

Abstract
This study examined the inflammation, lung function impairment, and immune protection associated with either wild-type or interferon (IFN)-γ-deficient Tc1- or Tc2-CD8 effector cells responding to influenza pneumonia. The adoptive transfer of influenza hemagglutinin-specific Tc1 effectors afforded protection and elicited only minimal impairment of lung function. IFN-γ-deficient Tc1 effector cells were equally protective, but were associated with an eosinophil influx and slightly more lung function impairment early in the response. Relative to Tc1, Tc2 effector cells were less protective, elicited an eosinophil influx and a greater impairment of lung functions. IFN-γ-deficient Tc2 effector cells were not protective and were associated with the severest impairment of lung function throughout the response, an accumulation of neutrophils, and extensive pulmonary vasculitis and alveolar hemorrhaging. Deletion of IFN-γ was associated with a delay in effector cell recruitment and the elicitation of a more intense inflammatory response that resulted in more severe lung function impairment in the recipients of either Tc1 or Tc2 IFN-γ-deficient effector cells. Thus, during influenza infections, IFN-γ production by the responding CD8 T cells is associated with effector cell recruitment and mitigation of the associated inflammation and of the resulting impairment in lung functions but is not necessary for optimal protection.
Redundancies within the adaptive immune system provide for alternative protective response mechanisms to infectious agents. Pulmonary influenza virus infections elicit both CD8 and CD4 T-cell-mediated responses as well as B-cell-mediated humoral responses. 1 Viral-specific CD8 T cell responses using perforin and/or Fas-dependent pathways are the predominant means of viral clearance in primary infections. 2,3 In the absence of cytotoxic CD8 T cells, CD4 T-cell-dependent B cell responses, specific for the production of neutralizing antibodies, can clear a primary influenza virus infection although at a less efficient rate. 4,5 Although these protective responses rely on disparate effector mechanisms, the recruitment of T cells to the site of infection occurs in both cases. Although T cell recruitment is essential for these protective responses, it is becoming increasingly evident that some T cells are also associated with the development of influenza-related immunopathological sequelae. 6,7
The inflammatory sequelae associated with the resolution of influenza virus infections is attributed in part to the activity of host immune cells at the site of infection. 8 After the adoptive transfer of CD4-Th1 cells, clearance of an influenza infection is associated with a diminished level of inflammatory sequelae, whereas the transfer of CD4-Th2 cells is not protective and elicits an exacerbated pulmonary eosinophilia. 7 Infection of CD8 T-cell transgenic RAG-1 knockout mice with a high-dose inoculum of influenza virus (10 7 pfu) induces an exacerbated pulmonary inflammation, loss of normal lung structure, and eventual death of the animal. The administration of 100 pfu elicits significantly less immunopathology and the animals eventually recover from infection. 8 These studies suggest that the viral burden and the responding viral-specific T cells are critical factors in the development of influenza virus-associated immunopathology.
Differentiation of cytotoxic CD8 T cells along the Th1- and Th2-like cytokine secretion pattern results in the production of CD8-Tc1 and CD8-Tc2 subsets. 9 We have found in adoptive transfer studies using influenza hemagglutinin (HA)-specific CD8 effector T cells that CD8-Tc1 cells are more efficient at viral clearance than CD8-Tc2 cells despite their equivalent in vitro cytolytic activity before transfer. 10 The CD8-Tc1 effectors are rapidly recruited into the lung and migrate to the epithelial sites of infection, whereas CD8-Tc2 effectors are associated with delayed cellular migration and localization in the lung at sites distant from the infected airway epithelium. The differences observed in chemokine receptor mRNA expression of these CD8 effector subsets may account for these differences in cell recruitment. Despite these findings, characterization of the CD8-Tc1- and CD8-Tc2-associated immunopathology, the effect of the cytokines associated with these CD8 effector subsets on the inflammatory sequelae and the extent to which the subsequent CD8-Tc1- or CD8-Tc2-mediated inflammation impairs critical lung function during resolution of influenza viral infection remains unresolved.
In this investigation, we used an adoptive transfer model to further our understanding of how the recruitment of CD8-Tc1- and CD8-Tc2 HA-specific effectors impact the progression of influenza-related pneumonitis. Our model was designed to compare the immune-mediated inflammatory sequelae attributable to responding wild-type (+/+) or interferon (IFN)-γ-deficient (−/−) CD8 effector T cell subsets with the subsequent impairment of lung functions observed in the recipient groups of mice. Influenza virus HA-specific CD8 effector T cells from +/+ and IFN-γ−/− mice were polarized in vitro before adoptive transfer into influenza infected mice. The CD8-Tc1+/+ and CD8-Tc1-IFN-γ−/− effectors provided effective protection against viral infection and did not induce extensive lung damage or prolonged impairment of lung functions in recipient mice. Although early recruitment of the Tc1+/+ effectors was more efficient than seen for the Tc1-IFN-γ−/− effectors, both effector subpopulations were present in the lung at effective anti-viral levels later in the infection. The recipients of the CD8-Tc2+/+ effectors displayed a delayed partial protection against viral infection that was accompanied by a significant level of lung damage and by a protracted period of recovery of lung functions. In contrast, recipients of CD8-Tc2-IFN-γ−/− effectors did not clear the virus and they continued to display the most severe indications of lung damage and compromise lung function whereas recipients of all other CD8 effectors exhibited signs of recovery. The Tc2+/+ effectors were more efficiently recruited into the lung than Tc2-IFN-γ−/− effectors throughout the infection process although neither group cleared the virus during the course of the experiment. These results indicate that the HA-specific CD8-Tc1 effectors were of greater benefit to the host in resistance to influenza infection, regardless of their ability to produce IFN-γ. On the other hand, the ability of the effectors to produce even small amounts of IFN-γ was crucial for the enhancement of the protective capabilities of CD8-Tc2 effectors and for mitigating the severity of the inflammatory sequelae attributable to the presence of the CD8-Tc2 effector cells in the lung.
Materials and Methods
Mice
BALB/c Thy 1.1+ mice were bred at and obtained from the Animal Breeding Facility at the Trudeau Institute, Saranac Lake, NY. Clone 4 TCR transgenic mice expressing the Vβ8.2/Vα10 chains in the TCR of CD8 T cells specific for the influenza virus HA2 transmembrane peptide at residues 518 to 528 (IYSTVASSL) 11 were originally provided by Dr. Linda Sherman (The Scripps Research Institute, La Jolla, CA). The mice were subsequently bred at and obtained from the Trudeau Institute’s Animal Breeding Facility. These mice were backcrossed for eight generations onto a BALB/c+/+ Thy1.2+ background and onto a BALB/c IFN-γ−/− Thy1.2+ background.
Preparation of Tc1 and Tc2 Effector Cells
The preparation of Tc1 and Tc2 effector cells was performed as previously described. 10 Briefly, an enriched CD8 T cell population was prepared from splenocyte and lymph node preparations obtained from the clone 4 HA-specific transgenic BALB/c+/+ or IFN-γ−/− Thy 1.2+ donor mice. The cell preparations were passed over nylon wool followed by complement-mediated antibody depletion using anti-CD4 (RL172.4), anti-heat-stable antigen (J11D) and anti-major histocompatibility complex class II (D3.137, M5114, CA4) monoclonal antibodies (mAbs). The CD8 T cell population harvested from the Percoll gradients (Sigma Chemical Co., St. Louis, MO) were 90 to 95% CD8+Vβ8+ T cells. Antigen presenting cell (APC) preparations were prepared from T-cell-depleted splenocytes using complement, anti-Thy 1.2 (HO13.14, F7D5), anti-CD4 (RL172.4) and anti-CD8 (3.155) mAb treatment followed by lipopolysaccharide (25 [m]g/ml) and dextran sulfate (25 μg/ml) stimulation for 48 hours. The HA peptide (11 μmol/L) was loaded onto the APC population for 30 minutes at 37°C after which the cells were mitomycin treated for 40 minutes at 37°C. After three washes, the APC populations were cultured in RPMI 1640 media (Irving Scientific Co., Santa Ana, CA) supplemented with penicillin, streptomycin, glutamine, 2-mercapto ethanol Hepes, and 10% fetal calf serum. Effector Tc1 cells were generated by stimulation of CD8 clone 4 transgenic T cells (2 × 10 5 cells/ml) with HA-peptide-loaded APCs (2 × 10 5 cells/ml) in the presence of interleukin (IL)-2 (10 ng/ml), IL-12 (2 ng/ml), and anti-IL-4 mAb (10 μg/ml). Effector Tc2 cells were likewise generated by co-culture of CD8 T-cell- and HA-peptide-loaded APC (both at 2 × 10 5 cells/ml) populations in the presence of IL-2 (10 ng/ml), IL-4 (5.6 ng/ml), and anti-IFN-γ mAb (10 μg/ml). At day 4 of culture, the effector populations were 99% CD8+Vβ8+ T cells.
Analysis of Cytokine Production by Tc Effector Cells
The production of IFN-γ, IL-4, and IL-5 by Tc1 and Tc2+/+ and IFN-γ−/− effector populations was assessed 4 days after their generation. Effector populations (2 × 10 5 cells/ml) were restimulated with mitomycin-treated P815 APCs with or without previous loading of the HA peptide. The cytokine-specific enzyme-linked immunosorbent assays were performed as previously described. 12 RNase protection assays were carried out as previously described. 10 The probe set mCK-3 (PharMingen, San Diego CA) was used to measure specific cytokine mRNA levels. The bands were detected using the densitometric feature of the Quantity One software (Bio-Rad Laboratories, Hercules, CA) and normalized against the housekeeping gene L32.
Cytotoxicity Assay
The cytolytic activity of each of the effector populations was assessed 4 days after their generation. P815 APCs were incubated or not with HA peptide (11 μmol/L) at 37°C for 30 minutes. The target APCs were then incubated with 3.7 mBq of 51Cr for 1 hour at 37°C. After three washes, the P815 target APCs (10 4 cells/well) were incubated with CD8 effector cells at the indicated effector:target (E:T) ratios. After a 4-hour incubation at 37°C the supernatants were collected and the level of radioactivity was detected by γ counting. The percentage of cytotoxicity was calculated as follows: 100 × (cpm experimental − cpm spontaneous)/cpm total − cpm spontaneous).
Viral Infection of Mice and Adoptive Cell Transfer
Influenza virus (A/PR/8/34) was grown in the allantoic fluid of 10-day-old embryonated chicken eggs. BALB/c Thy1.1+ mice were inoculated intranasally with 10 LD50 in 100 μl (10−3 dilution of the viral stock) while under light halothane anesthesia. Within 1 hour after viral infection, 4-day-old +/+ or IFN-γ−/− Tc1 or Tc2 HA transgenic Thy1.2+ cells (10 7 cells by intravenous tail injection in 0.5 ml of phosphate-buffered saline) were adoptively transferred. The cells were washed twice before adoptive transfer.
Lung Function Measurements
Respiration rates were measured using a whole body plethysmograph. Mice were confined within the chamber so that respirations per minute could be calculated from steady strip-chart recordings. Blood gases were measured from arterial blood samples taken from the ventral tail artery. 13 Animals were warmed for 10 minutes in an enclosed warming chamber at 37 to 40°C to produce vasodilatation of the ventral tail artery. The artery was carefully lacerated and arterial blood was collected into heparinized capillary tubes. Blood gas analysis was done on an IL 1620 series blood-gas analyzer (Instrumentation Laboratories, Lexington, MA).
Recovery of Cells from Infected Lungs
Five mice from each experimental group were killed on day 3 and day 5 after infection. The difference between the starting and final body weight of each mouse was recorded as the change in body weight during the course of the experiment. At the designated time points, the mice were placed under deep halothane anesthetization and exsanguinated by perforation of the abdominal aorta. The trachea was exposed and cannulated and the lungs were lavaged with two 1-ml volumes of cold 0.85% NaCl. The total cell recovery was recorded and an aliquot for differential cell counts was taken and stained using Diff-Quik (Baxter, Miami, FL). In a second series of experiments, the whole lung was removed and passed through a mesh screen to obtain a single cell suspension. The cells were then washed in Hanks’ balanced salt solution (HBSS), resuspended in 2 ml of 80% Percoll, and overlaid with 2 ml of 40% Percoll. The gradient was then centrifuged for 20 minutes at 1,700 rpm on a Sorval H1000B rotor (Du Pont, Wilmington, DE). The lymphocyte population recovered at the 80 to 40% interface of the gradient was washed in HBSS and use for fluorescence-activated cell sorting analysis.
Flow Cytometry
Analysis by flow cytometry of the lymphocyte population recovered in the lung homogenate was performed using a biotin-conjugated mAb specific for mouse CD90.2 (anti-Thy 1.2, clone 53-2.1) (PharMingen, San Diego, CA), and allophycocyanin-streptavidin (Caltag Laboratories, Burlingame, CA). All plot analyses were gated on live lymphocyte populations. Analysis of the stained cells was done on a FACScan using Cell Quest software (Becton Dickinson, San Jose, CA).
Lung Histology
Lung tissue sections were cut from the left lobe of whole lungs fixed in phosphate-buffered 10% formalin and embedded in paraffin. Two 4-μm thick sections from different regions of the left lobe were taken from each animal in each group. Sections were stained with hematoxylin and eosin for histopathological evaluation by an independent observer in a blinded and randomized manner. Immunohistochemical staining for influenza viral antigen was done using frozen tissue sections cut from cryopreserved lungs. Tissue sections were fixed in 4% formaldehyde for 10 minutes. The viral antigen was detected using a biotin-conjugated goat polysera specific for H1N1 influenza viruses (Accurate Chemical & Scientific Corp., Westbury, NY) and an avidin-conjugated alkaline phosphatase-staining reagent (Vectastain ABC Kit; Vector Laboratories, Burlingame, CA).
Viral Recovery
Virus recovered from the lungs of influenza infected mice was detected by plaque assay on Madin-Darby canine kidney cells. Lungs were removed from infected mice on days 3, 5, and 6 after infection and snap-frozen in serum-free minimal essential medium supplemented with 0.2% bovine serum albumin, 20 mmol/L Hepes, 100 U/ml penicillin, and 100 μg/ml streptomycin. Snap-frozen lungs were stored at −70°C until analyzed. Madin-Darby canine kidney cells were grown in 12-well plates (Becton Dickinson) to 90% confluency in minimal essential medium supplemented with 10% fetal calf serum, 20 mmol/L Hepes, 100 U/ml penicillin, and 100 μg/ml streptomycin. The snap-frozen lungs were homogenized and 100 μl samples from 10-fold dilutions of the homogenates were used to inoculate the Madin-Darby canine kidney monolayers. After incubation of the monolayers for 1 hour at 35°C, a 1-ml overlay of minimal essential medium containing 0.2% bovine serum albumin, 20 mmol/L Hepes, 100 U/ml penicillin, 100 μg/ml streptomycin, 100 μg/ml DEAE Dextran (Sigma Chemical Co.), 1 μg/ml TPCK trypsin (Life Technologies, Inc., Grand Island, NY), and 0.6% agar (Becton Dickinson) was added. The assay was then incubated for 4 to 5 days at 35°C after which 1 ml of 20% acetic acid was added to each well. The assay was incubated overnight and then stained with 0.2% crystal violet on the next day. The limit of detection in the assay was 20 viral particles per lung.
Statistics
The data are expressed as the means ± SD. At each time point, six groups of mice containing five mice per group were taken. Statistically significant differences between the means ± SD of the treatment groups were determined using either a Student’s t-test or a Kruskal-Wallis one analysis of variance on ranks followed by the Dunnett’s method of all pairwise multiple comparisons. Differences were considered significant if P < 0.05.
Results
Cytokine Production by CD8 T Cell Effectors
Analysis of cytokine production by 4-day-old effector cells revealed that the Tc1+/+ effectors produced significant levels of IFN-γ when restimulated by HA peptide-loaded target cells whereas the Tc2+/+ effectors produced only low levels of IFN-γ under similar conditions (Figure 1A) ▶ . The significant production levels of IL-4 and IL-5 were equivalent in the Tc2+/+ and Tc2-IFN-γ−/− effectors, whereas production of these cytokines by Tc1+/+ and Tc1-IFN-γ−/− effectors was not detected (Figure 1, B and C) ▶ . The in vitro production levels of IFN-γ, IL-4, and IL-5 by the various effector subsets was HA-peptide-specific and was proportional to the number of peptide-loaded target cells. Quantification of mRNA levels for Th1-associated cytokines 10 by RNase protection assay indicated that Tc1+/+ and Tc1-IFN-γ−/− effectors produced equivalent levels of tumor necrosis factor-α and lymphotoxin-α (LT-α) mRNA. The cytokine mRNA levels in the Tc1+/+ and Tc1-IFN-γ−/− effectors, normalized to the expression level of the L32 housekeeping gene, were 45.10% versus 45.63% for LT-α and 38.42% versus 39.21% for tumor necrosis factor-α mRNA, respectively.
Figure 1.
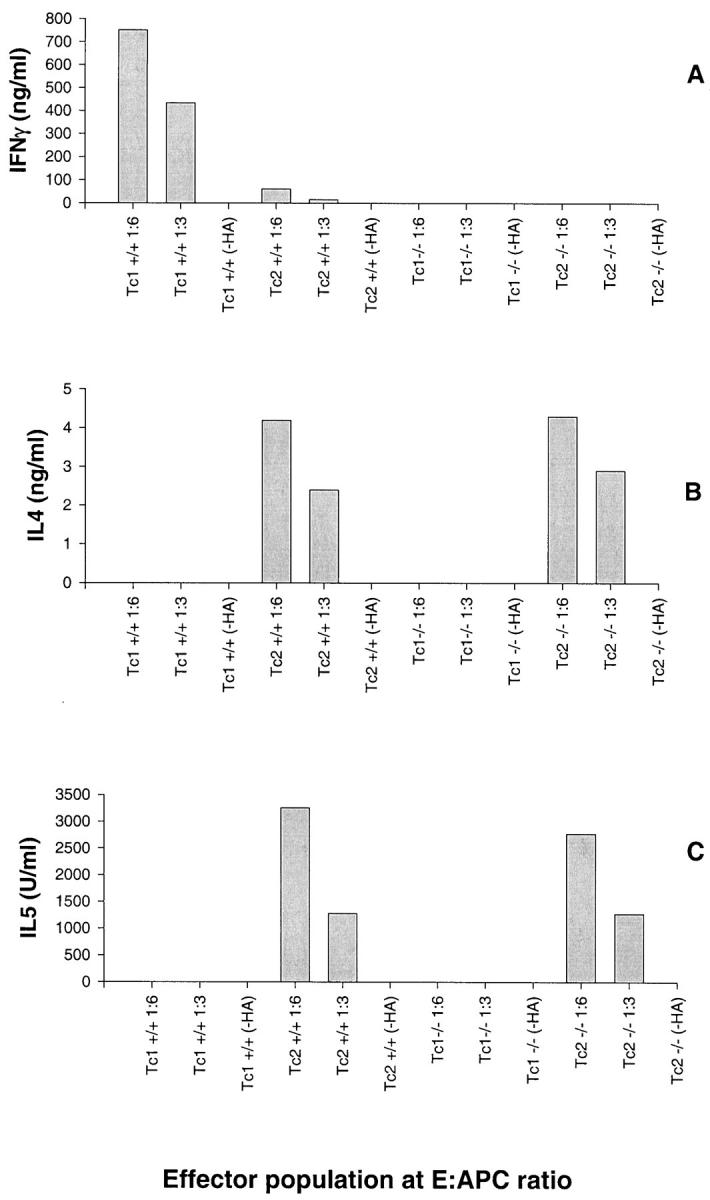
In vitro cytokine release from effector populations after restimulation with HA-peptide loaded or not loaded (−HA) P815 target APCs. Cytokine levels were measured by enzyme-linked immunosorbent assay after a 24-hour stimulation. Results are the means of duplicate wells from one representative experiment of three independently performed experiments.
Cytolytic Activity of CD8 T Cell Effectors
Four days after their generation, both the Tc1 and Tc2+/+ effector populations had equivalent in vitro cytolytic activity levels and both were slightly greater than that of the Tc1-IFN-γ−/− effectors (Figure 2) ▶ . All three of these populations showed increased target cell lysis in proportion to the number of effector cells present. The Tc2-IFN-γ−/− effectors maintained a low level of cytolytic activity against all titrations of HA peptide-loaded target cells.
Figure 2.

In vitro cytolytic activity of effector populations. 51Cr release from HA peptide-loaded P815 target APCs (10 4 cells per well) was assessed after a 4-hour incubation at 37°C with effector cell populations. These results are representative of three independently performed experiments.
Change in Body Weight
At day 3 after infection, the loss in body weight in the recipients of Tc1+/+ (−3.6 ± 0.3 g), Tc2+/+ (−3.3 ± 0.4 g), and Tc1-IFN-γ−/− (−3.6 ± 0.4 g) effectors was significantly greater (P < 0.05) than that recorded for the infected control group, (−2.8 ± 0.3 g). By day 5 after infection, only the loss in body weight observed in the recipients of the Tc1+/+ (−3.2 ± 0.3 g) and Tc2+/+ (−3.4 ± 1.2 g) effectors was significantly less than (P < 0.05) the weight loss recorded for the infected control mice (−5.0 ± 0.8 g).
Viral Recovery from Influenza Infected Mice
Three days after viral infection and adoptive cell transfer, the infected control mice had a viral burden of 3.1 ± 0.5 × 10 7 pfu of virus in their lungs (Figure 3) ▶ . At this time, the viral burden in the lungs of the recipients of the Tc1+/+ effectors (1.36 ± 0.8 × 10 6 pfu) and the Tc1-IFN-γ−/− effectors (2.7 ± 1.0 × 10 6 pfu) was <10% of that observed in the infected control mice. The viral burden in the lungs of the recipients of the Tc2+/+ effectors (8.1 ± 6.7 × 10 6 pfu) and that observed in the recipients of the Tc2-IFN-γ−/− effectors (1.9 ± 0.9 × 10 7 pfu) were equivalent to 25 and 60%, respectively, of the viral burden in the lungs of the infected control mice at day 3. By day 5, the viral burden in the lungs of the recipients of Tc1+/+ effectors (1.6 ± 3.3 × 10 3 pfu) and Tc1-IFN-γ−/− effectors (2.24 ± 3.30 × 10 2 pfu) was reduced to <0.0001% of the viral burden in the lungs of the infected control mice (2.12 ± 2.70 × 10 7 pfu). At this time, recipients of Tc2+/+ effectors had ∼3% (5.9 ± 6.8 × 10 5 pfu) of the viral burden of the infected control mice whereas the recipients of the Tc2-IFN-γ−/− effectors still had at least 60% (1.32 ± 1.74 × 10 7 pfu) of the viral burden observed in the infected control mice. At 6 days after infection, none of the recipients of the Tc1-IFN-γ effectors had any detectable virus in their lungs and only one recipient of the Tc1+/+ effectors had a detectable viral burden in its lung. The recipients of the Tc2+/+ effectors had 2% (7.04 ± 9.37 × 10 3 pfu) of the viral burden observed in the infected control mice (3.9 ± 3.6 × 10 5 pfu) whereas the viral burden in the lungs of the recipients of the Tc2-IFN-γ−/− effectors was slightly greater than in the infected control mice at this time.
Figure 3.

Viral recovery from lungs of infected mice after cell transfer. Madin-Darby canine kidney cell monolayers, at 90% confluency, were inoculated with 100 μl of 10-fold dilutions of lung homogenates from animals at days 3, 5, and 6 after infection/cell transfer. Plaque assays were incubated for 4 days at 35°C and then counted. The results are the means ±SD of triplicate wells for each dilution of five mice per group at each time point in one experiment. The limit of detection was 20 virus particles per lung.
Cellular Accumulation Levels in BALF
The recovery of neutrophils in the BALF of the Tc1-IFN-γ−/− recipients was greater than the recovery from the Tc1+/+ recipients on both days 3 and 5 (Table 1) ▶ . In contrast, equivalent numbers of neutrophils were recovered from the BALF of Tc2+/+ and Tc2-IFN-γ−/− recipients only on day 3. The intensity of the neutrophil accumulation in all infected groups at day 5, with the exception of the Tc2-IFN-γ−/− recipients was drastically reduced. The neutrophil accumulation level in the recipients of Tc2-IFN-γ−/− effectors was significantly greater than in all other groups at this time.
Table 1.
Neutrophil and Eosinophil Cell Counts from the BALF Obtained from Infected Mice after Adoptive Cell Transfer
| Cells transferred* | Neutrophils (×105)† | Eosinophils (×105) | ||
|---|---|---|---|---|
| Day 3 | Day 5 | Day 3 | Day 5 | |
| Tc1+/+ | 14.3 ± 4.7 | 0.4 ± 0.3 | 0.4 ± 0.3 | 0.1 ± 0.2 |
| Tc2+/+ | 23.4 ± 6.0 | 2.8 ± 2.0 | 3.3 ± 1.5‡ | 7.2 ± 2.3‡ |
| Tc1-IFN-γ−/− | 27.4 ± 5.4 | 2.0 ± 0.9 | 1.9 ± 1.4‡ | 6.6 ± 3.6‡ |
| Tc2-IFN-γ−/− | 19.2 ± 4.6 | 23.1 ± 7.2‡ | 1.5 ± 0.6‡ | 1.6 ± 0.9‡ |
| Flu, no cells | 20.1 ± 3.6 | 4.2 ± 2.0 | 0.0 ± 0.0 | 0.02 ± 0.04 |
| No flu, no cells | 0.03 ± 0.02 | 0.01 ± 0.01 | 0.0 ± 0.0 | 0.0 ± 0.0 |
*The results are the means ± standard deviations for five mice in each group per time point for one experiment.
†Differential cell counts multiplied by the total cell counts were used to calculate the number of recovered neutrophils and eosinophils in the BALF of each mouse.
‡P < 0.05. Results are statistically significant relative to the flu infected control group at each time point, according to Student’s t-test or Kruskal-Wallis analysis of variance and Dunnett’s pairwise multiple comparisons.
The recovery of eosinophils from the BALF of Tc2+/+, Tc1-IFN-γ−/−, and Tc2-IFN-γ−/− recipients was significantly greater than from the infected control mice on both day 3 and day 5 (Table 1) ▶ . The number of eosinophils recovered from the Tc1+/+ recipients on these days was minimal and not significantly different from the infected control mice. The recovery of eosinophils from the BALF of the Tc2+/+ and Tc1-IFN-γ−/− recipients increased substantially from day 3 to day 5, whereas in the remaining groups, the level of eosinophil recovery remained close to day 3 levels.
Recruitment of Thy1.2+ Cells into the Infected Lungs
Three days after infection and effector cell-transfer, the recruitment of effector cells was greatest in the recipients of Tc1+/+ effectors (Table 2) ▶ . The recruitment of Tc2+/+ effector cells was half that seen for the Tc1+/+ recipients. The recipients of both IFN-γ−/− subsets had equivalent effector recruitment levels that were significantly lower relative to their respective +/+ recipients. By day 5 after infection and effector cell transfer, the recruitment of Tc1+/+ effectors was significantly greater than that observed in any of the other recipient groups. The recruitment of Tc2-IFN-γ−/− effectors was significantly less than that observed for any other recipient group. The recruitment of effector cells by the recipients of Tc1-IFN-γ−/− and Tc2+/+ effectors to the lung was not significantly different.
Table 2.
Recruitment of Thy1.2+ Cells into the Lung
| Effector cells transferred* | Day 3 (×105) | Day 5 (×105) |
|---|---|---|
| Tc1+/+ | 0.68 ± 0.37 | 3.42 ± 1.11 |
| Tc1-IFN-γ−/− | 0.08 ± 0.02 | 1.18 ± 0.84 |
| Tc2+/+ | 0.34 ± 0.11 | 0.97 ± 0.06 |
| Tc2-IFN-γ−/− | 0.09 ± 0.02 | 0.45 ± 0.06 |
| Flu, no cells transferred | 0.00 ± 0.00 | 0.13 ± 0.06 |
*The results are the means ± standard deviations for four mice in each group per time point for one experiment.
Location of Viral Antigen in the Lung
Three days after infection, the epithelium of the airways, up to and including the terminal bronchial epithelium of all groups of mice, stained positive for influenza antigen. Only occasionally was influenza antigen staining seen in the alveoli and then, only in alveolar macrophages. At day 5, the infected control mice stained for influenza antigen in all bronchial epithelium (arrows), the epithelium of the alveoli closest to the terminal bronchioles (arrowheads) and the alveolar macrophages of the distal alveoli (Figure 4E) ▶ . The recipients of either the Tc2+/+ (Figure 4C) ▶ or Tc2-IFN-γ−/− (Figure 4D) ▶ effectors had a similar pattern of staining. In mice given either Tc1+/+ (Figure 4A) ▶ or Tc1-IFN-γ−/− (Figure 4B) ▶ effectors, influenza antigen staining was limited primarily to the bronchial epithelium down to the terminal bronchial epithelium. Only occasional influenza antigen staining was seen in the alveoli of either of these two groups of mice. Thus, the virus seemed to spread from the bronchial epithelium to the alveolar epithelium in the infected control mice and in the mice given Tc2+/+ or IFN-γ−/− effectors whereas the spread of the virus was limited to the bronchial epithelium in the mice given the Tc1+/+ or IFN-γ−/− effectors.
Figure 4.
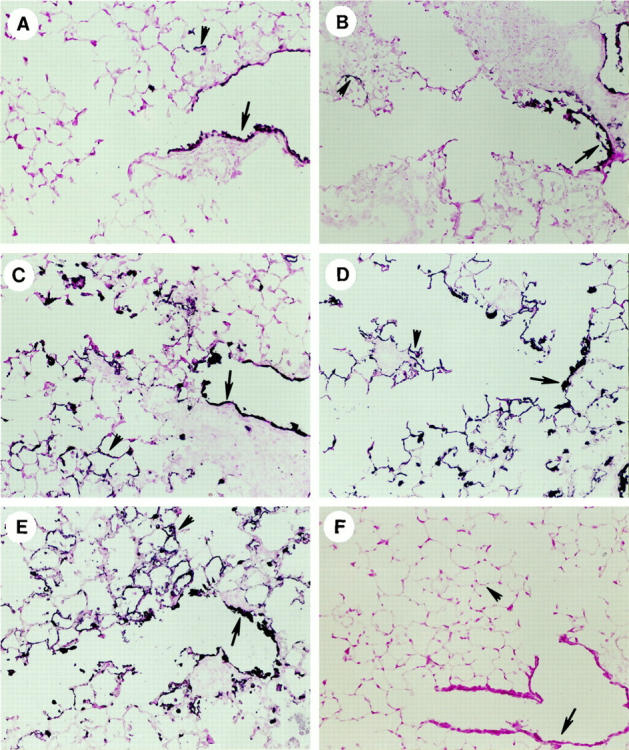
Lungs sections from the left lobe of mice were taken 5 days after infection/cell transfer. The tissue sections were cut from cryopreserved lungs and stained for the presence of influenza viral antigen. Viral antigen was present in the bronchial epithelium (arrows) and in the alveolar epithelium (arrowheads). Tissue sections were cut from the recipients of Tc1+/+ (A), Tc1-IFN-γ (B), Tc2+/+ (C), Tc2-IFN-γ−/− (D) effectors, from infected control mice (E) and from uninfected control mice (F). Original magnification, ×200.
Pulmonary Histology
No lesions were observed in the lungs from the uninfected control mice that did not receive any effector cells (Figure 5A) ▶ . At 5 days after infection and effector cell transfer, the principal morphological lesions in the examined lung sections of infected control mice were marked degeneration, necrosis, and exfoliation of the luminal surface epithelium lining the large and small conducting airways (ie, axial airway, and preterminal and terminal bronchioles) throughout the lung (Figure 5B) ▶ . Regenerative epithelium, characterized by basophilic, squamoid to low cuboidal basal cells with occasional mitotic figures, partially lined some of these altered airways. Associated with the airway epithelial lesions was a moderate mononuclear inflammatory cell infiltrate in the interstitial space surrounding the airways and blood vessels. The infiltrating inflammatory cells were predominantly lymphocytes and monocytes, with considerably lesser numbers of neutrophils and eosinophils. Although the inflammatory response was principally perivascular and peribronchiolar, there was mild extension of this cellular inflammation into the tissue and airspaces of the surrounding alveolar parenchyma. There was also a mild increase of alveolar macrophages in the affected parenchymal regions.
Figure 5.
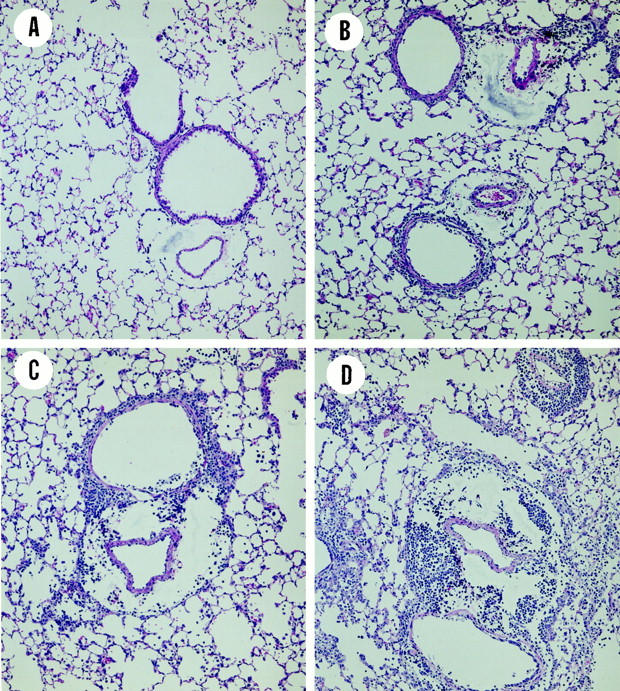
Lung sections cut from the left lobe of mice taken 5 days after adoptive transfer and influenza infection. The tissue sections were cut from 4% formalin-fixed lungs, mounted, and then stained with H&E. A: Uninfected control mice without adoptive transfer. B: Influenza infected control mice without adoptive transfer. C: Infected mice given Tc1+/+ effectors. D: Infected mice given Tc1-IFN-γ−/− effectors. Original magnification, ×100.
A similar, but more severe perivascular and peribronchiolar mononuclear cell infiltrate with more extension into the surrounding alveolar parenchyma, especially alveolar ducts, was present in the lung sections of recipients of Tc1+/+ and Tc1-IFN-γ−/− effectors (Figure 5, C and D ▶ , respectively). The extent and severity of the virus-induced airway epithelial lesions were similar among the Tc1+/+ and Tc1-IFN-γ−/− recipients. In addition, there was a slightly greater inflammatory response in the recipients of the Tc1-IFN-γ−/− effectors.
Although the necrotizing injury to the surface epithelium lining the conducting airways of the lung was extensive and similar in the Tc1+/+ and Tc2+/+ recipients, the associated inflammatory cell response was conspicuously different between these two groups. In contrast to the mononuclear (lymphocytes and monocytes) inflammatory cell response in the Tc1+/+ recipients, Tc2+/+ recipients had a marked perivascular and peribronchiolar cell infiltrate composed principally of numerous large macrophages, epithelioid cells, eosinophils, and widely scattered multinucleated giant cells (Figure 6, A and B) ▶ . Lymphocytes, monocytes, and neutrophils were also present, but in lesser numbers. This inflammatory response often extended into the adjacent alveolar parenchyma (ie, alveolitis with associated type two cell hyperplasia) and sometimes to the outer pleural surface (ie, pleuritis with mesothelial cell hyperplasia). Fibrinoid necrosis of the walls of the pulmonary vasculature, most notably in the pulmonary arteries (Figure 6, A–D ▶ , arrows) and widely scattered perivascular and alveolar hemorrhage (Figure 6, A and D ▶ , arrowhead) were additional features of the Tc2+/+ and Tc2-IFN-γ−/− recipients.
Figure 6.
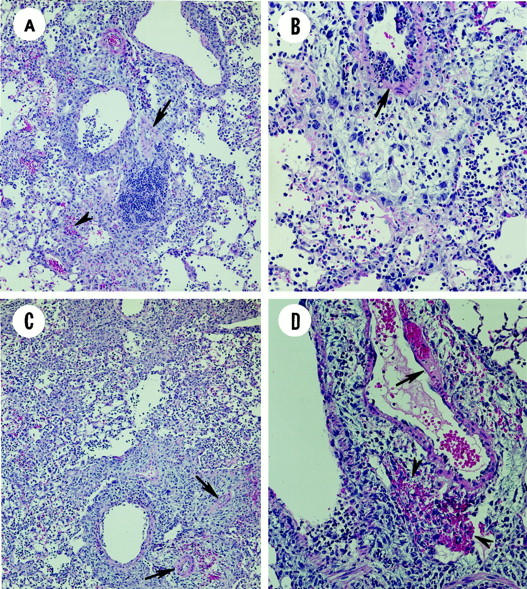
Lung sections cut from the left lobe of mice taken 5 days after adoptive transfer and influenza infection. Tissue sections were cut from lungs fixed in 4% formalin, mounted, and then stained with H&E. A: Mice given Tc2+/+ effectors (original magnification, ×100). B: Mice given Tc2+/+ effectors (original magnification, ×200). C: Mice given Tc2-IFN-γ{minus]/− effectors (original magnification, ×100). D: Mice given Tc2-IFN-γ−/− effectors (original magnification, ×200).
The Tc2-IFN-γ−/− recipients (Figure 6, C and D) ▶ were generally more severely affected than similarly treated Tc2+/+ recipients, both in the extent and severity of these induced lung lesions. In some areas of the lungs of the Tc2-IFN-γ−/− recipients, the alveolar airspaces were completely effaced by the infiltration and accumulation of large macrophages, epithelioid cells, eosinophils, and hypertrophic/hyperplastic type two cells. Multinucleated macrophages and giant cells were often widely scattered in these affected regions. Accumulation of fibrinous material was also observed in some of the severely affected alveolar airspaces.
Respiratory Rates
All of the mice that received +/+ or IFN-γ−/− effectors exhibited an approximate 20% increase in their respiratory rates immediately after infection and effector cell transfer (Figure 7, A and B) ▶ . Between day 1 and 2, all of these mice displayed a decrease in their respiration rates. The respiration rate of the infected control mice increased to its maximum level during this same time (Figure 7C) ▶ . After day 2, the respiration rates of the recipients of Tc1+/+ and Tc1-IFN-γ−/− (Figure 7A) ▶ , and Tc2+/+ (Figure 7B) ▶ effectors increased to and were then maintained within a maximal range until day 5. Recipients of Tc1+/+ and Tc1-IFN-γ−/− effectors displayed more rapid increases to slightly higher respiration rates than did the Tc2+/+ recipients. Only the Tc2-IFN-γ−/− recipients displayed a diminution in their respiration rate relative to levels that were equivalent to those observed with the infected control mice after (Figure 7, B and C) ▶ .
Figure 7.
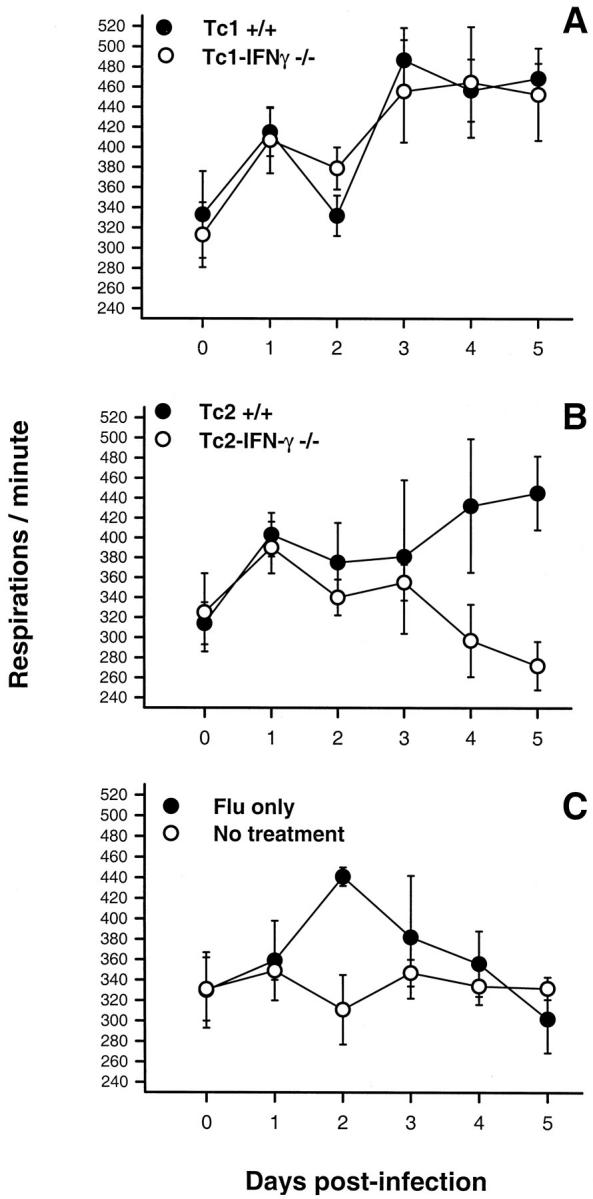
Respiration rate of animals after infection and adoptive cell transfer. A: Infected mice that received Tc1+/+ or Tc2+/+ effectors. B: Infected mice that received Tc1-IFN-γ−/− or Tc2-IFN-γ−/− effectors. C: Infected and noninfected mice that did not receive any cells. Results are the means ± standards of five mice per group at each time point for one experiment.
Arterial pO2
Arterial blood pO2 levels in the recipients of Tc1+/+ effectors were equivalent to that found for the uninfected control mice throughout the course of the experiment (Figure 8A) ▶ . A significantly depressed pO2 level was observed at day 3 in the Tc1-IFN-γ−/− recipients. The pO2 levels in these animals increased thereafter to levels equivalent to that recorded in the uninfected controls and Tc1+/+ recipients by day 6. At day 3 the recipients of Tc2+/+ effectors had significantly lower pO2 levels than any of the other groups (Figure 8B) ▶ . The arterial pO2 levels in these animals significantly increased by day 5 and were maintained to day 6. Although the Tc2-IFN-γ−/− recipients had only a slightly reduced pO2 level at day 3, their pO2 levels by day 5 were the lowest of the recorded levels for this group during the experiment. Although a slight increase in their pO2 levels was noted for these animals between day 5 and day 6, they still remained significantly depressed relative to the uninfected control mice.
Figure 8.
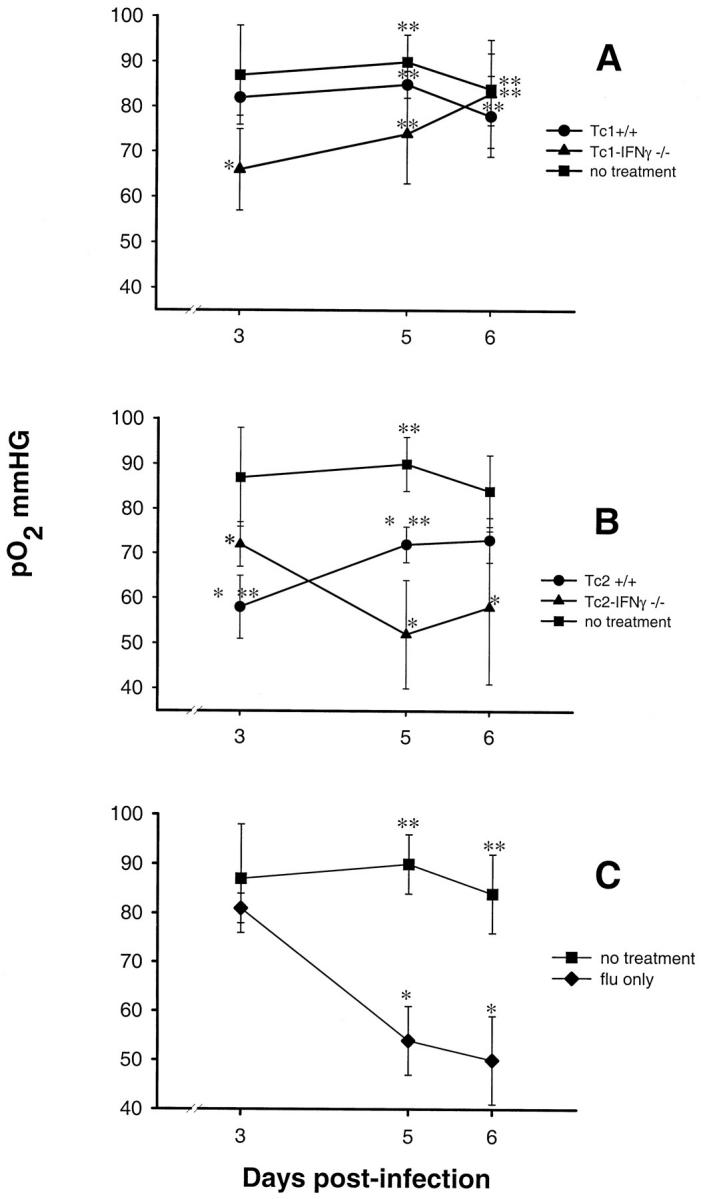
Arterial pO2 levels of infected mice after adoptive cell transfer. Arterial pO2 levels were assessed from arterial blood samples taken from the ventral tail artery of the mice killed at days 3, 5, and 6. A: Infected mice that received Tc1+/+ or Tc1-IFN-γ−/− effectors. B: Infected mice that received Tc2+/+ or Tc2-IFN-γ−/− effectors. C: Infected mice that did not receive any cells. All plots have the pO2 levels of uninfected animals that did not receive any cells as a standard reference. Significant difference in values relative to the uninfected/nontransferred control mice; *, P < 0.05, or relative to the infected/nontransferred control mice; **, P < 0.05. Statistically significant differences were calculated by Kruskal-Wallis analysis of variance and Dunnett’s pairwise multiple comparisons for A and B. Statistically significant differences were calculated by Student’s t-test for C. The results are the means ±SD for five mice in each group per time point for one experiment.
Discussion
The evidence presented in our study indicated that IFN-γ production associated with responding CD8 effector cells mitigated the inflammatory sequelae affiliated with the presence of these effectors in the lung. This in turn was reflected in a diminution of the subsequent level of impairment in lung function. Critical differences in the inflammatory sequelae and impaired lung functions were more evident between the recipients of Tc2+/+ and Tc2-IFN-γ−/− effectors. This mitigating role for IFN-γ was most apparent with responding CD8-Tc2+/+ effector cells than with the responding Tc1+/+ effector cells.
The comparable level of mRNA expression for the Tc1-associated cytokines, tumor necrosis factor-α, and LT-α 10 suggested that polarization of the Tc1+/+ and Tc1-IFN-γ−/− effectors was equivalent. Likewise, the similar IL-4 and IL-5 production levels in the Tc2+/+ and Tc2-IFN-γ−/− effectors suggested equivalent polarization of these subsets. The low level of IFN-γ production in the Tc2+/+ effector preparations has been detected previously. 10,14 It has been suggested that small numbers of contaminating Tc1 effector cells may account for this. 12 Adoptively transferred Thy1.2+ CD8 Tc1 and Tc2 cells recovered from the spleen 3 days after influenza infection still retained the same IFN-γ and IL-4 secretion pattern as the polarized effector cells before their transfer (unpublished observations). In earlier studies, the recovery of Tc1 and Tc2 CD8 memory cells from the lymphoid tissues of uninfected adoptively transferred hosts demonstrated that the cytokine secretion pattern of the polarized effector cells persisted for up to 13 weeks after adoptive transfer. 12
Although the Tc1+/+ and Tc2+/+ effectors had equivalent in vitro cytolytic activity levels, the mice that received Tc2+/+ effectors exhibited a delayed pattern of viral clearance. In vivo factors not accounted for in the in vitro assays may have differentially regulated the ability of the CD8-Tc1 and CD8-Tc2 effector cells to lyse infected target cells in the lung. This pattern of viral clearance is similar to that found previously. 10 The deletion of the IFN-γ gene was coincident with a slight reduction in the in vitro cytolytic activity of the Tc1-IFN-γ−/− effectors relative to the Tc1+/+ effectors. This circumstance was also true of the Tc2+/+ and Tc2-IFN-γ−/− effectors except that the coincident loss of cytolytic activity in the Tc2-IFN-γ−/− effectors was always substantially greater than that seen in the Tc1-IFN-γ−/− effectors. The low in vitro cytolytic activity of the Tc2-IFN-γ−/− effectors was reflected in the significant amount of virus recovered from the recipients of these cells. The means by which the loss of the IFN-γ gene results in the diminution of the cytolytic activity in the CD8 effector cells remains unresolved.
The recovery of eosinophils in this study was confined to the recipients of effector CD8 T cells whereas the recovery of neutrophils was noted in the infected recipient mice as well as in the infected controls. This suggested that the eosinophilia was dependent on the recruitment into the lung of the transferred CD8 effector T cells whereas host-derived factors 15-18 in the lung were sufficient for the recruitment of neutrophils. At day 5, the recipients of Tc2+/+ and Tc1-IFN-γ−/− effectors had equivalent eosinophil recovery levels despite their opposing polarity. In addition, although both Tc2+/+ and Tc2-IFN-γ−/− effectors produce similar amounts of the potent eosinophil chemoattractant, IL-5, their disparate levels of eosinophil recovery remain unexplained. The evidence from this study suggests that networks of complex regulatory pathways involving resident lung cells and recruited CD8 effector T cells are involved in the recruitment of eosinophils into the inflamed lungs.
The trend in physical deterioration of some mice and the recovery of others can be extrapolated to lung function and viral burden. Respiration rates increased in all infected groups shortly after infection as a means of compensating for the loss of functional alveolar surface area normally available for gas exchange. Although the recipients of Tc2+/+ effectors displayed a significantly low arterial pO2 level at day 3, these mice were able to sustain a continual increase in their compensatory respiration rate after day 2. This resulted in the gradual increase of their arterial pO2 levels to slightly less than the maximum seen for the Tc1+/+ and IFN-γ−/− recipients by day 5. During this same time period all three of these groups exhibited diminishing viral burdens in their lungs. The recipients of Tc2-IFN-γ−/− effectors and the infected control mice both failed to maintain their compensatory respiration rates during the course of the experiment. The respiration rates of both of these groups fell to levels below the normal resting respiration rate of the uninfected controls. In addition, both of these infected groups of mice displayed equivalent high viral burdens and decreasing arterial pO2 levels. The viral-induced damage in the infected control mice and the combination of the viral-induced and immune-associated lung damage in the recipients of the Tc2 -IFN-γ−/− effectors compromised the lung function of these mice. The increasing loss of functional alveolar surface area in these infected mice would result in an inadequate gas exchange and lead to the inability of these mice to maintain their compensatory respiration rate and the eventual death of the animal.
The pulmonary dysfunction that was evident in the Tc2+/+ and Tc2-IFN-γ−/− recipients correlated with the histological findings for these two groups. Both of these recipient groups exhibited indications of fibrinoid necrosis in their pulmonary vasculature as well as perivascular and alveolar hemorrhaging. These pathological features were more severe in the Tc2-IFN-γ−/− recipients. Abnormalities in gas exchange functions of the lung have been suggested to result from pulmonary capillary obliteration/thrombosis and ventilation/perfusion mismatching. 19 In view of the histological and lung function findings, these two factors may well have contributed to the pulmonary dysfunction seen in these recipient mice. In addition, the extent of viral replication into the alveolar spaces of the infected control mice and in the recipients of the Tc2+/+ and IFN-γ−/− effectors will contribute to the pulmonary dysfunction.
Ascribing a level of responsibility for the inflammatory sequelae and associated impairment of lung function during influenza virus infections to CD8-Tc1 or -Tc2 subsets had not been addressed before this study. In our study, the physical deterioration in the recipients of the Tc2+/+ and Tc2-IFN-γ−/− effectors was similar to that seen after adoptive transfer of HA-specific CD8 T cells into transgenic mice that express influenza HA on their type 2 pneumocytes. 6 As the HA antigen in the transgenic model is never cleared, its presence continuously stimulates the recruitment of the adoptively transferred HA-specific cytolytic CD8 T cells. This results in the ongoing destruction of the HA-expressing type 2 pneumocytes in the alveolar epithelium and causes the inflammation and impaired lung function in this model. In our influenza model, the inflammatory sequelae are initially established by the viral infection but subsequently exacerbated by the functional characteristics of the recruited HA-specific CD8 effector cells. In the recipients of Tc1+/+ and Tc1-IFN-γ−/− effectors, a relatively modest inflammatory response and a brief impairment of lung function accompanied the rapid clearance of viral antigen. These effector cells clearly restricted the viral infection to the level of the bronchial epithelium up to and including the epithelium of the terminal bronchioles. This effectively preserved the functional integrity of the alveolar space in these animals. The inefficient viral clearance observed in the recipients of Tc2+/+ and Tc2-IFN-γ−/− effectors allowed for the expansion of the viral infection into the alveolar epithelium and thus for the involvement of the alveoli in the inflammatory sequelae. The resultant loss of functional alveolar surface area was manifested by the greater impairment of lung function observed in these mice.
The recipients of the Tc1+/+ or Tc1-IFN-γ−/− effectors had equivalent or greater numbers of effector cells in their lungs relative to that found in the recipients of Tc2+/+ or Tc2-IFN-γ−/− effectors. Despite this, the later effector cells elicited a greater inflammatory response. This suggested that it was not the number of effector cells but rather the characteristics of the effector cells that were associated with the development of the inflammatory sequelae. The absence of IFN-γ production by the Tc1-IFN-γ−/− effectors resulted in only a small difference in viral clearance and in a slightly more intense inflammation in that it caused eosinophils to accumulate.
In contrast, the small amount of IFN-γ affiliated with the Tc2+/+ effectors played a significant role in the response of these effectors. The absence of IFN-γ in the Tc2-IFN-γ−/− effectors was associated with a decrease in the rate of viral clearance and an exacerbation of the inflammatory sequelae and lung function impairment relative to that observed in the Tc2+/+ associated response. The evidence in this study indicates that IFN-γ mitigates the inflammatory sequelae in the lung that are associated with the CD8-Tc1 and -Tc2 cells responding to influenza virus infections. This mitigating role for IFN-γ was more evident with the Tc2+/+ effectors. The evidence from this study suggests that this mitigating role is associated with the efficiency that these cells are recruited to the site of infection and with their cytolytic capabilities. The means by which IFN-γ had a greater influence on the Tc2 response is not known and is the subject of future investigation. Nevertheless, this study indicates that IFN-γ and viral-specific cells, particularly CD8-Tc2 effectors, recruited to the lung during influenza infections play significant roles in development of lung damage and dysfunction.
Acknowledgments
We thank Jean Brennan, Tammy Morgan, Michael Tighe, and Sharon Szady for technical assistance in the execution of this study.
Footnotes
Address reprint requests to James A. Wiley, Trudeau Institute, P.O. Box 59, Saranac Lake, NY 12983. E-mail: jwiley@trudeauinstitute.org.
Supported in part by National Institutes of Health grants AI7935 and AI36263 and in part by an American Lung Association grant from the American Lung Association of New York State awarded to J. A. W.
References
- 1.Murphy BR, Webster RG: Orthomyxoviruses. Fields BN Knipe DM eds. Virology. 1990, :pp 1091-1152 Raven Press, New York [Google Scholar]
- 2.Doherty PC, Topham DJ, Tripp RA, Cardin RD, Brooks JW, Stevenson PG: Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol Rev 1997, 159:105-117 [DOI] [PubMed] [Google Scholar]
- 3.Topham DJ, Tripp RA, Doherty PC: CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol 1997, 159:5197-5200 [PubMed] [Google Scholar]
- 4.Eichelberger M, Allan W, Zijlstra M, Jaenisch R, Doherty PC: Clearance of influenza virus respiratory infection in mice lacking class 1 major histocompatibility complex-restricted CD8+ T cells. J Exp Med 1991, 174:875-880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bender BS, Croghan T, Zhang L, Small PA, Jr: Transgenic mice lacking class 1 major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 1992, 175:1143-1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enelow RI, Mohammed AZ, Stoler MH, Liu AN, Young JS, Lou Y-H, Braciale TJ: Structural and functional consequences of alveolar cell recognition by CD8+ T lymphocytes in experimental lung disease. J Clin Invest 1998, 102:1653-1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham M, Braciale VL, Braciale TJ: Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J Exp Med 1994, 180:1273-1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moskophidis D, Kioussis D: Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J Exp Med 1998, 188:223-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sad S, Marcotte R, Mosmann TR: Cytokine-induced differentiation of precursor mouse CD8+ T cells into cytotoxic CD8+ T cells secreting Th1 or Th2 cytokines. Immunity 1995, 2:271-279 [DOI] [PubMed] [Google Scholar]
- 10.Cerwenka A, Morgan TM, Harmsen AG, Dutton RW: Migration kinetics and final destination of type 1 and type 2 CD8 effector cells predict protection against pulmonary virus infection. J Exp Med 1999, 189:423-434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan DJ, Liblau R, Scott B, Fleck S, McDevitt HO, Sarvetnick N, Lo D, Sherman LA: CD8+ T cell-mediated spontaneous diabetes in neonatal mice. J Immunol 1996, 157:976-983 [PubMed] [Google Scholar]
- 12.Cerwenka A, Carter LL, Reome JB, Swain SS, Dutton RW: In vivo persistence of CD8 polarized T cell subsets producing type 1 or type 2 cytokines. J Immunol 1998, 155:97-105 [PubMed] [Google Scholar]
- 13.Fields BT, Cunningham DJ: A tail artery technique for collecting one-half milliliter of blood. Lab Anim Sci 1976, 26:505-506 [PubMed] [Google Scholar]
- 14.Fowler DH, Breglio J, Nagel G, Eckhaus MA, Gress RE: Allospecific CD8+ Tc1 and Tc2 populations in graft-versus-leukemia and graft-versus-host disease. J Immunol 1996, 157:4811-4821 [PubMed] [Google Scholar]
- 15.Rossi DL, Hurst SD, Xu Y, Wang W, Menon S, Coffman RL, Zlotnik A: Lungkine, a novel CXC chemokine, specifically expressed by lung bronchoepithelial cells. J Immunol 1999, 162:5490-5497 [PubMed] [Google Scholar]
- 16.Gupta S, Feng L, Yoshimura T, Redick J, Fu S-M, Rose CE, Jr: Intra-alveolar macrophage-inflammatory peptide 2 induces rapid neutrophil localization in the lung. Am J Respir Cell Mol Biol 1996, 15:656-663 [DOI] [PubMed] [Google Scholar]
- 17.Carp H: Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med 1982, 155:264-275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janoff A: Elastase in tissue injury. Annu Rev Med 1985, 36:207-216 [DOI] [PubMed] [Google Scholar]
- 19.Agusti AGN, Roca J, Gea J, Wagner PD, Xaubet A, Rodriguez-Roisin R: Mechanisms of gas-exchange impairment in idiopathic pulmonary fibrosis. Am Rev Respir Dis 1991, 143:219-225 [DOI] [PubMed] [Google Scholar]