

Abstract
The Amelogenesis Imperfecta (AI) are a group of clinically and genetically heterogeneous disorders that affect enamel formation. To date, mutations in 4 genes have been reported in various types of AI. Mutations in the genes encoding the 2 enamel proteases, matrix metalloproteinase 20 (MMP20) and kallikrein 4 (KLK4), have each been reported in a single family segregating autosomal-recessive hypomaturation AI. To determine the frequency of mutations in these genes, we analyzed 15 Turkish probands with autosomal-recessive hypo-maturation AI for MMP20 and KLK4 gene mutations. No KLK4 mutations were found. A novel MMP20 mutation (g.16250T>A) was found in one family. This missense mutation changed the conserved active-site His226 residue of the zinc catalytic domain to Gln (p.H226Q). Zymogram analysis demonstrated that this missense mutation abolished MMP20 proteolytic activity. No MMP20 mutations were found in the remaining 14 probands, underscoring the genetic heterogeneity of hypomaturation AI.
Keywords: amelogenesis imperfecta, MMP20, proteolytic activity, hypomaturation, enamelysin
INTRODUCTION
The Amelogenesis Imperfecta (AI) are a group of clinically and genetically heterogeneous disorders that affect enamel development, resulting in abnormalities of amount, composition, and/or structure of enamel. Although enamel appearance (hypoplastic, hypomature, or hypocalcified) and mode of inheritance (autosomal-dominant, autosomal-recessive, or X-linked) have historically been used to classify AI into 14 subtypes (Witkop, 1988), current classifications of AI are problematic (Hart et al., 2002a; Aldred et al., 2003; Nusier et al., 2004). Development of a more robust nosology for AI will require a greater understanding of how enamel development is altered. This will necessitate identification of the genetic etiology of various forms of AI, and an understanding, at the molecular level, of how mutations of these genes affect enamel development.
To date, mutations in 4 genes—AMELX, ENAM, KLK4, and MMP20— have been reported to cause AI in humans. Most cases of X-linked AI result from mutations in the AMELX gene that codes for amelogenin, the most abundant protein in enamel (Hart et al., 2002a,b; Wright et al., 2003; Kim et al., 2004). Depending on the specific mutation, the phenotype associated with AMELX mutations can range from smooth hypoplastic to hypomineralized/hypomaturation (Hart et al., 2002b; Wright et al., 2003; Kim et al., 2004). ENAM mutations are etiologic for some autosomal forms of hypoplastic AI, including both dominantly and recessively inherited forms (Rajpar et al., 2001; Kida et al., 2002; Mardh et al., 2002; Hart PS et al., 2003; Hart TC et al., 2003; Kim et al., 2005b). Only one mutation each has been reported in genes encoding the enamel proteinases, matrix metalloproteinase 20 (MMP20) and kallikrein 4 (KLK4). These both result in a hypomaturation AI phenotype that is transmitted as an autosomal-recessive trait (Hart et al., 2004; Kim et al., 2005a).
In this study, members of 15 Turkish families identified through ascertainment of probands with hypomaturation AI segregating as an autosomal-recessive trait were evaluated for mutations in the MMP20 and KLK4 genes.
MATERIALS & METHODS
Pedigrees and Diagnosis
Individuals affected with hypomaturation AI were identified in the dental clinics at the School of Dentistry, Istanbul University. Informed consent was obtained according to the Declaration of Helsinki. Available family members received an oral examination, including dental radiographs, to determine if they had AI. Fifteen families were identified with hypomaturation AI, characterized by opaque, whitish, yellowish to brownish enamel with radiographically evident decreased contrast between dentin and enamel. The thickness of the enamel was not affected, but because the enamel was often softer than normal, it could be abraded on the incisal/occlusal surfaces. Eleven of the 15 families reported a history of consanguinity. Complete medical histories were reviewed to rule out syndromic disorders.
Mutation Analysis
Genomic DNA was isolated from whole blood with the QIAamp DNA Blood Maxi Kit (Qiagen Inc., Valencia, CA, USA). The oligonucleotide primers and conditions used to amplify and sequence all exons, including introns/exon splice sites for KLK4 and MMP20 amplification, were described previously (Hart et al., 2004; Kim et al., 2005a). Amplification products were sequenced on an ABI 3100 automated DNA sequencer. Sequence data were compared with reference sequences (MMP20: NT_033899) (KLK4: AF228497) using the Sequencher program (Gene Codes Corporation, Ann Arbor, MI, USA). One hundred healthy Turkish control individuals were also sequenced for MMP20.
Protein Expression
The full-length human MMP20 cDNA containing open reading frame (ORF) was generated by PCR, with MMP20 specific primers: 5′-CTA CTG TGA GGG GAT GAA GG-3′ (SS) and 5′-TTT CTA TTT AGC AAC CAA TCC-3′ (AS), AccuPrime™ Pfx polymerase (Invitrogen, Carlsbad, CA, USA), and human tooth cDNA library as a template. The PCR product was cloned into the TA vector (Invitrogen), sequence-verified, and subcloned into the pcDNA3.1 mammalian expression vector (Invitrogen). The mutant MMP20 cDNA (g.16250T>A) was generated with primers (5′-GTT TAC CGT TGC TGC TCA AGA ATT TGG CCA TGC C-3′ [SS] and 5′-GGC TAG GCC AAA TTC TTG AGC AGC AAC GGT AAA C-3′ [AS], where bold represents the T>A mutated sequence) and the QuikChange Site-Directed Mutagenesis kit (Stratagene Inc., La Jolla, CA, USA), according to the manufacturer's instructions. Clones were sequence-verified. The wild-type and mutant MMP20 cDNAs in the pcDNA3.1 mammalian expression vector were transfected into HEK293 cells by means of the lipofectamine plus transfection kit (Invitrogen). HEK293 cells transiently transfected with wild-type and mutant human MMP20 expression constructs were cultured in Dulbecco's Modified Eagle Media (DMEM), containing antibiotics without serum, for 48 hrs, to express human MMP20 proteins. The conditioned media were harvested and concentrated by ultrafiltration through an Amicon YM10 membrane (Millipore Inc., Bedford, MA, USA) for the Western blot and zymogram analyses. The expression vector pCDNA3.1 alone was transfected into HEK293 cells to serve as a negative control.
Western Blotting
The concentrated media were mixed with 4x NuPAGE SDS sample loading buffer (Invitrogen) containing β-mercaptoethanol, boiled for 5 min, and run in 12% NuPAGE Bis-Tris gel with MOPS-SDS running buffer (Invitrogen). Following electrophoresis, the gel was trans-blotted onto a PVDF membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and analyzed by Western blotting with an MMP20-specific polyclonal antibody (provided by Dr. James Simmer), at 1:1000 dilution, and HRP-conjugated anti-chicken IgG antibody (Sigma-Aldrich Co., St. Louis, MO, USA) at 1:3000 dilution.
Zymography
Zymograms were performed on 4-16% SDS-polyacrylamide gradient gels containing casein. The concentrated media were mixed with 2 vol of zymogram sample loading buffer (Bio-Rad Laboratories) and incubated at 37°C for 10 min. Protein samples were run on a zymogram gel at a constant current of 20 mA. Following electrophoresis, the gel was washed (x3) for 15 min in 50 mL of 2.5% Triton X-100 solution (2.5% Triton X-100 in 50 mM Tris-HCl buffer [pH 7.4]). Gels were incubated at 37°C for 2 days in 50 mM Tris buffer (pH 7.4) containing 5 mM CaCl2 and 1 mM MgCl2.
RESULTS
Clinical Findings
Probands from 15 families were identified with hypomaturation AI. Examination of the probands' first-degree relatives (n = 65) identified hypomaturation AI in five siblings and one parent. The pedigree of one family with a mutation is shown in Fig. 1. The proband from this family had hypomaturation AI and an anterior open bite (Fig. 2). Caries lesions were evident on the maxillary permanent central incisors as well as in primary and permanent molars. His affected sibling also had hypomaturation AI, an anterior open bite, and caries on primary and permanent posterior teeth (Fig. 3).
Figure 1.
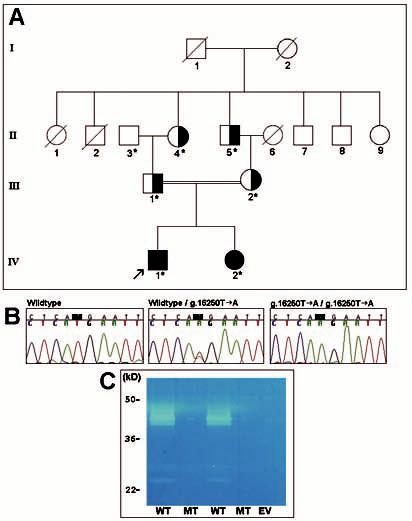
Pedigree, mutation, and biochemical analyses of family segregating autosomal-recessive hypomaturation AI. (A) Pedigree of family. Individuals from whom a blood sample was obtained are shown by an asterisk. Individuals clinically affected with hypomaturation AI are indicated by fully blackened symbols. These individuals are homozygous for the MMP20 g.16250T>A mutation. Carriers of the MMP20 g.16250T>A mutation are shown by partially blackened symbols. The enamel phenotype of all carriers is normal. (B) Mutational analysis of MMP20. The left panel shows the wild-type sequence for part of MMP20 exon 5 (from II-3). The middle panel shows the sequence from the same region of exon 5 from a carrier (III-1), showing the heterozygous missense mutation (g.16250T>A), which results in a p.His226Gln substitution. The right panel shows the sequence from the proband (IV-1), showing the homozygous change. (C) Enzymography of recombinant human wild-type and mutant MMP20. Recombinant human wild-type and mutant MMP20 cDNA were transiently transfected into the human kidney 293 cell line. The culture-conditioned media were collected, concentrated, and run on 4-16% SDS-polyacrylamide gradient gels containing casein. Following electrophoresis and washing, the gel was incubated at 37°C for 2 days in 50 mM Tris buffer (pH 7.4) containing 5 mM CaCl2 and 1 mM MgCl2. Lanes marked by WT indicate recombinant human wild-type MMP20, lanes marked by MT indicate recombinant human mutant MMP20, and EV indicates expression vector only.
Figure 2.
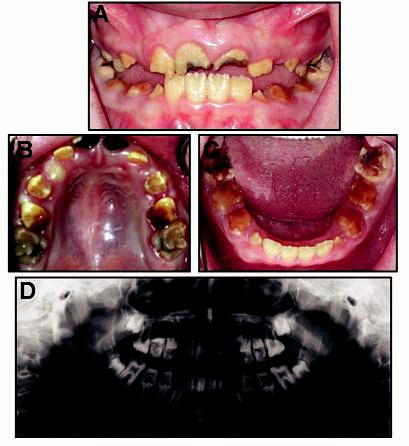
Clinical photographs and radiograph of proband (IV-1 in Fig. 1). (A) Frontal view. Proband is in mixed dentition, showing anterior open bite and hypomaturation phenotype. (B) Maxillary occlusal view. Caries is evident on the central incisors and posterior teeth. (C) Mandibular occlusal view. Note extensive attrition of posterior teeth, and caries is evident on the molars. (D) Panoramic radiograph showing mixed dentition and reduced mineralization of the enamel.
Figure 3.
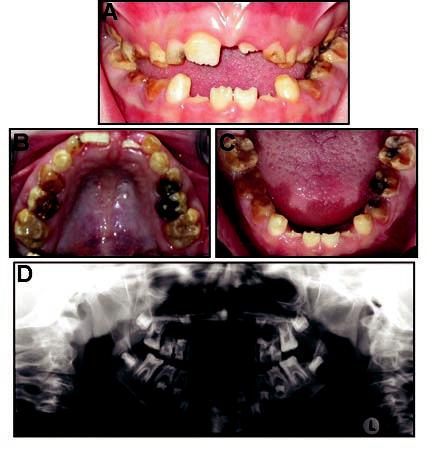
Clinical photographs and radiograph of the affected sibling (IV-2 in Fig. 1). (A) Frontal view. Proband is in mixed dentition, showing anterior open bite and hypomaturation phenotype. (B) Maxillary occlusal view. Caries is evident on posterior teeth. (C) Mandibular occlusal view. Caries is evident on posterior teeth. (D) Panoramic radiograph showing mixed dentition and reduced mineralization of the enamel.
Mutation Results
No KLK4 gene mutations were found in any probands. One family (Fig. 1) was found to have 4 MMP20 sequence alterations: g.18661T>C, g.18679C>A, g.31605G>A, and g.16250T>A. The first 3 genetic variants were found in the SNP database, indicating that they are polymorphisms present in at least 1% in the population, and therefore are unlikely to have any pathological effects (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=snp): rs1784423, rs1784424, and rs1784440. The last alteration, an exon 5 single-base substitution (g.16250T>A), changes the DNA codon for the amino acid histidine (CAT) to glutamine (CAA) (p.H226Q; Fig. 1B). His226 is an important catalytic site component of MMP20. The proband and his affected sister were both homozygous for this mutation, consistent with consanguinity. The parents, paternal grandfather, and maternal grandfather were all carriers of the mutation. Sequence analysis of 100 healthy Turkish controls (200 chromosomes) did not reveal this sequence alteration, indicating that this g.16250T>A mutation is not a common variant in the Turkish population.
Functional Studies
Recombinant human wild-type and mutant MMP20 cDNAs were transiently transfected into HEK293 cells, and the culture-conditioned media were collected and subjected to electrophoresis. Western blot analysis with an MMP20-specific antibody revealed that mutant and wild-type MMP20 cDNA produced a protein of approximately 45 kDa. This band was not detected in conditioned media from the negative control transfectant (data not shown). Casein zymograms performed to determine the protease activity of wild-type and mutant MMP20 revealed that the wild-type MMP20 protein cleaved the casein substrate (Fig. 1C), whereas the mutant protein was unable to cleave the substrate, consistent with a loss of proteolytic function.
DISCUSSION
These findings represent the second MMP20 mutation, confirming that mutation of this gene is etiologic for some cases of autosomal-recessive hypomaturation AI (Table). Individuals homozygous for the exon 5 g.16250T>A mutation have hypomaturation AI and anterior open bite. Enamel anomalies were observed in both the primary and secondary dentitions. Heterozygous carriers of the mutation do not have an enamel phenotype.
Table.
Mutations Resulting in Autosomal-recessive Hypomaturation AI
| Gene | Mutation Location |
Genomic DNAa | cDNAb | Proteinc | Reference |
|---|---|---|---|---|---|
| KLK4 | exon 4 | g.2142G>A | c.458G>A | p.W153X | Hart et al., 2004 |
| MMP20 | intron 6 | g.30561A>T | c.954-2A>T; IVS6-2A>T | p.0 or p.I319X or p.I319fs338X | Kim et al., 2005a |
| MMP20 | exon 5 | g.16250T>A | c.678T>A | p.H226Q | This report |
Reference sequences NT_033899.6 (MMP20) and AF228497 (KLK4).
Reference sequences O60882.
MMP20, also called enamelysin (OMIM604629), is a member of the matrix metalloproteinase family, a group of zinc-dependent metallopeptidases involved in extracellular matrix remodeling (Nagase and Woessner, 1999; Simmer and Hu, 2002). MMP20 is unique among the MMP family because of its highly restricted expression. In developing teeth, MMP20 is expressed primarily during the secretory to late-transition stages of amelogenesis (Bartlett et al., 1998). With its specificity for amelogenin, the major organic component of the enamel matrix, MMP20 is considered a predominant enzyme for the processing of enamel matrix. Human MMP20 consists of 483 amino acids and shares extensive (89%) amino acid sequence identity with porcine MMP20 (Llano et al., 1997; Bartlett et al., 1998). MMP20 has the characteristic domain structure of most other MMPs, including a signal peptide, a propeptide that maintains enzyme latency, a catalytic domain with a zinc-binding site, and a hinge region that links the catalytic domain to the C-terminal hemoplexin-like domain (Llano et al., 1997; Bartlett and Simmer, 1999). Computer modeling of pig enamelysin with the use of several crystal structures determined that 3 highly conserved amino acids—His226, His230, and His236—are the active sites of the zinc catalytic domain (Ryu et al., 2000). The g.16250T>A substitution mutation identified in this report changes the conserved active site His226 residue of the zinc catalytic domain (p.H226Q). The glutamine substitution destroys this metal-binding site and abolishes catalytic activity. Results of the biochemical assays in which the g.16250T>A mutant MMP20 demonstrates a complete lack of MMP20 activity corroborate these computational predictions. A similar clinical phenotype (hypomaturation AI) is also seen with homozygous mutations in KLK4 lacking the Ser207 active site of the catalytic triad (Hart et al., 2004).
MMP20 processes amelogenin to produce biologically active fragments that function during the initial developmental stages of enamel (Ryu et al., 1999). The complete loss of MMP20 proteolytic activity may prevent proper processing and degradation of enamel matrix proteins, resulting in qualitative defects in enamel malformation. The mouse mmp20 null model indicates that MMP20 performs enamel matrix protein cleavages that are essential for processing and removal of enamel matrix proteins during the secretory and maturation stages of enamel development (Caterina et al., 2002; Bartlett et al., 2004). mmp20 null mice do not process enamel matrix protein properly, yielding a severe AI phenotype, although hemizygous mice have no phenotype. Interestingly, the mature enamel in the mmp20 null mouse contained less bulk mineral (53%) and less weight percent of mineral (range, 7-16%) compared with controls, and was significantly (∼ 37%) softer than normal enamel. AI-affected teeth of mmp20 null mice show attrition of the molar occlusal surfaces that is related to the time of eruption. A similar trend was observed in the affected siblings in the current report who showed such attrition on the primary molars (Figs. 2, 3). The murine knock-out model suggests that there is no compensation for mmp20 function, indicating that it is necessary for proper enamel development. Clinical findings in individuals homozygous for the MMP20 g.16250T>A mutation indicate an analogous situation in human enamel development. The finding that heterozygous carriers (MMP20 g.16250T>A/wild-type) display no clinically apparent anomalies of enamel are similar to those observed in hemizygous mice (null/wild-type), suggesting that one functional MMP20 allele is sufficient for normal enamel development.
Similar to the g.16250T>A mutation, homozygosity of the previously identified MMP20 g.30561A>T mutation responsible for recessive pigmented hypomaturation AI is also associated with anterior open bite (Kim et al., 2005a). Whether this is a biologic or serendipitous relationship is unknown. The intron 6 g.30561A>T mutation is predicted to cause aberrant splicing, but due to the restricted expression of MMP20, the effect at the mRNA level was not determined. The mutant mRNA generated by aberrant splicing likely undergoes nonsense-mediated decay, and individuals homozygous for the mutation would not produce functional MMP20, representing the equivalent of a null allele. Heterozygous carriers of the mutation did not show an altered enamel phenotype.
This study demonstrates that a mutation in the MMP20 active site results in hypomaturation AI. These findings corroborate those of Kim et al. (2005a), that loss of function protease mutations underlie at least some forms of autosomal-recessive hypomaturation AI. However, 14 of the 15 families did not have detectable mutations in KLK4 or MMP20, suggesting that other genes account for the majority of hypomaturation AI in this Turkish population. While it is possible that other types of KLK4 and MMP20 mutations may be involved, such as promoter or intronic mutations or deletions that encompass whole exons, results of homozygosity linkage mapping studies in these families were not consistent for linkage in the regions spanning either the KLK4 (chromosome 16p13) or MMP20 (chromosome 11q22.3-q23) locus (data not shown). Identification of these genes and the continued cataloging of mutations underlying different forms of AI will provide a greater understanding of the molecular basis of enamel anomalies. This understanding should facilitate the development of a classification scheme that relates clinical phenotypes and underlying molecular mechanisms that may aid dentists in determining the most appropriate treatment course.
ACKNOWLEDGMENTS
This work was supported by the intramural program of the NIDCR/NIH, Department of Health and Human Services, Bethesda, MD, USA.
REFERENCES
- Aldred MJ, Savarirayan R, Crawford PJ. Amelogenesis imperfecta: a classification and catalogue for the 21st century. Oral Dis. 2003;9:19–23. doi: 10.1034/j.1601-0825.2003.00843.x. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Simmer JP. Proteinases in developing dental enamel. Crit Rev Oral Biol Med. 1999;10:425–441. doi: 10.1177/10454411990100040101. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Ryu OH, Xue J, Simmer JP, Margolis HC. Enamelysin mRNA displays a developmentally defined pattern of expression and encodes a protein which degrades amelogenin. Connect Tissue Res. 1998;39:101–109. doi: 10.3109/03008209809023916. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Beniash E, Lee DH, Smith CE. Decreased mineral content in MMP-20 null mouse enamel is prominent during the maturation stage. J Dent Res. 2004;83:909–913. doi: 10.1177/154405910408301204. [DOI] [PubMed] [Google Scholar]
- Caterina JJ, Skobe Z, Shi J, Ding Y, Simmer JP, Birkedal-Hansen H, et al. Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem. 2002;277:49598–49604. doi: 10.1074/jbc.M209100200. [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Simmer JP, Wright JT. A nomenclature for X-linked amelogenesis imperfecta. Arch Oral Biol. 2002a;47:255–260. doi: 10.1016/s0003-9969(02)00005-5. [DOI] [PubMed] [Google Scholar]
- Hart PS, Aldred MJ, Crawford PJ, Wright NJ, Hart TC, Wright JT. Amelogenesis imperfecta phenotype-genotype correlations with two amelogenin gene mutations. Arch Oral Biol. 2002b;47:261–265. doi: 10.1016/s0003-9969(02)00003-1. [DOI] [PubMed] [Google Scholar]
- Hart PS, Michalec MD, Seow WK, Hart TC, Wright JT. Identification of the enamelin (g.8344delG) mutation in a new kindred and presentation of a standardized ENAM nomenclature. Arch Oral Biol. 2003;48:589–596. doi: 10.1016/s0003-9969(03)00114-6. [DOI] [PubMed] [Google Scholar]
- Hart PS, Hart TC, Michalec MD, Ryu OH, Simmons D, Hong S, et al. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet. 2004;41:545–549. doi: 10.1136/jmg.2003.017657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Gorry MC, Michalec MD, Ryu OH, Uygur C, et al. Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet. 2003;40:900–906. doi: 10.1136/jmg.40.12.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida M, Ariga T, Shirakawa T, Oguchi H, Sakiyama Y. Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J Dent Res. 2002;81:738–742. doi: 10.1177/0810738. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hu YY, Lin BP, Boyd C, Wright JT, et al. Amelogenin p.M1T and p.W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta. J Dent Res. 2004;83:378–383. doi: 10.1177/154405910408300505. [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Hart TC, Hart PS, Ramaswami MD, Bartlett JD, et al. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet. 2005a;42:271–275. doi: 10.1136/jmg.2004.024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lin BP, Kiziltan B, Gencay K, Simmer JP, et al. ENAM mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res. 2005b;84:278–282. doi: 10.1177/154405910508400314. [DOI] [PubMed] [Google Scholar]
- Llano E, Pendas AM, Knauper V, Sorsa T, Salo T, Salido E, et al. Identification and structural and functional characterization of human enamelysin (MMP-20) Biochemistry. 1997;36:15101–15108. doi: 10.1021/bi972120y. [DOI] [PubMed] [Google Scholar]
- Mardh CK, Backman B, Holmgren G, Hu JC, Simmer JP, Forsman-Semb K. A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2) Hum Mol Genet. 2002;11:1069–1074. doi: 10.1093/hmg/11.9.1069. [DOI] [PubMed] [Google Scholar]
- Nagase H, Woessner JF., Jr Matrix metalloproteinases. J Biol Chem. 1999;274:21491–21494. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- Nusier M, Yassin O, Hart TC, Samimi A, Wright JT. Phenotypic diversity and revision of the nomenclature for autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97:220–230. doi: 10.1016/j.tripleo.2003.08.007. [DOI] [PubMed] [Google Scholar]
- OMIM604629. Online Mendelian Inheritance in Man, OMIM™. Johns Hopkins University; Baltimore, MD: MIM Number:{604629}:{3/3/2003}. http://www.ncbi.nlm.nih.gov/omim/ [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet. 2001;10:1673–1637. doi: 10.1093/hmg/10.16.1673. [DOI] [PubMed] [Google Scholar]
- Ryu OH, Fincham AG, Hu CC, Zhang C, Qian Q, Bartlett JD, et al. Characterization of recombinant pig enamelysin activity and cleavage of recombinant pig and mouse amelogenins. J Dent Res. 1999;78:743–750. doi: 10.1177/00220345990780030601. [DOI] [PubMed] [Google Scholar]
- Ryu OH, Hsiung D, Hu CC, Sun X, Cao X, Bartlett JD, et al. The structure and function of enamelysin (MMP20) In: Goldberg M, Robinson C, editors. Chemistry and biology of mineralized tissues: proceedings of the Sixth International Conferences. American Academy of Orthopaedic Surgeons; Vittel, France: 2000. pp. 363–367. [Google Scholar]
- Simmer JP, Hu JC. Expression, structure, and function of enamel proteinases. Connect Tissue Res. 2002;43:441–449. doi: 10.1080/03008200290001159. [DOI] [PubMed] [Google Scholar]
- Witkop CJ., Jr Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol. 1988;17:547–553. doi: 10.1111/j.1600-0714.1988.tb01332.x. [DOI] [PubMed] [Google Scholar]
- Wright JT, Hart PS, Aldred MJ, Seow K, Crawford PJ, Hong SP, et al. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect Tissue Res. 2003;44(Suppl 1):72–78. [PubMed] [Google Scholar]