotype was observed in the lungs of these mice at this time point (Figure 2C) ▶ . Associated with these areas of hemorrhage were hemosiderin-laden macrophages, as observed in adjacent tissue sections stained with Prussian blue, indicating chronic hemorrhage (Figure 2D) ▶ . In contrast, all bleomycin-treated Pg−/− mice examined (n = 7) showed extensive areas of fibrosis but no areas of hemorrhage (Figure 2E) ▶ . In these animals, abnormal collagen deposition was observed in the interstitium associated with the areas of fibrosis (Figure 2F) ▶ . Pg+/− mice showed a mixed phenotype of hemorrhage, observed in the WT mice, and collagen content and fibrotic lesions, found in the Pg−/− mice (Figure 2G) ▶ . Chronic hemorrhage observed in these mice was confirmed by Prussian blue staining of adjacent sections, which showed the presence of hemosiderin-laden macrophages in areas of hemorrhage (Figure 2H) ▶ .
Figure 2.

Photomicrographs of representative lungs from WT (n = 6; A−D), Pg−/− (n = 7; E and F), and Pg+/− (n = 7; G and H) mice 14 days after saline or bleomycin treatment. A−C and E−G were stained with Masson’s trichrome; D and H were stained with Prussian blue. A: Saline-treated WT lung showing a bronchiole (b) and artery (a) from the bronchioarterial bundle surrounded by normal deposition of collagen (arrow) and normal alveoli; original magnification, ×100. B: Bleomycin-treated WT lung showing focal areas of fibrosis (f) and proteinaceous exudate (arrows) in the alveolar spaces; original magnification, ×100. C: Another area in the lung in B, showing extensive intra-alveolar hemorrhage (RBCs) and fibrin deposition (arrow); original magnification, ×100. D: Adjacent section to the lung in C showing hemosiderin-laden macrophages (arrows) associated to the area of extensive alveolar hemorrhage; original magnification, ×100. E: Bleomycin-treated Pg−/− lung showing extensive fibrosis (f). No intra-alveolar hemorrhage was observed in the lungs of these animals; original magnification, ×100. F: Higher magnification of the lung in E, showing extensive deposition of collagen (arrows) associated with fibrotic areas (f); original magnification, ×400. G: Bleomycin-treated Pg+/− lung showing both areas of intra-alveolar hemorrhage (h) and fibrosis (f); original magnification, ×100. H: Adjacent section to the lung in G, showing hemosiderin-laden macrophages (arrows) associated with areas of extensive hemorrhage (h) but not with fibrotic areas (f); original magnification, ×100.
Bleomycin also induced extensive fibrosis in the lungs of u-PA−/− mice after 14 days of treatment, although collagen deposition, as visualized with Masson’s trichrome staining, did not appear as extensive as observed in the lungs of Pg−/− mice (Figure 3A) ▶ . Similar to Pg−/− mice, no hemorrhage was found in any of these animals. u-PAR−/− mice exhibited focal areas of fibrosis as observed in the WT mice (Figure 3B) ▶ . Similar to Pg−/− mice, none of these animals showed signs of chronic intra-alveolar hemorrhage as confirmed by the absence of positive cells in adjacent sections stained with Prussian blue (not shown). In contrast, t-PA−/− mice presented a mixed phenotype of hemorrhage found in the WT and Pg−/− animals and extensive fibrosis found in Pg−/− and u-PA−/− mice (Figure 3C) ▶ . The fibrotic lesions in all of the animals studied showed the typical thickening of the alveolar wall and the presence of abnormal deposition of collagen in the interstitium. The differences in the extent of abnormal collagen deposition observed in the different genotypes correlated with the accumulation of collagen in these mice as measured by hydroxyproline levels (Figure 1) ▶ . Interestingly, the hemorrhage phenotype observed in the WT, Pg−/−, and t-PA−/− mice correlated with the early deaths observed in these animals, Table 1 ▶ . Moreover, earlier death exhibited in the Pg+/− and t-PA−/− mice could also be due to their enhanced accumulation of fibrotic lesions compared to the WT mice. In that regard, early deaths of u-PA−/− mice could be the result of the extensive fibrosis observed in these animals as early as day 7 after bleomycin treatment. However, no early deaths were found in Pg−/− or u-PAR−/− mice.
Figure 3.

Photomicrographs of representative lungs (n = 6 for each genotype) from mice 14 days after bleomycin treatment stained with Masson’s trichrome. A: u-PA−/− lung showing extensive area of fibrosis (f); original magnification, ×100. B: u-PAR−/− lung showing focal areas of fibrosis (f) surrounded by normal alveoli; original magnification, ×100. C: t-PA−/− lung showing areas of fibrosis (f) surrounded by areas of extensive intra-alveolar hemorrhage (h); original magnification, ×100.
Table 1.
Early Deaths in Mice after Bleomycin Treatment
| Genotype | Average day of early death | % of animals that died early |
|---|---|---|
| WT | 8.7 | 33 |
| Pg+/− | 6 | 46 |
| u-PA−/− | 7.8 | 62 |
| t-PA−/− | 7 | 75 |
Leukocyte Levels in the Lungs of Bleomycin-Treated Mice
Studies using thioglycollate 14 as a model of inflammation have shown quantitative differences between WT and Pg−/− mice in macrophage levels in the peritoneum. Therefore, macrophage levels during the acute stage after bleomycin treatment of Pg−/−, u-PA−/−, u-PAR−/−, t-PA−/−, and WT mice were studied. Leukocytes were isolated from collagenase-digested whole lungs at days 0 (no treatment, baseline), 3, 5, 7, and 9 after saline or bleomycin treatment. This method yielded samples containing >95% leukocytes as confirmed by positive CD45 immunostaining (data not shown). The leukocytes isolated in this way represented those present in the alveolar spaces as well as in the interstitium of the lung at the time of collection.
No significant increase was observed in the number of macrophages present in the lungs of mice at 3 days after bleomycin treatment, although WT mice showed the highest levels of all of the genotypes studied (Table 2) ▶ . Macrophage levels at this time point were comparable to those found in the saline-treated counterparts of Pg−/−, u-PA−/−, and t-PA−/− mice, and may represent the alveolar macrophages already present in the lung (Table 2) ▶ . The peak in the macrophage levels in the lungs of WT mice occurred 5 days after bleomycin treatment (Table 2) ▶ . This was also the peak time for the accumulation of macrophages in the lungs of Pg−/− (4.86 ± 0.40 × 106) and u-PA−/− (5.62 ± 0.70 × 106) mice treated with bleomycin. However, their levels were considerably less than those found in WT mice (8.68 ± 0.60 × 106), P = 0.001 relative to Pg−/− and P = 0.04 relative to u-PA−/− (Table 2) ▶ . These findings are consistent with those observed in Pg−/− mice using thioglycollate- and biopolymer-induced peritoneal inflammation models. 14 In the present study, at day 7 post-treatment, there was a decrease in the number and percentage of macrophages found in the lungs of Pg−/−, u-PA−/−, and WT mice (Table 2) ▶ . The decrease in macrophage counts at this time point coincided with an increase in the percentage of lymphocytes found in the lungs of bleomycin-treated mice (data not shown). This was not the case for t-PA−/− mice treated with bleomycin, in which macrophage numbers continued to increase reaching levels comparable to those of WT mice at 9 days after bleomycin treatment (Table 2) ▶ . No increase in the macrophage levels in the lungs of u-PAR−/− mice treated with bleomycin was observed between 3 and 7 days after bleomycin treatment. However, a significant increase in macrophage number was observed at day 9 after bleomycin treatment compared to their saline controls (6.22 ± 0.50 vs. 2.73 ± 0.70, P = 0.05) although at this late time point the level did not reach the highest observed level in WT mice (Table 2) ▶ .
Table 2.
Total Lung Macrophage Levels Post-Bleomycin Treatment (×106, n = 6, mean ± SE)
| Genotype | Day 0* | Day 3 | Day 5 | Day 7 | Day 9 |
|---|---|---|---|---|---|
| WT | 2.87 ± 0.52 | 5.25 ± 0.90 | 8.68 ± 0.60 | 5.42 ± 0.70 | 5.45 ± 0.50 |
| Pg−/− | 2.47 ± 0.43 | 3.10 ± 0.30 | 4.86 ± 0.40 | 3.50 ± 0.60 | 4.68 ± 1.00 |
| u-PA−/− | 3.85 ± 0.30 | 2.30 ± 0.70 | 5.62 ± 1.00 | 5.25 ± 0.80 | 4.28 ± 0.60 |
| u-PAR−/− | 4.80 ± 0.60 | 3.87 ± 0.70 | 3.40 ± 0.40 | 3.95 ± 0.70 | 6.22 ± 0.50 |
| t-PA−/− | 3.81 ± 0.61 | 3.87 ± 0.40 | 5.15 ± 0.80 | 6.47 ± 0.50 | 7.48 ± 0.30 |
*Baseline macrophage levels, representing macrophages present in lungs of animals that did not receive any treatment.
Fibrin Deposition in Bleomycin-Treated Mice
Because one of the first events after acute lung injury is the extravasation of plasma proteins such as fibrinogen, 32 accumulation of fibrin in the intra-alveolar spaces and interstitium of the lung of bleomycin- or saline-treated mice was analyzed. Therefore, the extent and localization of fibrin(ogen) in the lungs of these mice were studied at 3, 7, and 14 days after treatment. No fibrin(ogen) deposits were observed in the lungs of WT animals treated with saline at any time point studied (data not shown). In contrast, as described earlier, 11,12 Pg−/− control mice showed spontaneous fibrin deposition in the lungs throughout the time points studied. Bleomycin-treated lungs of WT mice showed few areas of fibrin(ogen) deposits at 3 and 7 days after bleomycin treatment, which, for the most part, were resolved by day 14 (Figure 4A) ▶ . In sharp contrast, Pg−/− mice showed extensive fibrin (ogen) deposition as early as day 3, peaking at day 7, and continuing even at day 14 after bleomycin treatment (Figure 4, B and C) ▶ . The areas of fibrin(ogen) deposition in the Pg−/− mice were associated with regions of fibrosis (Figure 4C) ▶ . u-PA−/− mice also showed extensive areas of fibrin(ogen) deposition in the lung interstitium which were associated with areas of fibrosis (data not shown). Additionally, t-PA−/− mice also exhibited areas of fibrin(ogen) deposits, especially in the vasculature of the lung, where fibrin thrombi could be observed (data not shown).
Figure 4.
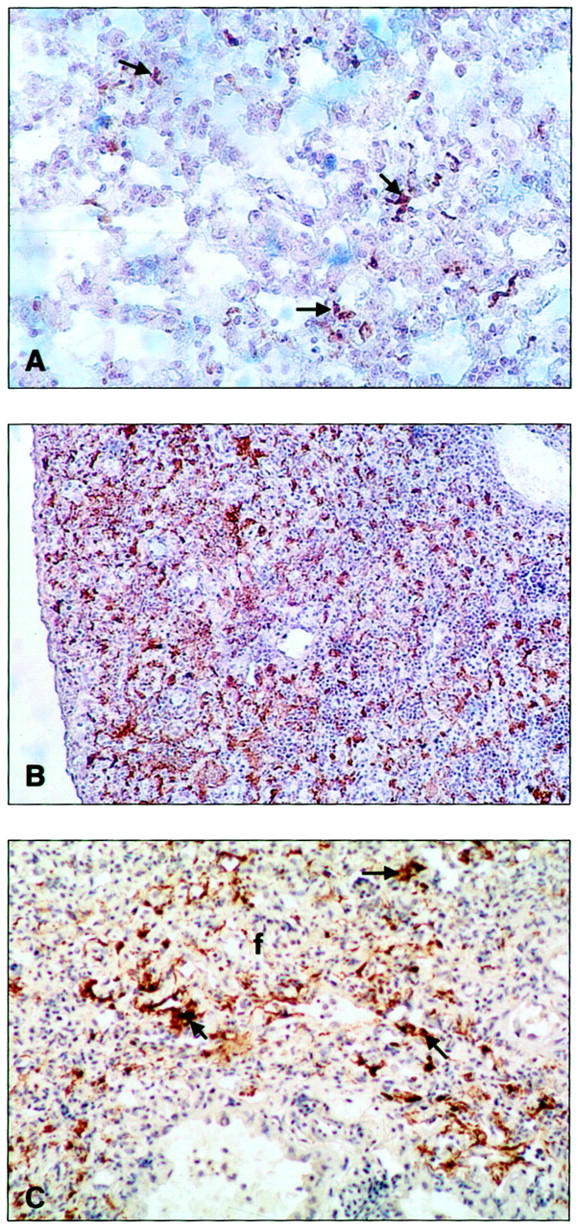
Photomicrographs of representative mice lungs (n = 3 for each genotype) treated with bleomycin and immunostained with an anti-fibrin (ogen) antibody. A: WT lung 14 days after treatment showing few areas of fibrin deposition (arrows); original magnification, ×100. B: Pg−/− lung 7 days after treatment showing extensive fibrin deposition (brown aggregates); original magnification, ×40. C: Pg−/− lung 14 days after treatment showing extensive fibrin deposition (arrows) associated with areas of fibrosis (f); original magnification, ×200.
MMP-12 Synthesis and Activation in the Lungs of Bleomycin-Treated Mice
Because Pm is a known activator of pro-MMPs 24,33 the expression, localization, and activation of MMPs in whole lung extracts of saline- or bleomycin-treated mice were analyzed. Of particular interest was MMP-12, also known as macrophage metalloelastase. 34 The pro-enzyme form of MMP-12 (54 kd) was present in whole lung extracts from both WT and Pg−/− mice as early as 5 days after treatment with bleomycin (Figure 5) ▶ . However, the active, 22-kd form of the enzyme was observed only in WT animals at this time point (Figure 5) ▶ . Neither the proenzyme nor the active form was observed in either genotype by day 14 after bleomycin treatment (Figure 5) ▶ . Immunohistochemical studies using the same antibody to localize MMP-12 showed that the enzyme is present in the alveolar wall of WT mice associated with areas of hemorrhage in these animals (Figure 6A) ▶ . Specifically, MMP-12 seemed to be localized in the capillary component of the alveolar wall (Figure 6B) ▶ . The specific MMP-12-positive cell type in the capillary of the alveolar wall remains to be determined. However, macrophages are potential candidates, because they are the only cell type known to produce this enzyme. 34 In contrast, MMP-12 was not present in areas of fibrosis in the WT or Pg−/− mice (data not shown).
Figure 5.

Western blot showing MMP-12 expression in representative whole lung extracts from WT (n = 3/time point) and Pg−/− (n = 3/time point) mice 5 and 14 days after bleomycin treatment. Relative molecular weights are shown on the left.
Figure 6.

Photomicrographs of representative sections of lungs from bleomycin-treated WT mice (n = 3) 14 days after treatment immunostained with an anti-MMP-12 antibody. A: Deposition of MMP-12 is observed in the alveolar walls (arrows) associated with areas of extensive hemorrhage (h); original magnification, ×100. B: MMP-12 is concentrated in the capillaries (arrows) in the wall of the alveoli (a); original magnification, ×400.
Discussion
Pulmonary fibrosis, the end stage of acute lung injury, is characterized by impaired fibrinolysis. 35-38 However, the role of components of the fibrinolytic system in lung injury and repair is not well understood.
Several animal models have been developed to study the mechanisms of lung repair after acute and chronic inflammatory injury. Cytotoxic drugs are the most widely used to induce pulmonary fibrosis and among these the antibiotic bleomycin has been the most extensively studied. 39-41 Intratracheal administration of bleomycin in mice and rats causes both the inflammatory and fibrotic responses observed in patients with pulmonary fibrosis. 42 In this study, bleomycin was used to study the role of the fibrinolytic system in lung repair after injury and the development of pulmonary fibrosis.
The first phase of the lung response to injury is characterized by severe inflammation. Initial recruitment of inflammatory cells into the alveolar spaces is brought about by chemoattractant agents derived from the injured lung tissue. Injury causes increased permeability of pulmonary epithelium and endothelium, resulting in extravasation of plasma proteins and, ultimately, formation of fibrin clots in the alveolar lumen. 35 Pertinent to this study, Pm-catalyzed fibrinolysis generates a series of high molecular weight fibrin degradation products (FDPs) that can serve as chemoattractants to leukocytes. 21,22 Therefore, the inflammatory response could be augmented with the recruitment of new inflammatory cells by these potent FDPs. Peak levels of macrophages occurred at day 5 for WT, u-PA−/−, and Pg−/− mice, although levels were significantly diminished in u-PA−/− and Pg−/− mice relative to WT mice. A delayed, but significant, increase in macrophage levels was observed in t-PA−/− mice, whereas the delayed response in u-PAR−/− mice never reached the peak levels observed in t-PA−/− and WT mice. The low macrophage levels in u-PAR−/− mice during the early stages of repair may be due to a necessity for an intact uPA/uPAR system for macrophage motility. 43 On the other hand, low macrophage levels in u-PAR−/− mice could be related to a u-PA independent function for this protein. It has been demonstrated that a lack of u-PAR results in compromised β2-integrin activation, which has a deleterious effect on leukocyte-endothelial cell adhesion and resultant transendothelial migration. 44 Additionally, ICAM-1, a ligand for β2-integrins, has been identified in bleomycin-injured lungs in rats, 45 and, therefore, u-PAR/β2-integrin interactions may play a significant role in inflammatory cell migration in response to bleomycin-induced lung injury. The eventual increase in macrophage levels in u-PAR−/− mice may be the result of activation of other adhesion receptors independent of the u-PAR/β2-integrin system during the later stages of the inflammatory response. These observations implicate u-PA-mediated activation of plasminogen in contributing to the enhanced influx of macrophages into the lung after acute injury.
In the current studies, areas of alveolar hemorrhage were observed in WT and t-PA−/− mice but not in u-PA−/−, u-PAR−/−, and Pg−/− mice. The hemorrhagic areas were characterized by the presence of hemosiderin-laden macrophages, indicative of chronic hemorrhage in the lungs of these mice. Similar phenotypes have been observed in studies of myocardial infarction with mice deficient for components of the fibrinolytic system. 16 In these latter studies, Pg−/− and u-PA−/− mice did not show the ventricular hemorrhage that was present in WT counterparts, as well as in t-PA−/− mice. Taken together, these investigations support the involvement of the u-PA/Pg system in mediating the hemorrhagic phenotype observed in these two animal models (Figure 7) ▶ . This is underscored by the clinical observation that intra-alveolar hemorrhage has been shown to occur during the alveolar edema phase of patients with acute respiratory distress syndrome. 46
Figure 7.

Proposed model for the role of the fibrinolytic system in acute lung injury.
Because macrophages are found to be associated with areas of hemorrhage and are known to produce matrix metalloproteases (MMP), an analysis of MMP expression and activation in bleomycin-injured lungs of WT and Pg−/− mice was made. Clinically, MMPs have been shown to be present in the lungs of patients with diffuse alveolar damage and idiopathic pulmonary fibrosis. 47 MMP-2 (gelatinase A) and MMP-12 (macrophage metalloelastase) have been shown to increase significantly during the acute phase of bleomycin-induced pulmonary fibrosis in mice and rats. 48,49 Additionally, Pm has been shown to participate in the first step in the activation of matrix metalloproteases. 24 Specifically, MMP-12 has been shown to be activated by Pm. 16 MMP-12 is produced by activated macrophages in mice and is capable of degrading elastin and collagen type IV, both components of the basement membrane. 50 The present studies demonstrate that proMMP-12 is converted to its active form in lung tissue from WT mice, and not in Pg−/− mice. This activation appears to occur early after bleomycin treatment, during the peak time of macrophage levels in the lungs of WT mice, and is associated only with areas of hemorrhage. Other studies using MMP-12−/− mice have revealed that macrophages from these mice have a diminished capacity to degrade extracellular matrix protein, rendering them unable to penetrate basement membranes. 50 Previous studies using t-PA−/−, u-PA−/−, and Pg−/− mice in a myocardial infarction model revealed that the activation of various MMPs, including MMP-12, in macrophages was dependent on an intact u-PA/Pg system. 16 It was proposed that hemorrhage was caused by the activation of matrix metalloproteinases due to u-PA-mediated activation of Pg in macrophages present at the lesion site. Additionally, clinical studies have shown that the secretion of MMP-12 by macrophages is associated with damaged areas in the lungs. 51 The current studies demonstrate that mice with an intact u-PA/Pg system respond to acute oxidative lung injury by increasing macrophage levels with resultant Pm-mediated activation of matrix metalloproteases and eventual basement membrane denudation and leaky capillary formation (Figure 7) ▶ .
One of the prominent phenotypes in bleomycin-induced pulmonary fibrosis is extensive abnormal collagen deposition in the interstitium. It was previously shown that overexpression of PAI-1 resulted in increased deposition of collagen in the lungs of bleomycin treated mice, whereas PAI-1−/− mice had levels comparable to those of PBS-treated control mice. 26 This suggests that Pm activity plays a key role in the regulation of collagen accumulation in the lungs after acute lung injury. Histologically, this study demonstrated enhanced fibrotic lesion development in bleomycin-treated Pg−/−, Pg+/−, t-PA−/−, and u-PA−/− mice relative to WT and u-PAR−/− mice. Additionally, hydroxyproline levels, a measure of collagen content, in the lungs of these mice were all enhanced at 14 days after drug treatment. Pg−/− mice demonstrated 1.7-fold levels of hydroxyproline over that observed in WT mice. Plasminogen levels in the blood of Pg+/− mice have been shown to be approximately one-half of that observed in WT mice (35 ± 2 μg/ml vs. 84 ± 8 μg/ml), 11 and even at those levels a dramatic effect on collagen deposition was observed in Pg+/− mice which was similar to that observed for Pg−/− mice. The levels of hydroxyproline found in the u-PA−/− mice were similar to those observed in WT animals at 14 days, even though the lungs appeared more fibrotic on histological examination. These findings contrast with those of Lardot and others 52 using a crystalline silica model of alveolitis that progressed to fibrosis in u-PA−/− mice. In that study an increase in hydroxyproline levels in the u-PA−/− mice was found compared to their WT counterparts. This correlated with the development of fibrotic lesions in these mice 30 days after treatment. However, it was also noted that the fibrinolytic response varied depending on the agent used to promote alveolitis. This differential response may be related to selective tissue responses to different agents and may explain the discrepancy observed in hydroxyproline levels in u-PA−/− mice from these two studies. It is also well known that collagen deposition is a balance between processes of synthesis and degradation that is regulated by a complex network of cytokines, ie, transforming growth factor-β (TGF-β), interleukin-1α (IL-1α), and tumor necrosis factor-α (TNF-α). 32 In the case of u-PA−/− mice, the absence of u-PA could result in altered expression of important mediators of collagen synthesis independent of plasmin activity. For example, studies have indicated that the release of IL-1 by macrophages is inhibited by PAI-I. 53 IL-1α, which is known to increase fibroblast collagen synthesis, has been shown to be increased in bleomycin-induced pulmonary fibrosis. 54 Additionally, studies have shown that u-PA amplifies TNF-α secretion of THP-1 mononuclear phagocytes independent of plasmin activity. 55 Therefore, u-PA could potentially play diverse roles in the repair process after acute lung injury.
Bleomycin-induced lung injury has been shown to result in the accumulation of fibrin in the alveolar and interstitial spaces of the lung. 56 These fibrin deposits colocalized with the abnormal collagen deposited in the lung interstitium, which was associated with fibroproliferative lesions. Additionally, fibrin has been shown to serve as a scaffolding molecule for the migration and proliferation of fibroblasts. 57,58 Therefore, the inability of Pg- and Pg activator-deficient mice to clear fibrin deposited into the alveolar spaces and interstitium would potentially result in the proliferation and migration of fibroblasts into the space once occupied by the alveoli, leading to the development of pulmonary fibrosis and end stage honeycomb appearance of the lung (Figure 7) ▶ .
In conclusion, the studies reported here have shown that the repair process after acute oxidative lung injury depends on the balance between deposition and breakdown of matrix molecules such as fibrin and collagen. As a result, differences in the expression of components of the fibrinolytic system alter this balance by locally affecting fibrinolysis, the inflammatory response, and activation of matrix metalloproteinases at the site of lung injury.
Acknowledgments
We thank Drs. E. Plow and F. J. Castellino for their thoughtful review and comments concerning this manuscript, David Hellard for technical assistance with mice, the Division of Pathology and Laboratory Medicine of the Cleveland Clinic Foundation for assistance in the histological studies, and Suzanne Turner of the Cardiology Graphics Department of the Cleveland Clinic Foundation for assisting in the preparation of this manuscript.
Footnotes
Address reprint requests to Victoria A. Ploplis, Ph.D., Dept. of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556. E-mail: ploplis.3@nd.edu.
Supported by National Institutes of Health (National Heart, Lung and Blood Institute) RO1 grant HL63682–01 (to V. A. P.) and postdoctoral fellowship no. 9804562 from the American Heart Association, Midwest Affiliate (to C. M. S.).
References
- 1.National Center for Health Statistics: Births and deaths: United States. Monthly Vital Statistics Report. Atlanta, Centers for Disease Control, 1997, vol. 46, no. 1, suppl. 2 [PubMed]
- 2.Katzenstein A-L, Askin FB: Idiopathic interstitial pneumonia/idiopathic pulmonary fibrosis. Surgical Pathology of Non-Neoplastic Lung Disease. 1990, :pp 58-96 W.B. Saunders/Harcourt Brace Jovanovich, Inc., Philadelphia [Google Scholar]
- 3.Katzenstein A-L, Askin FB: Acute lung injury patterns: diffuse alveolar damage, acute interstial pneumonia, bronchiolitis obliterans-organizing pneumonia. Surgical Pathology of Non-Neoplastic Lung Disease. 1990, :pp 9-57 W. B. Saunders/Harcourt Brace Jovanovich, Inc., Philadelphia [Google Scholar]
- 4.Crouch E: Pathology of pulmonary fibrosis. Am J Physiol 1990, 259:L159-L184 [DOI] [PubMed] [Google Scholar]
- 5.Tomashefski JF, Jr: Pathology of Pulmonary Disease. 1994:pp 125-138 J. B. Lippincott Company, Edited by MJ Saldana. Philadelphia
- 6.Dail DH: Metabolic and other disorders. Dail DH Hammar SP eds. Pulmonary Pathology. 1988, :pp 543-548 Springer-Verlag, Inc., New York [Google Scholar]
- 7.Snider GL: Interstitial pulmonary fibrosis: which cell is the culprit? Am Rev Resp Dis 1983, 127:535-539 [DOI] [PubMed] [Google Scholar]
- 8.Hammar SP: Idiopathic interstitial fibrosis. Dail DH Hammar SP eds. Pulmonary Pathology. 1988, :pp 483-510 Springer-Verlag, Inc., New York [Google Scholar]
- 9.Kuhn III C: Pathology. Pulmonary Fibrosis. Edited by SH Phan, RS Thrall. New York, Marcel Dekker, Inc., 1995, pp 59–83
- 10.Collen D: On the regulation and control of fibrinolysis. Thromb Haemost 1980, 43:77-89 [PubMed] [Google Scholar]
- 11.Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, Collen D: Effects of disruption of the plasminogen gene in mice on thrombosis, growth, and health. Circulation 1995, 92:2585-2593 [DOI] [PubMed] [Google Scholar]
- 12.Bugge TH, Flick MJ, Daugherty CC, Degen JL: Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev 1995, 9:794-807 [DOI] [PubMed] [Google Scholar]
- 13.Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K: Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med 1996, 2:287-292 [DOI] [PubMed] [Google Scholar]
- 14.Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF: Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood 1998, 91:2005-2009 [PubMed] [Google Scholar]
- 15.Ny A, Nordström L, Carmeliet P, Ny T: Studies of mice lacking plasminogen activator gene function suggest that plasmin production prior to ovulation exceeds the amount needed for optimal ovulation efficiency. Eur J Biochem 1997, 244:487-493 [DOI] [PubMed] [Google Scholar]
- 16.Carmeliet P, Moons L, Lijnen R, Baes M, Lemaître V, Tipping P, Drew A, Eeckhout Y, Shapiro S, Lupu F, Collen D: Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet 1997, 17:439-444 [DOI] [PubMed] [Google Scholar]
- 17.Bugge TH, Kombrinck KW, Xiao Q, Holmbäck K, Daugherty CC, Witte DP, Degen JL: Growth and dissemination of Lewis lung carcinoma in plasminogen-deficient mice. Blood 1997, 90:4522-4531 [PubMed] [Google Scholar]
- 18.Sabapathy KT, Pepper MS, Kiefer F, Möhle-Steinlein U, Tacchini-Cottier F, Fetka I, Breier G, Risau W, Carmeliet P, Montesano R, Wagner EF: Polyoma middle T-induced vascular tumor formation: the role of the plasminogen activator/plasmin system. J Cell Biol 1997, 137:953-963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan R: Physiological consequences of loss of plasminogen activation gene function in mice. Nature 1994, 368:419-424 [DOI] [PubMed] [Google Scholar]
- 20.Lijnen HR, Carmeliet P, Bouche A, Ploplis VA, Plow EF, Collen D: Restoration of thrombolytic potential in plasminogen-deficient mice by bolus administration of plasminogen. Blood 1996, 88:870-876 [PubMed] [Google Scholar]
- 21.Richardson DL, Pepper DS, Kay AB: Chemotaxis for human monocytes by fibrinogen-derived peptides. Br J Haemotol 1976, 32:507-513 [DOI] [PubMed] [Google Scholar]
- 22.Leavell KJ, Peterson MW, Gross TJ: The role of fibrin degradation products in neutrophil recruitment to the lung. Am J Resp Cell Mol Biol 1996, 14:53-60 [DOI] [PubMed] [Google Scholar]
- 23.Syrovets T, Tippler B, Ricks M, Simmet T: Plasmin is a potent and specific chemoattractant for human peripheral monocytes acting via a cyclic guanosine monophosphate-dependent pathway. Blood 1997, 89:4574-4583 [PubMed] [Google Scholar]
- 24.Nagase H: Activation mechanisms of matrix metalloproteinases. Biol Chem 1997, 378:151-160 [PubMed] [Google Scholar]
- 25.Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA: Matrix metalloproteinases: a review. Crit Rev Oral Biol Med 1993, 4:197-250 [DOI] [PubMed] [Google Scholar]
- 26.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH: Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest 1996, 97:232-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hart DA, Whidden P, Green F, Henkin J, Woods DE: Partial reversal of established bleomycin-induced pulmonary fibrosis by rh-urokinase in a rat model. Clin Invest Med 1994, 17:69-76 [PubMed] [Google Scholar]
- 28.Sisson TH, Hattori N, Xu Y, Simon RH: Treatment of bleomycin-induced pulmonary fibrosis by transfer of urokinase-type plasminogen activator genes. Hum Gene Ther 1999, 10:2315-2323 [DOI] [PubMed] [Google Scholar]
- 29.Dewerchin M, Van Nuffelen A, Wallays G, Bouche A, Moons L, Carmeliet P, Mulligan RC, Collen D: Generation and characterization of urokinase receptor-deficient mice. J Clin Invest 1996, 97:870-878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bugge TH, Suh TT, Flick MJ, Daugherty CC, Romer J, Solberg H, Ellis V, Dano K, Degen JL: The receptor for urokinase-type plasminogen activator is not essential for mouse development or fertility. J Biol Chem 1995, 270:16886-16894 [DOI] [PubMed] [Google Scholar]
- 31.McCoy RD, Davidson BL, Rossler BJ, Huffnagle GB, Janich SL, Laing TJ, Simon RH: Pulmonary inflammation induced by incomplete or inactivated adenoviral particles. Hum Gene Ther 1995, 6:1553-1560 [DOI] [PubMed] [Google Scholar]
- 32.McAnulty RJ, Laurent GJ: Collagen and its regulation in pulmonary fibrosis. Phan SH Thrall RS eds. Pulmonary Fibrosis. 1995, :pp 135-171 Marcel Dekker, Inc., New York [Google Scholar]
- 33.Nagase N, Enghild JJ, Suzuki K, Salvesen G: Stepwise activation of the precursor of matrix metalloproteinase 3 (stromelysin) by proteinases and (4-aminophenyl)mercurin acetate. Biochemistry 1990, 29:5783-5789 [DOI] [PubMed] [Google Scholar]
- 34.Shapiro SD, Griffin GL, Gilbert DJ, Jenkins NA, Copeland NG, Welgus HG, Senior RM, Ley TJ: Molecular cloning, chromosomal localization, and bacterial expression of a murine macrophage metalloelastase. J Biol Chem 1992, 267:4664-4671 [PubMed] [Google Scholar]
- 35.Idell S: Coagulation, fibrinolysis and fibrin deposition in lung injury and repair. Phan SH Thrall RS eds. Pulmonary Fibrosis. 1995, :pp 743-777 Marcel Dekker, Inc., New York [Google Scholar]
- 36.Idell S, James KK, Gilles C, Fair DS, Thrall RS: Abnormalities of pathways of fibrin turnover in lung lavage of rats with oleic acid and bleomycin-induced lung injury support alveolar fibrin deposition. Am J Pathol 1989, 135:387-399 [PMC free article] [PubMed] [Google Scholar]
- 37.Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, McLarty J, Fair DS: Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest 1989, 84:695-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Idell S, Peters J, James KK, Fair DS, Coalson JJ: Local abnormalities of coagulation and fibrinolytic pathways that promote alveolar fibrin deposition in the lungs of baboons with diffuse alveolar damage. J Clin Invest 1989, 84:181-193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myers JL: Surgical Pathology of Non-Neoplastic Lung Disease. Edited by A-L Katzenstein, FB Askin. Philadelphia, W.B. Saunders/Harcourt Brace Jovanovich, Inc., 1990, pp 97–127
- 40.Thrall RS, Scaliso PJ: Bleomycin. Phan SH Thrall RS eds. Pulmonary Fibrosis. 1995, :pp 231-292 Marcel Dekker, Inc., New York [Google Scholar]
- 41.Phan SH, Thrall RS, Williams C: Bleomycin-induced pulmonary fibrosis: effects of steroid on lung collagen metabolism. Am Rev Respir Dis 1981, 124:428-434 [DOI] [PubMed] [Google Scholar]
- 42.Adamson IY, Bowden DH: The pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Pathol 1974, 77:185-197 [PMC free article] [PubMed] [Google Scholar]
- 43.Gyetko MR, Chen G-H, McDonald RA, Goodman R, Huffnagle GB, Wilkinson CC, Fuller JA, Toews GB: Urokinase is required for the pulmonary inflammatory response to Cryptococcus neoformans. J Clin Invest 1996, 97:1818-1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT: Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med 1998, 188:1029-1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiner RE, Sasso DE, Gionfriddo MA, Syrber SI, Smilowitz HM, Vento J, Thrall RS: Early detection of bleomycin-induced lung injury in rat using indium-111-labeled antibody directed against intercellular adhesion molecule-1. J Nucl Med 1998, 39:723-728 [PubMed] [Google Scholar]
- 46.Bloor CM: Cardiogenic pulmonary edema. Saldana MJ eds. Pathology of Pulmonary Disease. 1994, :pp 117-123 J.B. Lippincott Company, Philadelphia [Google Scholar]
- 47.Hayashi T, Stetler-Stevenson WG, Fleming MV, Fishback N, Koss MN, Liotta LA, Ferrans VJ, Travis WD: Immunochemical study of metalloproteinases and their tissue inhibitors in the lungs of patients with diffuse alveolar damage and idiopathic pulmonary fibrosis. Am J Pathol 1996, 149:1241-1256 [PMC free article] [PubMed] [Google Scholar]
- 48.Swiderski RE, Dencoff JE, Florchinger CS, Shapiro SD, Hunninghake GW: Differential expression of extracellular matrix remodeling genes in a murine model of bleomycin-induced pulmonary fibrosis. Am J Pathol 1998, 152:821-828 [PMC free article] [PubMed] [Google Scholar]
- 49.Koslowski R, Knoch KP, Wenzel KW: Proteinases and proteinase inhibitors during the development of pulmonary fibrosis in rats. Clin Chem Acta 1998, 271:45-56 [DOI] [PubMed] [Google Scholar]
- 50.Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD: Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci USA 1996, 93:3942-3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shapiro SD: Elastolytic metalloproteinases produced by human mononuclear phagocytes: potential roles in destructive lung disease. Am J Respir Crit Care Med 1994, 150:S160-S164 [DOI] [PubMed] [Google Scholar]
- 52.Lardot CG, Huax FA, Broeckaert FR, Declerck PJ, Delos M, Fubini B, Lison DF: Role of urokinase in the fibrogenic response of the lung to mineral particles. Am J Resp Crit Care 1998, 157:617-628 [DOI] [PubMed] [Google Scholar]
- 53.Robson SC, Saunders R, Kirsch RE: Monocyte-macrophage release of IL-1 is inhibited by type-1 plasminogen activator inhibitors. J Clin Lab Immunol 1990, 33:83-90 [PubMed] [Google Scholar]
- 54.Asada K, Ogushi F, Tani K, Maniwa K, Ichikawa W, Endo T, Haung L, Nishioka Y, Sone S, Ogura T: The role of cell-associated interleukin-1 in bleomycin-induced pulmonary fibrosis. Tokushima J Exp Med 1996, 43:79-86 [PubMed] [Google Scholar]
- 55.Sitrin RG, Shollenberger SB, Strieter RM, Gyetko MR: Endogenously produced urokinase amplifies tumor necrosis factor-alpha secretion by THP-1 mononuclear phagocytes. J Leukoc Biol 1996, 59:302-311 [DOI] [PubMed] [Google Scholar]
- 56.Olman MA, Simmons WL, Pollman DJ, Loftis AY, Bini A, Miller EJ, Fuller GD, Rivera KE: Polymerization of fibrinogen in murine bleomycin-induced lung injury. Am J Physiol 1996, 271:L519-L526 [DOI] [PubMed] [Google Scholar]
- 57.Brown LF, Lanir N, McDonagh J, Tognazzi K, Dvorak AM: Fibroblast migration in fibrin gel matrices. Am J Pathol 1993, 142:273-283 [PMC free article] [PubMed] [Google Scholar]
- 58.Gray AJ, Bishop JE, Reeves JT, Laurent GJ: Aα and Bβ chains of fibrinogen stimulate proliferation of human fibroblasts. J Cell Sci 1993, 104:409-413 [DOI] [PubMed] [Google Scholar]