

Abstract
17β-estradiol (17β-E2) protects against H2O2-mediated depletion of intracellular ATP and lessens the degree of depolarization of mitochondrial membrane potential (Δψm) in cultured lens epithelial cells consequential to oxidative insult. We now report that 17β-E2 acts as a positive regulator of the survival signal transduction pathway, MAPK which, in turn, acts to stabilize Δψm, in effect, attenuating the extent of depolarization of mitochondrial membrane potential in the face of acute oxidative stress. The SV-40 viral transformed human cell line, HLE-B3 was treated with 17β-E2 over a time course of 60 min and phosphorylation of ERK1/2 was analyzed by Western blot. ERK1/2 was phosphorylated within 5–15 min in the presence of 17β-E2. Cell cultures were exposed to the MEK1/2 inhibitor, UO126, subsequent to H2O2±17β-E2 treatment and the Δψm examined using JC-1, a potentiometric dye which serves as an indicator for the state of mitochondrial membrane potential. UO126 treatment attenuated ERK1/2 phosphorylation irrespective of whether estradiol was administered. Mitochondrial membrane depolarization resulting from H2O2 stress was substantially greater in the presence of UO126. The greater the extent of depolarization, the less effective 17β-E2 treatment was in checking mitochondrial membrane depolarization, indicating that the relative degree of ERK phosphorylation influences mitochondrial stability with oxidative insult. The data support a positive correlation between 17β-E2 stimulation of ERK1/2 phosphorylation and mitochondrial stabilization that would otherwise cause a complete collapse of Δψm.
Keywords: ERK, Estrogen, Hydrogen peroxide, Human lens epithelial cell, Mitochondria
1. Introduction
The biological actions of estrogens are mediated by binding to one of two estrogen receptors (ERs), ERα and ERβ, which are members of the nuclear receptor superfamily, a family of ligand-related transcription factors (reviewed in Matthews and Gustafsson, 2003). The actions of 17β-estradiol (17β-E2) occur on binding the ER, and the nuclear pool of these receptors can then transactivate target genes (reviewed in Levin, 2001). In addition to its role in being a prominent transcription factor, studies of the antioxidant activity of 17β-E2 have demonstrated that estrogens do not necessarily require the classical receptor-dependent mechanism in order to exert their positive effects (Behl et al., 1997; Gridley et al., 1998). It has been recently shown that 17β-E2 protects against H2O2-mediated depletion of intracellular ATP in human lens epithelial cells (HLE-B3) (Wang et al., 2003) and that stabilization of the mitochondrial membrane potential (Δψm) by 17β-E2 plays a crucial role in protecting the lens epithelial cell from oxidative damage (Wang et al., 2003; Moor et al., 2004). The dependency on ER binding for this stabilization process to occur has yet to be definitively demonstrated.
Under conditions of oxidative stress, mitochondria undergo a loss of impermeability of the inner mitochondrial membrane which subsequently causes a complete collapse of mitochondrial membrane potential (Δψm) (Murphy et al., 1999). H2O2 can collapse Δψm in many cell types including lens epithelial cells (Wang et al., 2003), ultimately eliminating the driving force for ATP production and exacerbating free radical production (Dykens, 1994). The mechanism(s) by which 17β-E2 stabilizes the inner mitochondrial membrane to maintain Δψm during oxidative stress are unknown. The current study describes the activation of ERK1/2 by 17β-E2 in both HLE-B3 and secondary cultures of bovine lens epithelial cells (BLECs). We propose that, in spite of acute oxidative stress, the activation of this upstream target may be triggering an anti-apoptotic cascade of events in cultured lens epithelial cells that promotes the downstream stabilization of the inner mitochondrial membrane preventing the complete collapse of Δψm.
2. Materials and methods
2.1. Materials
1,3,5(10)-ESTRATRIEN-3, 17β-DIOL (17β-E2) was purchased from Steraloids, Inc. (Newport, RI). For use in our experiments, the hormone was dissolved in either 100% ethanol or a diluted (0.025%) solution of (2-Hydroxypropyl)-β-cyclodextrin (in water) from Sigma-Aldrich (St Louis, MO). Stock solutions of hormone were prepared fresh for each experiment and diluted in culture medium to a working concentration of 1 μM. 30% hydrogen peroxide (H2O2) was purchased from Fisher Scientific (Fair Lawn, NJ) and diluted in water and culture medium to obtain a final working concentration of 100 μM. 5,5′,6,6′-tetra-chloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1) was purchased from Molecular Probes (Eugene, OR). L-buthionine-[s,r]-sulfoximine (L-BSO), an inhibitor of glutathione biosynthesis was from Sigma-Aldrich (St Louis, MO). The MEK1/2 inhibitor, UO126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio] butadiene), rabbit anti-p44/42 MAP Kinase, mouse anti-phospho-p44/42 MAPK (Thr202/Tyr204), rabbit anti-Akt, rabbit anti-phospho-Akt (Ser473), rabbit anti-phospho-SAPK/JNK (Thr183/Tyr185) antibodies and horseradish peroxidase-conjugated (HRP) goat anti-rabbit and horse anti-mouse IgG were all obtained from Cell Signaling Technology®(Beverly, MA). Rabbit anti-actin and mouse anti-JNK antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All other chemicals and reagents were of analytical grade and were obtained from commercially available sources.
2.2. Cell culture
HLE-B3 cells, a human lens epithelial cell line immortalized by SV-40 viral transformation (Andley et al., 1994), were obtained from Usha Andley (Washington University School of Medicine, Department of Ophthalmology, St Louis, MO). Cells were maintained in Eagle’s minimal essential medium (MEM) supplemented with 20% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT), 2 mM L-glutamine, nonessential amino acids and 0.02 g/L gentamycin solution (Sigma Chemical Co., St Louis, MO) at 37 °C and 5%CO2/95%O2. Primary cultures of bovine lens epithelial cells (BLECs) were established from the aseptic dissection of bovine (Bostaurus) lenses and cultures were maintained in MEM supplemented with 10% bovine calf serum (BCS) (Hyclone Laboratories, Logan, UT), 2 mM L-glutamine, nonessential amino acids and 0.02 g/L gentamycin solution as previously described (Cammarata and Yorio, 1990; Cammarata et al., 2002). All experiments were performed with monolayers of BLECs that did not exceed passage 3. In order to deplete the cell cultures of estrogens, cells were maintained in 20% charcoal dextran-stripped FBS (Gemini Bio-Products, Woodland, CA) (CSFBS) MEM for 24–48 h then switched to 2% (CSFBS) MEM for up to 18 h with a final medium change to either 0.5% (CSFBS) MEM or serum-free MEM on the day of the experiments. In select experiments, cells were either kept in serum-free MEM for 18–24 h prior to estrogen treatment or pretreated with estrogen overnight and this was done in 2% (CSFBS) MEM followed by a medium change to 0.5% (CSFBS) MEM with fresh estrogen the next morning prior to further experimentation.
2.3. Cell lysis, electrophoresis and western blot
Total cell lysates were collected from HLE-B3 and BLEC cultures after treatments by rinsing adherent cells with ice-cold 1× phosphate buffered saline (PBS) pH 7.4 and then adding lysis buffer [25 mM HEPES, pH 7.4, 0.25 NaCl, 0.5% IGEPAL (NP-40), 0.2% Triton X-100, 1 mM EGTA, 1 mM EDTA, 0.5 mM DTT, 10 mM NaF, 0.1 mM Na3VO4 and a cocktail of protease inhibitors (Sigma-Aldrich, St Louis, MO)] directly to the monolayers for 30 min at 4 °C. Lysates were collected, sonicated for 5 s and sampled for protein concentration using the Bio-Rad protein assay buffer (Bio-Rad Laboratories, Hercules, CA). 3× SDS (Laemmli) buffer was added to the lysates which were subsequently boiled for 3 min and the proteins resolved by electrophoresis on 10% SDS-polyacrylamide gels (20 μg protein per lane). Proteins were transferred to nitrocellulose (Scheicher and Schuell, Keene, NH) and the membranes were blocked with 1% bovine serum albumen (BSA) and 0.02% NaN3 in Tween Tris-buffered saline (TTBS) for 15 min. Membranes were probed overnight at 4 °C with primary antibodies (see Materials), rinsed in TTBS (4×5 min washes) the next day and incubated in either goat anti-rabbit HRP conjugate or horse anti-mouse HRP conjugate for 1 h at room temperature. Required concentrations of antibodies were determined according to the manufacturer’s protocols. Membranes were again rinsed in TTBS (4×5 min washes) and proteins were detected using an ECL™ kit for chemiluminescence (Amersham Biosciences, Buckinghamshire, England). Probed membranes were exposed to Kodak BioMax Light Film (Kodak Scientific Imaging, Rochester, NY).
2.4. Measurement of glutathione (GSH)
The determination of GSH levels in both HLE-B3 and BLECs treated overnight (20 h) with or without 1 mM L-BSO was performed as previously described (Giblin et al., 1985). Briefly, the culture medium was removed and a 0.2 ml volume of ice cold 0.05 M EDTA pH 4.0–4.5 was added to the monolayer and the cells were gently scraped off with a cell scraper (Becton Dickinsin Labware, Franklin Lakes, NJ). The scraping process was repeated 3× with cells being collected into a 2 ml glass homogenizer on ice and homogenized. The homogenates were made in 5% in trichloroacetic acid, rehomogenized and centrifuged. The concentration of GSH was determined by the use of an automatic amino acid analyzer sensitive to 1 nmol and expressed as nmol/million cells.
2.5. JC-1 Staining and confocal microscopy
Following treatments, cells were stained with the cationic dye, JC-1, as previously described (Moor et al., 2004) to visualize the state of mitochondrial membrane potential. This is a potentiometric dye which exhibits potential dependent loss of J-aggregates (hyperpolarization) to accumulation of JC-1 monomers (depolarization) as indicated by a fluorescence emission shift from red to green. Briefly, culture medium was removed from adherent HLE-B3 and BLECs and the monolayers were rinsed 1×with Dulbecco’s Modified Eagle’s Medium (DMEM) without phenol red (Sigma-Aldrich, St Louis, MO). Cell monolayers were incubated with DMEM containing 10% serum and 5 μg/ml JC-1 at 37 °C for 30 min. Following the incubation, cells were rinsed 2× with DMEM and images were obtained using a confocal microscope (Zeiss LSM410) excited at 488 nm (for JC-1) and set to simultaneously detect green emissions (510–525 nm) and red emissions (590 nm) channels.
2.6. Statistical analysis
Analyses of confocal images were performed using MetaMorph software version 6.1 for Windows. Images were analyzed as two separate channels (red and green), background removed and each channel’s fluorescence intensity signal quantified for the entire image. Significant differences between groups were determined by an independent sample Student’s t-test (2-tailed) using SPSS version 12.0 for Windows. For all experiments, data are reported as mean±S.E. or S.D. as indicated and P values <0.05 were considered significant.
3. Results
3.1. 17β-E2 activation of MAPK signaling in HLE-B3 and BLECs
Identification of MAPK as activated by 17β-E2 was carried out by harvesting total cell lysates from quiescent HLE-B3 and BLEC cultures that had been maintained in charcoal stripped (i.e. estrogen depleted) serum followed by serum-starvation for at least 18 h prior to stimulation by either 1 μM 17β-E2 or 1% serum (used as a positive control for phosphorylation) for 0, 5 15, 30 and 60 min. For this set of experiments, complete serum starvation for 18 h was utilized in order to maximally diminish the basal cellular levels of phosphorylated ERK1/2 prior to estrogen or serum introduction, so that activation of phosphorylation could be optimally detected. Proteins were collected, run on SDS-PAGE gels and analyzed by Western blot (see Materials and Methods). In HLE-B3 (Fig. 1A) and BLECs (Fig. 1B), ERK1 (p44) and ERK2 (p42) proteins were rapidly phosphorylated (pERK1/2) within 5–15 min after the addition of 17β-E2. This phosphorylation peaked between 15 and 30 min and remained evident by 60 min. Serum stimulation also produced a rapid phosphorylation over the same time span in both cell types. Both membranes were additionally probed with ERK antibody, followed by actin antibody to show equal loading of protein (20 μg) for each time point.
Fig. 1.
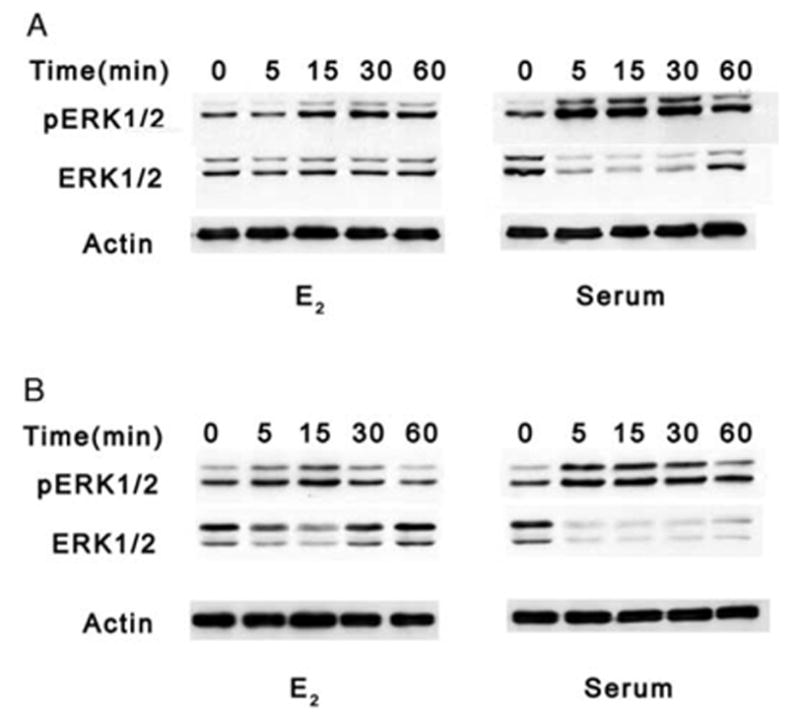
Western blot analyses of ERK1/2 phosphorylation in HLE-B3 and BLECs with E2 or serum. Total cell lysates (20 μg protein/lane) from quiescent HLE-B3 (A) and BLEC (B) cultures serum-starved for at least 18 h prior to stimulation by either 1 μM E2 or 1% serum for 0, 5, 15, 30, and 60 min. Membranes were stripped and reprobed with an antibody to actin as a control for protein loading. This experiment was repeated three times with similar results.
3.2. UO126 inhibition of MAPK signaling in HLE-B3 and BLECs during oxidative stress
UO126 is a highly selective inhibitor of MEK1 and MEK2 kinases which lie upstream of ERK1 and ERK2 in the MAPK signaling cascade (Favata et al., 1998). The inhibitor was utilized to prevent the downstream phosphorylation activity of ERK1/2 in both HLE-B3 (Fig. 2A) and BLECs (Fig. 3A) during oxidative stress in the presence and absence of 17β-E2. Estrogen-depleted cultures (see Materials and Methods) were pretreated with either 1 μM 17β-E2 or diluted vehicle [.025% solution of (2-Hydroxypropyl)-β-cyclodextrin (in water)] overnight in 2% (CSFBS) MEM. In this set of experiments, the culture medium was replaced with 0.5% (CSFBS) MEM with fresh 1 μM 17β-E2 or vehicle and 10 μM UO126 (+UO126) or DMSO vehicle (−UO126) for 1 h at 37 °C. Note that cells were cultured in the presence low serum (0.5%), not serum-free, in order to experimentally manipulate increased basal levels of the phospho-proteins of interest. As a result of influencing the cells by the introduction of a low level of serum, thereby assuring an elevated level of phospho-proteins of interest, the inhibitory effect of UO126 in both cell types could be observed. Cultures then received a bolus of 100 μM H2O2±17β-E2 and cells were subsequently harvested over a time course of 0, 15, 30 and 60 min and total cell lysates analyzed by Western blot. Control cultures were not exposed to H2O2 or 17β-E2 but only to UO126 or DMSO vehicle. UO126 treatment significantly reduced basal levels of pERK1/2 in HLE-B3 (Fig. 2A) and BLEC (Fig. 3A) control, H2O2-treated and H2O2+17β-E2-treated cultures compared to their DMSO-treated counterparts. The basal levels of pAkt (Figs. 2B and 3B) were unaffected with UO126 treatment in both HLE-B3 and BLECs, indicating specificity of the drug in the MAPK signaling cascade and not PI-3 kinase signaling cascade. Unexpectedly, however, the pSAPK/JNK levels (Figs. 2C and 3C) were diminished with UO126 treatment. Of further interest was the observation that differences in susceptibility to U0126 between the two cell species were detected, as the pSAPK/JNK upper and lower bands were eliminated by UO126 treatment in BLECs, whereas the phospho-p54 band (upper band) could still be detected in HLE-B3 cells.
Fig. 2.
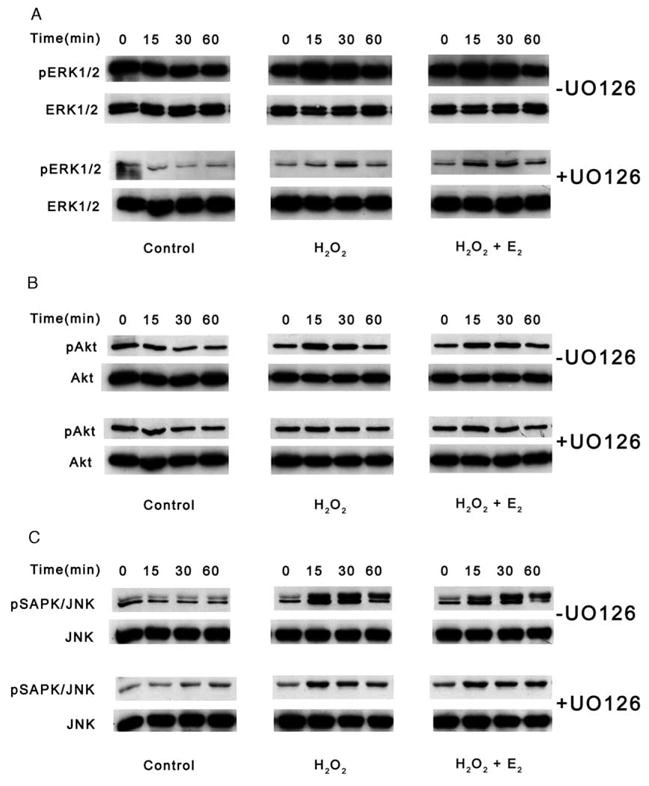
Western blot analyses of UO126 inhibition of MAPK signaling in HLE-B3 cells during an oxidative stress. Following a 1 h pretreatment with 10 μM UO126 (+UO126) or DMSO vehicle (−UO126), total cell lysates (20 μg protein/lane) were collected after 0, 15, 30 and 60 min of 100 μM H2O2±1 μM E2 exposure (middle and right columns) and analyzed for (A) ERK1/2, (B) Akt and (C) SAPK/JNK phosphorylation. Controls were not exposed to H2O2 or E2 (left column). This experiment was repeated twice with similar results.
Fig. 3.
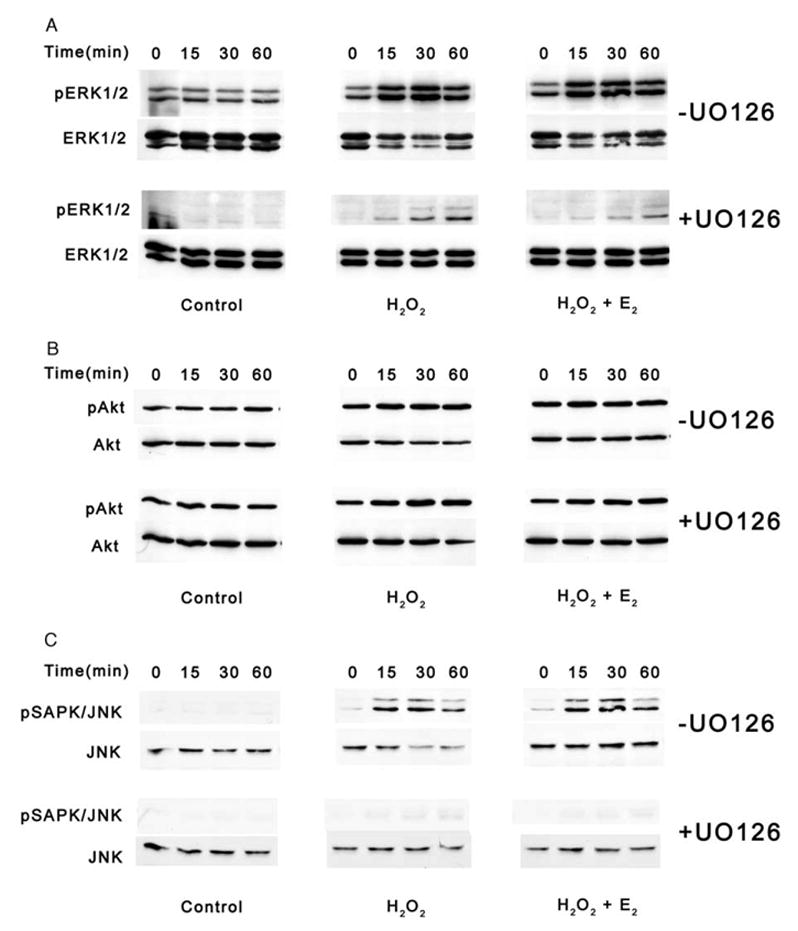
Western blot analyses of UO126 inhibition of MAPK signaling in BLECs during an oxidative stress. Following a 1 h pretreatment with 10 μM UO126 (+UO126) or DMSO vehicle (−UO126), total cell lysates (20 μg protein/lane) were collected after 0, 15, 30 and 60 min of 100 μM H2O2±1 μM E2 exposure (middle and right columns) and analyzed for (A) ERK1/2, (B) Akt and (C) SAPK/JNK phosphorylation. Controls were not exposed to H2O2 or E2 (left column). This experiment was repeated twice with similar results.
Oxidative stress as induced by the bolus addition of H2O2 resulted in noticeable increased levels of pERK1/2, pAkt and pSAPK/JNK within 15 min in both cell types not exposed to UO126 (Figs. 2 and 3 [−UO126]). As with the controls, UO126 treatment reduced the ERK1/2 (Figs. 2A and 3A) and SAPK/JNK (Figs. 2C and 3C) phosphorylation levels but not pAkt levels (Figs. 2B and 3B) in both cell species. In all cases, co-administration of 17β-E2 with H2O2 failed to further augment any of the phosphorylation events observed with H2O2 addition alone.
3.3. Effect of UO126 on Δψm in HLE-B3 cells
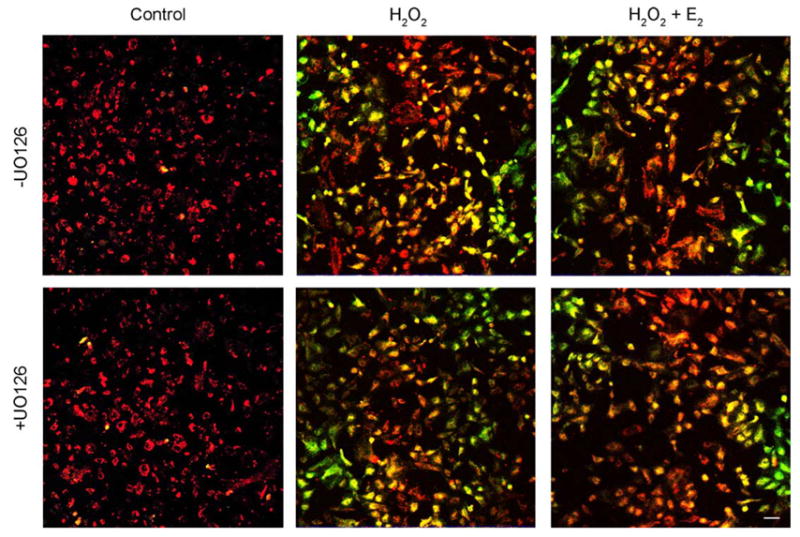
To ascertain whether a relationship could be established between the MAPK signaling cascade and Δψm, peroxide-stressed HLE-B3 cells in the presence and absence of UO126 were examined using the potentiometric dye, JC-1. JC-1 is a cationic dye that exhibits potential-dependent accumulation of JC-1 monomers from J-aggregates, indicated by a fluorescence emission shift from red (~590 nm) to green (~525 nm), as the mitochondrial permeability transition pore opens (i.e. depolarizes). Polarized mitochondria are marked by red fluorescence staining, while depolarization is marked by green fluorescence staining. Therefore, mitochondrial depolarization is indicated by an increase in the green/red fluorescence intensity ratio. Estrogen-depleted cultures (see Materials and Methods) were pretreated with either 1 μM 17β-E2 or ETOH vehicle overnight in 2% (CSFBS) MEM and subsequently, the culture medium was replaced with 0.5% (CSFBS) MEM and fresh 1 μM 17β-E2 or vehicle and 10 μM UO126 (+UO126) or DMSO vehicle (−UO126) for 1 h at 37C. Consequent to the treatment with MEK inhibitor, a bolus of 100 μM H2O2 was administered to the cell cultures, followed by JC-1 analysis at 0, 2 and 4 h (see Materials and Methods). Mitochondrial depolarization is shown after a 4 h peroxide treatment (Fig. 4). HLE-B3 cells showed marked depolarization with oxidative stress both in the presence and absence of U0126 administration and 17β-E2 reduced the extent of depolarization caused by H2O2 irrespective of the addition of MEK inhibitor. The degree of mitochondrial membrane depolarization over the 4 h time course of oxidative insult could be quantified by determining the ratio of average green to red fluorescence intensity (Fig. 7A). H2O2 caused significant progressive depolarization both in the presence and absence of UO126 and 17β-E2 treatment significantly diminished depolarization, both at the 2 and 4 h time points. Particularly noteworthy is the observation that UO126 treatment greatly exacerbated depolarization, as most evident by 4 h (0.304±0.011) compared to its untreated counterpart (0.171±0.022). 17β-E2 treatment reduced the extent of depolarization by H2O2 in both +UO126 treated cultures [0.173±0.013 (H2O2+E2) vs. 0.304±0.011 (H2O2)] and −UO126 cultures [0.048±0.004 (H2O2+E2) vs. 0.171±0.022 (H2O2)]. However, it was evident that the addition of UO126 diminished estradiol’s cytoprotective capacity to protect against mitochondrial depolarization [+UO126 (0.173±0.013) vs. −UO126 (0.048±0.004)].
Fig. 4.
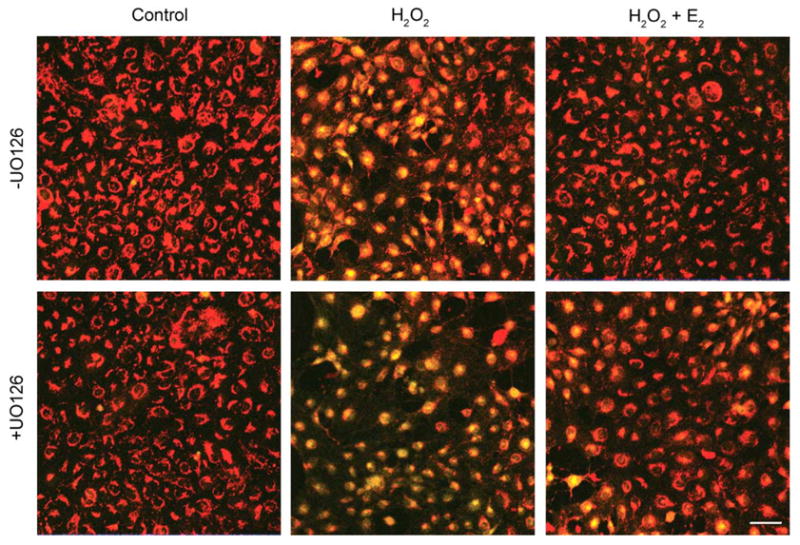
Confocal imaging of H2O2-induced mitochondrial membrane depolarization in HLE-B3 cells after 4 h. Following a 1 h pretreatment with 10 μM MEK1/2 inhibitor UO126 (+UO126) or DMSO vehicle (−UO126), cultures were treated with 100 μM H2O2±1 μM E2 (middle and right panels) and after 4 h stained with 5 μg/ml JC-1, a Δψm sensitive dye, for 30 min. Control cultures (left panels) received only UO126 or DMSO vehicle. Mitochondrial membrane depolarization is indicated by a shift from red to green fluorescence in H2O2-exposed cultures (middle panels). These images are typical of eight randomly chosen fields per treatment. Bar=20 μm.
Fig. 7.
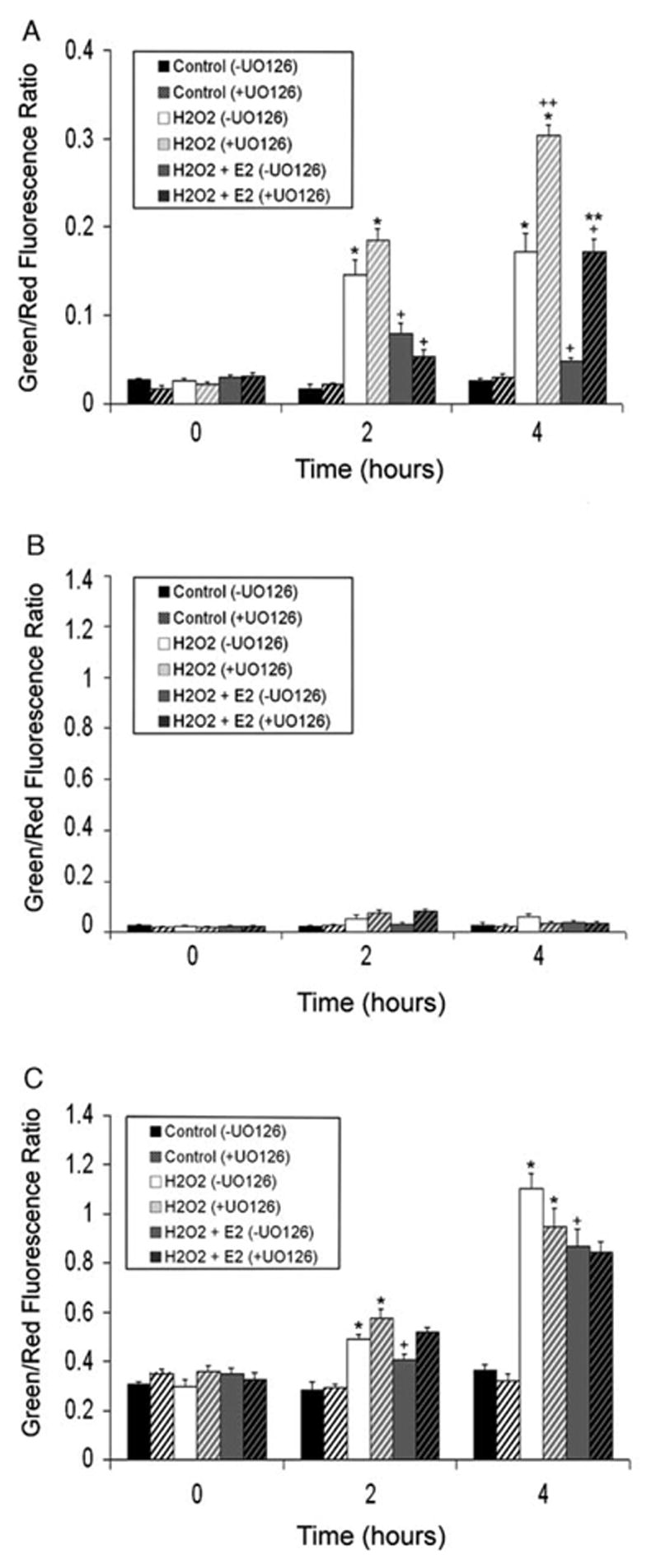
Quantitation of green and red fluorescence intensity from JC-1 images. Average green and red fluorescence values were calculated and ratios obtained. Bar graphs represent ratio data over the time course of 0, 2 and 4 h of 100 μM H2O2 exposure in (A) HLE-B3 cells, (B) BLECs and (C) BLECs+BSO. Data are presented as Mean±SE with n=8. (*) represents significantly different (P<0.001) from controls with the same treatment. (+) represents significantly different (P<0.02) from H2O2 with the same treatment. (**) represents significantly different (P<0.001) from H2O2+E2 (−UO126). (++) represents significantly different (P<0.001) from H2O2 (−UO126).
3.4. Effect of UO126 on Δψm in BLECs
Secondary cultures of BLECs were evaluated in parallel to the HLE-B3 cells in order to rule out the possibility that the putative association uncovered between activated MAPK signaling cascade and Δψm observed with HLE-B3 cells was an artifact of viral transformation. BLECs exhibited an unexpectedly high tolerance to oxidative stress by H2O2 regardless of MAPK inhibition by UO126 (Figs. 5 and 7B). There was no observed depolarization at 2 or 4 h following oxidative insult (Fig. 7B).
Fig. 5.
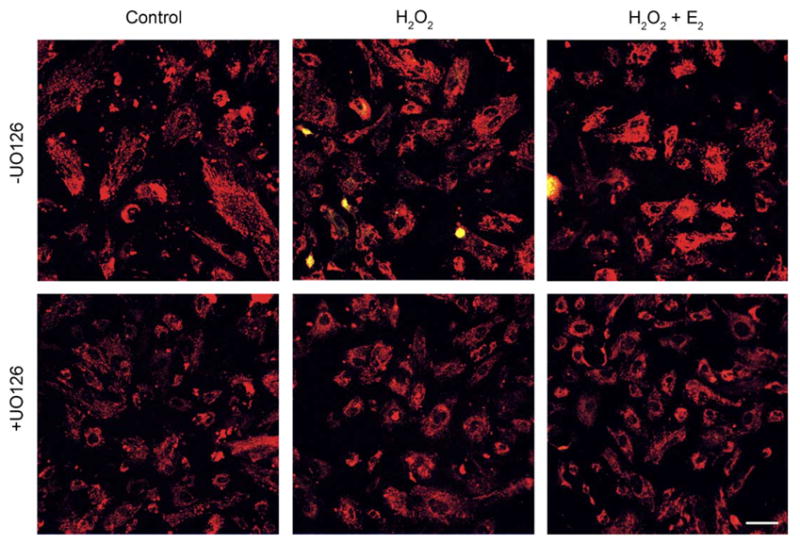
Confocal imaging of Δψm in BLECs exposed to H2O2 for 4 h. Following a 1 h pretreatment with 10 μM MEK1/2 inhibitor UO126 (+UO126) or DMSO vehicle (−UO126), cultures were treated with 100 μM H2O2±1 μM E2 (middle and right panels) and after 4 h stained with 5 μg/ml JC-1, a Δψm sensitive dye, for 30 min. Control cultures (left panels) received only UO126 or DMSO vehicle. No mitochondrial membrane depolarization was detected as no shift from red to green fluorescence occurred in H2O2-exposed cultures (middle panels). These images are typical of eight randomly chosen fields per treatment. Bar=20 μm.
To gain insight as to why BLECs were so tolerant to peroxide insult under conditions which markedly depolarized HLE-B3 cells, GSH levels were measured in both HLE-B3 and BLECs (Table 1). BLECs presented 3–4 times higher GSH levels per million cells (29.6±0.1) compared to HLE-B3 cells (8.9±0.5). Treatment of the cell cultures with 1 mM L-BSO, an inhibitor of GSH biosynthesis, extensively lowered GSH levels in BLECs and HLE-B3 cells (9.6±0.4 and 3.5±0.4), respectively. The sizeable reduction in intracellular GSH content with L-BSO treatment could not be attributed to a loss in total cell number as protein levels per million cells remained the same, irrespective of L-BSO treatment, for both BLECs and HLE-B3 cells. This data supported the notion that resistance to mitochondrial depolarization prompted by oxidative insult was linked to the relative ‘reducing potential power’ (i.e. GSH levels) of the cultured cell.
Table 1.
GSH content in human (HLE-B3) and bovine (BLEC) lens epithelial cells
| Treatment | GSH(nmol/million cells) | Protein(mg/million cells) |
|---|---|---|
| HLE-B3−BSO | 8.9±0.5 | 66±4 |
| HLE-B3+BSO | 3.5±0.4* | 61±3 |
| BLEC−BSO | 29.6±0.1+ | 206±7 |
| BLEC+BSO | 9.6±0.4** | 198±14 |
Data represented are mean±SD, n=3 flasks of cells.
Significantly different from HLE-B3−BSO (P<0.05).
Significantly different from HLE-B3−BSO (P<0.05).
Significantly different from BLEC−BSO (P<0.05).
We re-examined the relationship between MAPK signaling and Δψm, in GSH-depleted BLECs undergoing oxidative insult by H2O2. Similar to the experiments described above, estrogen-depleted cultures (see Materials and Methods) were pretreated with either 1 μM 17β-E2 or ETOH vehicle overnight in 2% (CSFBS) MEM. At the same time, 1 mM L-BSO was added to the medium for all cultures studied. The culture medium was subsequently replaced with 0.5% (CSFBS) MEM with fresh 1 μM 17β-E2 or vehicle, 1 mM L-BSO and 10 μM UO126 (+UO126) or DMSO vehicle (−UO126) for 1 h at 37°C. Cell cultures then received a bolus of 100 μM H2O2 over a time course of 0, 2 and 4 h. At each time point, cells were rinsed and incubated with 5 μg/ml JC-1 for 30 min at 37 °C, rinsed with DMEM and immediately imaged (see Materials and Methods). As a result of lessening intracellular GSH content in BLECs by treatment with L-BSO, marked depolarization was evident with or without UO126 treatment (Fig. 6). Both in the presence (+UO126) and absence (−UO126) of the MEK1/2 inhibitor, H2O2 caused severe and progressive depolarization (compare the average green/red fluorescence intensity ratio in Fig. 7A and C) over the 4 h time course. 17β-E2 was somewhat effective in reducing the extent of depolarization caused by H2O2 irrespective of whether the MEK inhibitor was added or omitted. In contrast to HLE-B3 cells (Fig. 7A), which showed a marked cytoprotection by 17β-E2 against mitochondrial membrane depolarization in the presence and absence of UO126, BLECs+L-BSO treated with 17β-E2 showed statistical significance against depolarization [(2 h) 0.408±0.017 (H2O2+E2) vs. 0.489±0.021 (H2O2); (4 h) 0.870±0.069 (H2O2+E2) vs. 1.103±0.063 (H2O2)] only in the absence of UO126. The combined manipulation of L-BSO+UO126+H2O2 likely overwhelmed the cell preventing 17β-E2 from fully exerting its stabilizing effect on Δψm observed in cultures not treated with UO126.
Fig. 6.
Confocal imaging of H2O2-induced mitochondrial membrane depolarization in BLECs pretreated for 20 h with 1 mM BSO, an inhibitor of GSH biosynthesis. Following a 1 h pretreatment with 10 μM MEK1/2 inhibitor UO126 (+UO126) or DMSO vehicle (−UO126), cultures were treated with 100 μM H2O2±1 μM E2 (middle and right panels) and after 4 h stained with 5 μg/ml JC-1, a Δψm sensitive dye, for 30 min. Control cultures (left panels) received only UO126 or DMSO vehicle. Mitochondrial membrane depolarization is indicated by a shift from red to green fluorescence in H2O2-exposed cultures (middle panels). These images are typical of eight randomly chosen fields per treatment. Bar=20 μm.
4. Discussion
Signaling pathways previously reported to be activated by 17β-E2-ER include the threonine/serine kinase, extracellular-regulated kinase (ERK1/2), a member of the mitogen activated protein kinase (MAPK) family, the phosphoinositol-3-hydroxy (PI-3) kinase and c-Jun NH2-terminal kinase (JNK), also a member of the MAPK family (reviewed in Levin, 2001). Data from studies utilizing the breast cancer cell line, MCF-7, have indicated rapid activation (i.e. phosphorylation) of ERKs by 17β-E2 which results from proximal kinase activations, which include Ras, Src, raf and MAP kinase kinase (MEK1/2) (Migliaccio et al., 1996). Activation of PI-3 kinase by 17β-E2-ER interacting with its regulatory subunit has been reported to stimulate Akt kinase (Simoncini et al., 2000). In ER-expressing CHO cells, it has been demonstrated that 17β-E2 can activate JNK (a stress-activated protein kinase) through ERβ, but inhibits this kinase through ERα (Razandi et al., 1999). In tissue studies, the ovarian hormones progesterone and estrogen have been shown to promote phosphorylation of ERKs and Akt in the cerebral cortex (Singh, 2000). In human lens epithelial cells, growth factors activate MAPK signaling. The addition of platelet-derived growth factor (PDGF) to serum-deprived HLE-B3 cells promoted activation of ERK1/2 and JNK proteins (Chen et al., 2004). Thus, there is ample data in the literature to support that 17β-E2 plays an important role in regulating signal transduction events via MAPK and other survival pathways. Our data is unique in that it advances these prior findings by demonstrating a positive correlation between ERK activation (i.e. phosphorylation) by 17β-E2 and mitochondrial stabilization against irreversible loss of the impermeability of the inner mitochondrial membrane. 17β-E2 promoted activation of MAPK signaling which, in turn, stabilized mitochondria against membrane potential collapse in spite of oxidative insult in the virally-transformed lens epithelial cell line, HLE-B3 and we reconfirmed that observation with normal lens epithelial cells (BLECs), thereby ruling out the possibility that our results were dictated by viral transformation. In our hands, using serum-deprived conditions, HLE-B3 (Fig. 1A) and BLECs (Fig. 1B) were both shown to be responsive to 17β-E2 and a rapid increase of pERK1/2 was observed. This analysis did not include an examination of Akt and JNK phosphorylation by 17β-E2 treatment and further studies are warranted and are currently in progress.
In a second set of experiments, cells were maintained in 0.5% serum rather than serum deprivation as described above. Under low serum-supplemented conditions (relative to serum deprivation), basal levels of pERK were clearly elevated for HLE-B3 (compare Fig. 1A with Fig. 2A), as were, albeit less so, BLECs (compare Fig. 1B with 3B). The introduction of H2O2 which acted as a mitogen and promoted increased levels of pERK1/2, pAkt (to a less significant degree) and pSAPK/JNK was not an unexpected result (Figs. 2 and 3) as ranges of H2O2 concentrations (20 μM-200 μM) have been shown to cause transient increases in kinase activation in other studies (Chen et al., 2004; Goswami et al., 2003; Zhang et al., 1998). We deliberately used the elevated levels of pERK in the presence of low serum supplementation to our experimental advantage. UO126 is a highly selective inhibitor of MEK1/2 kinases which lie upstream of ERK1/2 in the MAPK signaling cascade (Favata et al., 1998). Utilizing the MEK1/2 inhibitor, UO126, a link was established between 17β-E2-stimulated MAPK signaling and stabilization of mitochondrial membrane potential. With the pretreatment of cells with UO126, pERK phosphorylation was significantly diminished (Figs. 2A and 3A). Thus, we could demonstrate a clear diminution on the intracellular degree of pERK with the application of the MEK1/2 inhibitor and accordingly contrast and evaluate the state of Δψm between the untreated basal and MEK1/2 inhibitor-treated reduced levels of pERK in HLE-B3 cells. Mitochondrial membrane potential was analyzed using the potentiometric dye, JC-1. HLE-B3 cells are H2O2-sensitive at supraphysiological doses (Wang et al., 2003) and their limited tolerance to oxidative stress was further diminished in the presence of UO126 as mitochondria displayed significant depolarization (Figs. 4 and 7A). Therefore, by inhibiting MAPK signaling, a demonstrable increase in mitochondrial depolarization was uncovered. pAkt levels were unaffected in the presence of UO126, an anticipated result as this kinase is part of the PI-3 kinase signaling pathway (Figs. 2B and 3B) and this observation effectively ruled out a role for pAkt in mitochondrial stabilization. Unpredictably, JNK phosphorylation was lessened in the presence of U0126 (Figs. 2C and 3C). Our data does not, at present, allow us to rule out the possibility the pJNK levels also play some role and influence mitochondrial stabilization. The ERK1/2 signaling pathway is generally regarded as a survival pathway, whereas the JNK kinases are described as stress kinases that become activated during environmental stress or inflammation (reviewed in Kyriakis and Avruch, 2001). However, it has been documented that PDGF and serum (Chen et al., 2004) can activate JNK phosphorylation in lens epithelial cells. Further investigation into the activation of JNKs by mitogens in lens epithelial cells and the potential interrelationship of this phosphorylation event with Δψm appears warranted.
Studies utilizing bovine lens fractions (Chaney and Spector, 1984) showed significant levels of reduced glutathione. A highly active glutathione redox cycle has been shown to contribute in the protection of lens epithelial cells from oxidant injury (Reddy et al., 1980). Bovine lens epithelial cells contained substantially higher levels of GSH than HLE-B3 cells (Table 1). Unlike the human lens epithelial cells in culture, BLECs exhibited absolute tolerance to the supraphysiological dose of H2O2 used in these studies insofar as mitochondrial depolarization was not readily evident (Figs. 5 and 7B). The attenuation of GSH levels in BLECs by administration of L-BSO prompted marked depolarization of mitochondria (Figs. 6 and 7C) under conditions of oxidative stress that heretofore had been negligible (Figs. 5 and 7B). These data confirm that peroxide-induced depolarization of mitochondria in HLE-B3 cells is not an artifact of viral transformation as normal epithelial cells could be made to depolarize once the ‘reducing power’ of the cell (i.e. ability to cope with acute oxidative insult) was compromised by lowering the intracellular pool of GSH. However, the effects of UO126 treatment with BLECs were not as easily distinguishable as with the HLE-B3 cells (compare the 4 h time point between Fig. 7A and C). Moreover, while 17β-E2 treatment did afford some level of protection against depolarization in the absence of UO126, the combination of oxidative stress, L-BSO and UO126 in bovine lens epithelial cells was ultimately too harsh an insult, and protection against mitochondrial depolarization was not as readily apparent.
The data presented in this study establish a relationship between estradiol-stimulated activation of the MAPK survival pathway and estradiols’ capacity to act as an antioxidative cytoprotectant via mitochondrial stabilization of Δψm in virally-transformed and normal lens epithelial cell cultures. It remains to be determined what the defined mechanism of protection might be which links pERK levels with the degree of mitochondrial stabilization. It is well established that the stimulation of the mitogenic MAPK and PI-3 kinase pathways can inhibit apoptotic activity of the BAD protein by promoting phosphorylation at serine sites 112 and 136, respectively. Phosphorylation at these sites results in the binding of BAD to 14-3-3 proteins and the inhibition of BAD binding to Bcl-2 and Bcl-xL (Zha et al., 1996). Recently, in breast cancer cells, E2 was able to induce BAD phosphorylation through Ras/ERK/p90RSK1 and Ras/PI-3 kinase/Akt pathways (Fernando and Wimalasena, 2004). The prevention of BAD binding to Bcl-2 proteins in the mitochondria and their subsequent inability to form the permeability transition pore, would conceivably lead to a stabilization of Δψm. The ability of 17β-E2 to activate anti-apoptotic cascades via mitogenic signaling pathways with subsequent increase in pBad levels and whether this is the mechanism that links stimulation of pERK to downstream stabilization of Δψm is presently unknown. Whether estradiol-stimulated activation of this anti-apoptoic cascade is predominantly via the ERK1/2 signaling pathway or working synergically with PI-3 kinase/Akt or JNK kinases to contribute to downstream stabilization of Δψm is currently under investigation.
In sum, our data demonstrate that 17β-estradiol activates (i.e. phosphorylates) ERK and stabilizes mitochondrial membrane potential when human and GSH-depleted bovine lens cells are exposed to acute hydrogen peroxide stress. The effectiveness of estradiol in the presence of a MEK inhibitor and hydrogen peroxide is associated with the relative glutathione reducing power of the cell and markedly differs from one species to another. To the best of our knowledge, our data is the first to establish a heretofore unknown link between estradiol-stimulated activation of the MAPK signaling pathway and cytoprotection of the mitochondrial membrane potential. The protective mechanism(s) activated by 17β-E2 are likely to be multifactorial and function by both genomic and non-genomic pathways integrating at a point of mitochondrial control which regulates the mitochondrial defense state in order to advance anti-oxidative protection. Targeting mitochondrial function to reduce oxidative stress, thereby preventing activation of the permeability transition pore, distinguishes a novel concept which will contribute to innovative regimens for prevention or treatment of oxidative stress-related mitochondrial pathology associated with cataractogenesis, Alzheimer’s disease and cancer.
Acknowledgments
This work was supported by Public Health National Service Awards to F.J.G. (EY02027) and P.R.C. (EY05570). The authors wish to thank Victor Leverenz for his technical support and Dr. Lawrence X. Oakford for graphical assistance in the preparation of this manuscript. This work was first reported and presented at the 2005 ARVO meeting in Fort Lauderdale, Florida.
References
- Andley UP, Rhim JS, Chylack LT, Jr, Flemming TP. Propagation and immortalization of human lens epithelial cells in culture. Invest Ophthalmol Vis Sci. 1994;35:3094–3102. [PubMed] [Google Scholar]
- Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F. Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- Cammarata PR, Yorio T. L-buthionine sulfoximine-induced loss of glutathione does not elicit PGH synthase inactivation in cultured bovine lens epithelial cells. J Ocul Pharmacol. 1990;6 (1):43–49. doi: 10.1089/jop.1990.6.43. [DOI] [PubMed] [Google Scholar]
- Cammarata PR, Schafer G, Chen S, Guo Z, Reeves RE. Osmoregulatory alterations in taurine uptake by cultured human and bovine lens epithelial cells. Invest Ophthalmol Vis Sci. 2002;43 (2):425–433. [PubMed] [Google Scholar]
- Chaney WG, Spector A. HPLC analysis of lens GSH and GSSG. Curr Eye Res. 1984;3 (2):345–350. doi: 10.3109/02713688408997219. [DOI] [PubMed] [Google Scholar]
- Chen KC, Zhou Y, Xing K, Krysan K, Lou MF. Platelet derived growth factor (PDGF)-induced reactive oxygen species in the lens epithelial cells: the redox signaling. Exp Eye Res. 2004;78:1057–1067. doi: 10.1016/j.exer.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Dykens JA. Isolated cerebellar and cerebral mitochondria produce free radicals when exposed to elevated Ca+2 and Na+: implications for neurodegeneration. J Neurochem. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273 (29):18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Fernando RI, Wimalasena J. Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras-dependent nongenomic pathways requiring signaling through ERK and Akt. Mol Biol Cell. 2004;15:3266–3284. doi: 10.1091/mbc.E03-11-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami S, Sheets NL, Zavadil J, Chauhan BK, Bottinger EP, Reddy VN, Kantorow M, Cvekl A. Spectrum and range of oxidative stress responses of human lens epithelial cells to H2O2 insult. Invest Ophthalmol Vis Sci. 2003;44:2084–2093. doi: 10.1167/iovs.02-0882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giblin FJ, McCready JP, Reddan JR, Dziedzic DC, Reddy VN. Detoxification of H2O2 by cultured rabbit lens epithelial cells: participation of the glutathione redox cycle. Exp Eye Res. 1985;40 (6):827–840. doi: 10.1016/0014-4835(85)90128-9. [DOI] [PubMed] [Google Scholar]
- Gridley KE, Green PS, Simpkins JW. A novel, synergistic interaction between 17 beta-estradiol and gluta-thione in the protection of neurons against beta-amyloid 25-35-induced toxicity in vitro. Mol Pharmacol. 1998;54:874–880. doi: 10.1124/mol.54.5.874. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81 (2):807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Levin ER. Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol. 2001;91:1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- Matthews J, Gustafsson JA. Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Intervent. 2003;3 (5):281–292. doi: 10.1124/mi.3.5.281. [DOI] [PubMed] [Google Scholar]
- Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol–receptor complex in MCF-7 cells. EMBO J. 1996;15:1292–1300. [PMC free article] [PubMed] [Google Scholar]
- Moor AN, Gottipati S, Mallet RT, Sun J, Giblin FJ, Roque R, Cammarata PR. A putative mitochondrial mechanism for antioxidative cytoprotection by 17 beta-estradiol. Exp Eye Res. 2004;78:933–944. doi: 10.1016/j.exer.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J Cereb Blood Flow Metab. 1999;19:231–245. doi: 10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M. Ovarian hormones elicit phosphorylation of Akt and extracellular-signal regulated kinase in explants of the cerebral cortex. Endocrine. 2000;14 (3):407–415. doi: 10.1385/ENDO:14:3:407. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors derive from a single transcript: studies of ERα and ERβ expressed n CHO cells. Mol Endocrinol. 1999;13:307–319. doi: 10.1210/mend.13.2.0239. [DOI] [PubMed] [Google Scholar]
- Reddy VN, Giblin FJ, Matsuda H. Defence systems of the lens against oxidative damage. In: Srinvastava SK, editor. Red Blood Cells and Lens Metabolism. Elsevier, North-Holland; New York: 1980. pp. 139–154. [Google Scholar]
- Wang J, Simpkins JW, Dykens JA, Cammarata PR. Oxidative damage to human lens epithelial cells in culture: estrogen protection of mitochondrial potential, ATP, and cell viability. Invest Ophthalmol Vis Sci. 2003;44:2067–2075. doi: 10.1167/iovs.02-0841. [DOI] [PubMed] [Google Scholar]
- Zha J, Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL. Cell. 1996;87 (4):619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhang J, Jin N, Liu Y, Rhoades RA. Hydrogen peroxide stimulates extracellular signal-regulated protein kinases in pulmonary arterial smooth muscle cells. Am J Respir Cell Mol Biol. 1998;19:324–332. doi: 10.1165/ajrcmb.19.2.3209. [DOI] [PubMed] [Google Scholar]