

Abstract
Endothelial cell death is a hallmark of diabetic retinopathy. Its occurrence is required for the formation of acellular (devitalized) capillaries, lesions that produce irreversible retinal ischemia through their inability to support blood flow. The mechanisms underlying diabetic retinal endothelial cell injury and death remain largely unknown. The current study demonstrates that adherent leukocytes are temporally and spatially associated with retinal endothelial cell injury and death within 1 week of streptozotocin-induced experimental diabetes in rats. Moreover, the antibody-based neutralization of intercellular adhesion molecule-1 and CD18 is shown to prevent both leukocyte adhesion and retinal endothelial cell injury and death. These data highlight the central and causal role of adherent leukocytes in the pathogenesis of diabetic retinopathy. They also underscore the potential utility of anti-intercellular adhesion molecule1- and anti-CD18-based therapies in the treatment of diabetic retinopathy, a newly recognized inflammatory disease.
One of the most important lesions in diabetic retinopathy is the obliterated or acellular capillary. Acellular capillaries form when the cellular elements die, leaving behind naked basement membrane tubes. Blood does not traverse through these devitalized vessels. Acellular capillaries therefore lead to retinal ischemia and the up-regulation of vascular endothelial growth factor (VEGF), a process that triggers iris and retinal neovascularization. 1,2 Initially, endothelial cell death precedes acellular capillary formation. 3 However, with time, acellular capillaries develop and become widespread. Several hypotheses have been advanced regarding the mechanism(s) of endothelial cell death, but none has been definitively proven. 3-6
Leukocytes adhere to the retinal vascular endothelium early in experimental diabetic retinopathy. 7 Leukocyte adhesion, like endothelial cell death, has its onset before any clinical pathology is apparent. Further, its occurrence coincides with the development of blood-retinal barrier breakdown and capillary nonperfusion. 7 Intercellular adhesion molecule-1 (ICAM-1) and CD18 have been shown to be operative in these events. 7,8 The expression of both molecules is increased in diabetes, and the specific inhibition of ICAM-1 or CD18 prevents diabetic retinal leukocyte adhesion and blood-retinal barrier breakdown.
Previous work in non-ophthalmic tissues has demonstrated that adherent leukocytes can mediate endothelial cell and parenchymal injury. 9,10 Studies have also colocalized leukocytes with dead and dying endothelial cells in the diabetic retina. 4 However, the primacy of leukocytes in the development of diabetic retinal endothelial cell death is in doubt. 5,6 To address this issue, the current study directly examined the causal role of leukocytes in the development of diabetic retinal endothelial cell injury and death. The temporal association between leukocyte adhesion and endothelial cell injury and death was assessed, and leukocyte adhesion was then disrupted using antibody-based ICAM-1- and CD18-neutralizing reagents. The effect of leukocyte adhesion blockade on retinal endothelial cell injury and death was then determined.
Materials and Methods
Experimental Diabetes
All animal experiments followed the Association for Research in Vision and Ophthalmology guidelines and were approved by the Animal Care and Use Committees of the Children’s Hospital and the Joslin Diabetes Center. After an overnight fast, pathogen-free Long-Evans rats weighing 200 to 250 g (Charles River) received single 60 mg/kg intraperitoneal (i.p.) injections of streptozotocin (Sigma, St. Louis, MO) in 0.05 mol/L citrate buffer (pH 4.5). Control nondiabetic animals received citrate buffer alone. Animals with blood glucose levels greater than 250 mg/dl 48 hours after streptozotocin injection were deemed diabetic. Before each experiment and sacrifice, the diabetic state was reconfirmed. The animals selected for study possessed blood glucose levels >250 mg/dl at all time points tested. Blood glucose was measured with an automated test strip employing the glucose oxidase method (One Touch Meter, Lifescan, Milpitas, CA). The rats were fed standard laboratory chow and were allowed free access to food and water in an air-conditioned room with a 12-hour light/12-hour dark cycle. All animals were sacrificed at the conclusion of the experiment, 9 days after streptozotocin injection.
Lectin Labeling of Adherent Retinal Leukocytes
The animals were anesthetized with intramuscular xylazine hydrochloride (8 mg/kg; Phoenix Pharmaceutical, St. Joseph, MO) and ketamine hydrochloride (50 mg/kg; Parke-Davis, Morris Plains, NJ). The chest cavity was carefully opened and a 14-gauge perfusion cannula was introduced into the left ventricle. Drainage was achieved using a 16-gauge needle placed in the right atrium. The animals were perfused with 250 ml phosphate buffered saline (PBS) per kg body weight (BW) over 2 minutes to remove erythrocytes and non-adherent leukocytes. After PBS perfusion, fixation with 1% paraformaldehyde and 0.5% glutaraldehyde was achieved using 200 ml/kg perfusate over approximately 3 minutes. The PBS perfusion was performed at a physiological pressure, since the pumping heart provided the motive force. All subsequent perfusions were postmortem and were performed at 100 mmHg pressure. Nonspecific binding was blocked with 1% albumin in PBS (total volume 100 ml/kg BW) followed by perfusion with fluorescein-isothiocyanate (FITC)-coupled Concanavalin A lectin (20 μg/ml in PBS, pH 7.4, 5 mg/kg BW; Vector Labs, Burlingame, CA). Concanavalin A was used to label adherent leukocytes and vascular endothelial cells. Residual unbound lectin was removed with a 1% albumin in PBS perfusion for 1 minute followed by a PBS perfusion for 4 minutes. The retinae were carefully removed and flat mounts prepared using a fluorescence anti-fading medium (Southern Biotechnology, Birmingham, AL). The retinae were then imaged using a fluorescence microscope (Zeiss Axiovert, Oberkochen, Germany; FITC filter). Retinae in which the peripheral collecting vessels of the ora serrata were not visible were discarded. The total number of leukocytes in the retinal arterioles, venules, and capillaries was then determined.
In Vivo Leukocyte Imaging in the Diabetic Retina
Leukocyte dynamics in the retinal microcirculation were evaluated using carboxyfluorescein diacetate succinimidyl ester (CFDASE; Molecular Probes, Eugene, OR) labeling and a scanning laser ophthalmoscope (SLO; Rodenstock Instrument, Munich, Germany). Immediately before the observation of leukocyte dynamics, each rat was anesthetized with intramuscular xylazine hydrochloride (4 mg/kg; Phoenix Pharmaceuticals, St. Joseph, MO) and ketamine hydrochloride (25 mg/kg; Parke-Davis). The pupil of the left eye was dilated with 1% tropicamide (Alcon, Humancao, Puerto Rico). CFDASE was dissolved in dimethylsulfoxide (20 mg/ml), and 10 mg/kg of this solution was injected through a jugular vein catheter at a rate of 0.25 ml/minute. The fundus was observed with the SLO using the argon blue laser as the illumination source and the standard fluorescein angiography filter in the 20° or 40° field setting. The images were recorded on videotape at the rate of 30 frames/second. The video recordings were analyzed on a computer equipped with a video digitizer (Radius, San Jose, CA). The digitized video images were viewed in real time at 640 × 480 pixels with an intensity resolution of 256 steps. Leukocytes were defined as being adherent to the vascular endothelium if they remained stationary for at least 30 seconds. The adherent leukocytes were expressed as the number of cells within a circle around the optic disk measuring three disk diameters in diameter.
CD18 Immunofluorescence
To confirm the identity of the sticking cells within the vasculature, CD18 immunofluorescence was performed. CD18 is a leukocyte marker expressed on monocytes and neutrophils. 11 Sticking leukocytes were labeled with fluorescein isothiocyanate-linked Concanavalin A as described above. Flat mounts were permeabilized with 1% Triton X-100 (Sigma) in PBS for 24 hours and nonspecific binding was blocked with 1% albumin in PBS. The retinae were then incubated with a biotinylated anti-rat CD18 antibody (1:100; Pharmingen, San Diego, CA) overnight at 4°C. After five washes with PBS, the tissues were exposed to streptavidin-coupled CyChrome (1:500; Pharmingen) for 2 hours at 25°C, and mounted with an anti-fading reagent (Southern Biotechnology, Irvine, CA) after three PBS washes.
Propidium Iodide (PI) Labeling
Dead and injured endothelial cells were labeled in vivo using PI (Molecular Probes). After the induction of deep anesthesia with 50 mg/kg i.p. sodium pentobarbital, PI (1 mg/ml in PBS) was injected intravenously via the tail vein at a concentration of 5 μmol/kg (0.668 ml/200 mg BW). The solution was allowed to circulate for 20 minutes, after which it was followed by fixation via whole body perfusion and lectin labeling as described above. Retinal flat mounts were examined by fluorescence microscopy as described above. Labeled endothelial cells were distinguished from surrounding cells, especially pericytes, by focusing through the tissue to discern the distinct cellular outline and nuclear shape of the endothelial cells.
Systemic ICAM-1 and CD18 Blockade
Confirmed diabetic animals were randomized to receive i.p. injections of 1 mg/kg of a mouse anti-rat ICAM-1 neutralizing antibody (clone 1A29; R&D Systems, Minneapolis, MN) or 1 mg/kg of an isotype control non-immune mouse IgG1 (R&D Systems). In separate experiments, diabetic rats received either 1 mg/kg of a mouse anti-rat CD18 F(ab′)2 (WT.3) (Associates of Cape Cod Inc., Falmouth, MA) or a purified mouse anti-human non-immune F(ab′)2 (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). All blocking reagents were injected at a final concentration of 1 mg/ml in sterile PBS. The animals were treated 3 times weekly for 1 week. Cell death and retinal leukostasis were determined 1 week after the first injection of the blocking reagent.
Statistical Analysis
All results are expressed as mean ± SD. The data were analyzed by Whitney-Mann-U test with post hoc comparisons tested using Fisher’s protected least significant difference procedure. Differences were considered statistically significant when the P values < 0.05.
Results
Leukocyte Adhesion Is Increased in the Diabetic Retina
Adherent retinal leukocytes were labeled in situ with FITC-linked Concanavalin A. Preliminary experiments using various FITC-linked lectins revealed that Concanavalin A gave the most homogeneous staining in the retinal vasculature. After leukocyte labeling, whole body perfusion was performed under physiological perfusion pressure to remove non-adherent blood elements from the retinal circulation. Retinal flat mounts were prepared and the adherent leukocytes were counted in the arterioles, venules, and capillaries. Compared to the nondiabetic control retinae, a 2.2-fold (n = 9, P < 0.005), 3.8-fold (n = 9, P < 0.001) and 3.6-fold (n = 8, P < 0.001) increase in adherent leukocytes was seen in the diabetic retinal arterioles, venules, and capillaries, respectively (Figure 1) ▶ .
Figure 1.
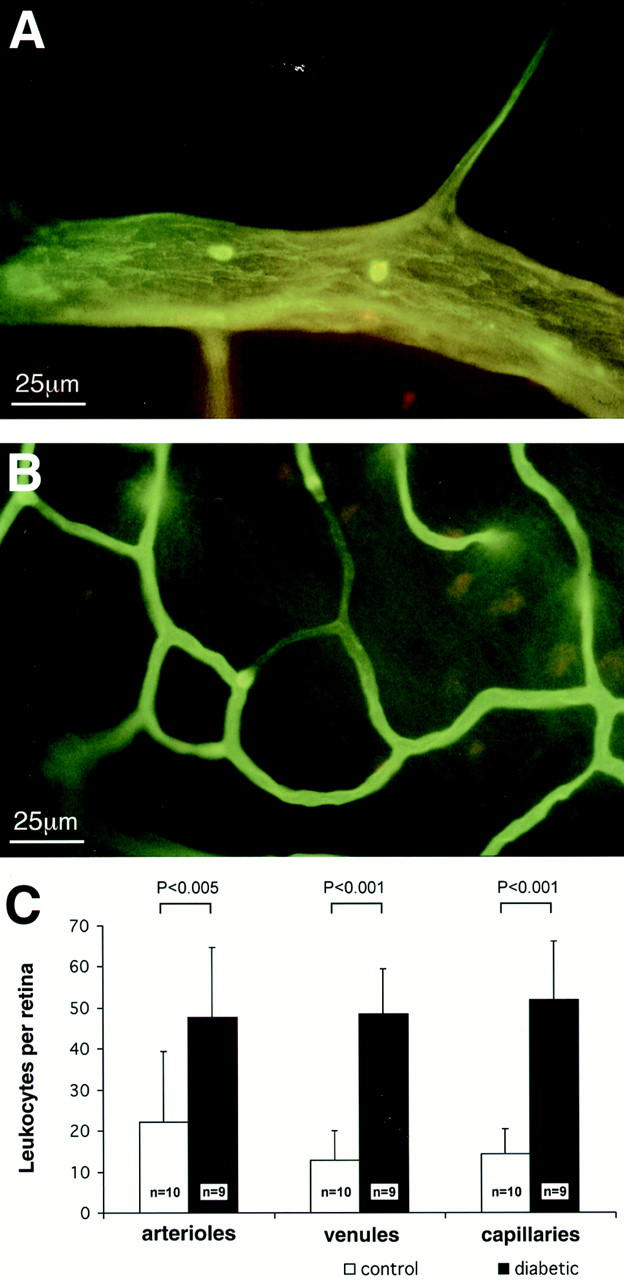
Lectin staining highlights adherent leukocytes within the retinal vasculature. A: In this example, an adherent leukocyte is visible in the arteriole of a diabetic rat. B: Impedance of flow is occasionally seen downstream from adherent leukocytes in capillaries. C: The collated graphical data show a marked increase in the number of adherent leukocytes in the diabetic retinae.
Arteriolar leukocyte adhesion is rare. To confirm that the adhesion observed with the perfusion method was not artifactual, an alternative in vivo method of leukocyte labeling was used. Briefly, leukocytes in larger vessels were visualized in situ with CFDASE, and a SLO was then used to image leukocyte dynamics in vivo. Arteriolar leukocyte adhesion was confirmed using this method (Figure 2) ▶ . A quantitative assessment of leukocyte adhesion was completed using the SLO. The adherent leukocyte density was increased in the arterioles and venules of 1-week diabetic rats. The arterioles contained 4.4 ± 1.9 vs. 1.5 ± 0.9 adherent leukocytes in the diabetic versus nondiabetic retinae, respectively (n = 9, P < 0.01). The venules contained 2.9 ± 1.2 vs. 1.2 ± 1.0 adherent leukocytes in the diabetic and non-diabetic retinae, respectively (n = 9, P < 0.01). The -fold increases observed with the two methods were comparable.
Figure 2.
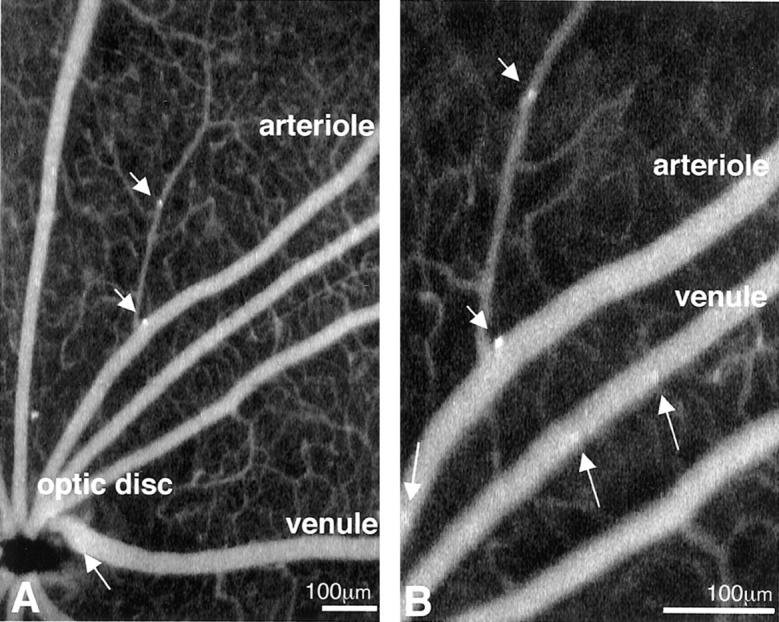
In vivo leukocyte fluorography confirms the adhesion of leukocytes to retinal arterioles. A: A CFDASE fundus image of a diabetic rat (40° field setting) shows that leukocytes adhere not only to venules (arrow), but also to arterioles (arrowheads). B: The same fundus image in the 20° field setting, taken 30 seconds later, confirmed the static nature of the leukocyte. While many leukocytes flow through the retinal vessels (arrows), two leukocytes adhere to the walls of retinal arterioles (arrowheads). The direction of the flow of blood was used to distinguish arterioles from venules.
Previous work has shown that neutrophil and monocyte numbers are increased in the vasculature of diabetic humans and rats. 4,12 To confirm the identity of the Concanavalin A-labeled adherent cells, CD18 immunofluorescence in retinal flat mounts was performed. CD18 expression is primarily restricted to neutrophils and monocytes. 13 Concanavalin A/CD18 double labeling identified the adherent cells as being CD18-positive leukocytes (Figure 3) ▶ .
Figure 3.

Anti-CD18 surface staining of a static leukocyte within a vessel. Green fluorescence from the FITC-linked Concanavalin A (A) and red fluorescence from the anti-CD18 antibody (B) identify the leukocytes as being CD18-positive when the images are superimposed (C).
Retinal Vascular Endothelial Cell Injury and Death in Diabetic Retina
Endothelial cell injury and death were investigated using PI, a molecule that is excluded from uninjured viable cells. PI fluoresces when it leaks through injured cell membranes and binds to DNA and RNA, primarily identifying cells undergoing either necrosis or apoptosis. 13 PI was injected intravenously and endothelial cell injury and death were quantified in retinal flat mounts. Little or no PI staining was observed in nondiabetic retinae. In contrast, endothelial PI staining was markedly increased in the retinal arterioles (23.0 ± 10.8 cells/retina), venules (31.8 ± 7.6 cells/retina), and capillaries (27.6 ± 6.1 cells/retina) of diabetic eyes (Figure 4) ▶ . In both small and large caliber vessels, PI-positive cells were often found in clusters (Figure 4) ▶ . A cohort of 2-month diabetic animals was also studied and revealed a similar level of leukocyte adhesion and endothelial cell injury (data not shown). These data confirmed that endothelial cell injury and death are ongoing during the course of diabetes, a result previously shown by others. 3,4
Figure 4.
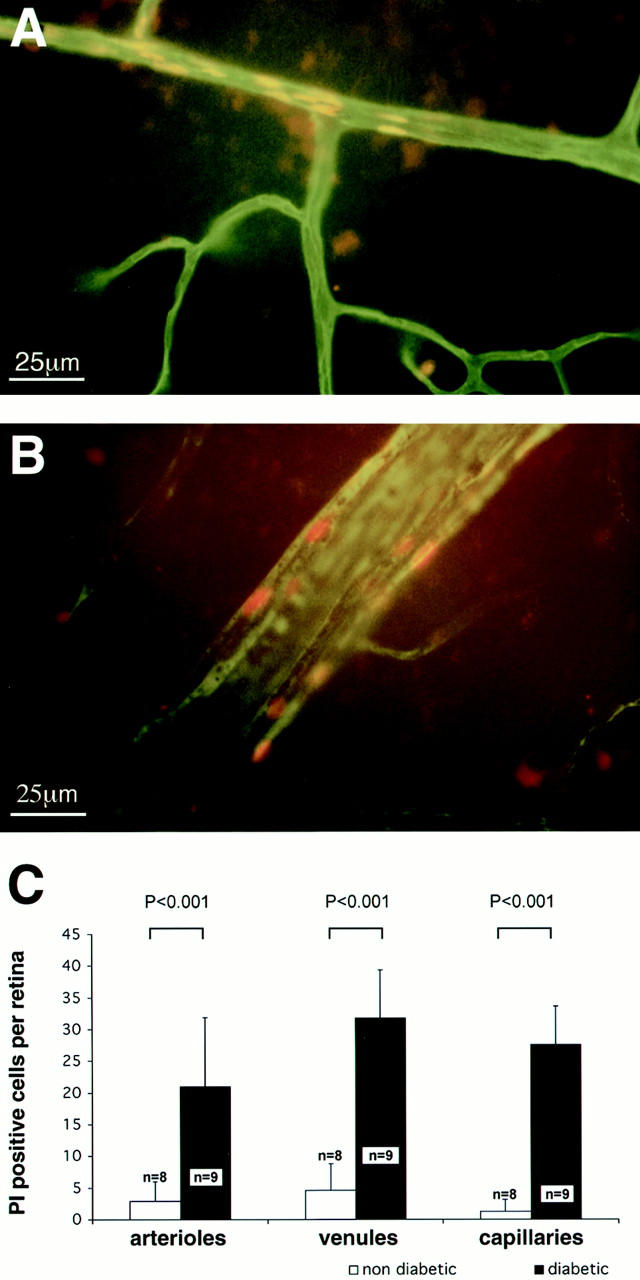
Endothelial cell injury and death identified with propidium iodide (PI). A: PI-positive cells show sharply demarcated borders in a venule located in the mid-periphery of the diabetic retina. B: Capillaries, like larger vessels, exhibit clusters of PI-positive cells. Original magnification, ×400. C: The collated data show markedly increased numbers of PI-positive endothelial cells in the diabetic retinae.
ICAM-1 and CD18 Blockade Prevent Leukocyte Adhesion and Endothelial Cell Injury and Death
To assess the role of leukocyte adhesion in retinal endothelial cell injury and death, in vivo adhesion blockade experiments were performed. Diabetic animals were randomized to receive either 1 mg/kg of an ICAM-1-neutralizing antibody or 1 mg/kg of a CD18-neutralizing F(ab′)2. For each experiment, a separate cohort of diabetic animals received 1 mg/kg of a corresponding non-immune isotype control. The reagents were administered 3 times weekly following the onset of diabetes. All animals were analyzed 1 week after the first injection of the blocking reagent. The ICAM-1 antibody reduced diabetes-associated leukocyte adhesion 48.6% (n = 10, P < 0.001) in arterioles, 41.2% (n = 10, P < 0.001) in venules, and 70% (n = 10, P < 0.001) in capillaries (Figure 5) ▶ . Similarly, when compared to the controls, the ICAM-1 antibody reduced endothelial cell PI staining 90.1% (n = 10, P < 0.001) in venules, 90.8% (n = 10, P < 0.001) in arterioles, and 89.9% (n = 10, P < 0.001) in capillaries.
Figure 5.

ICAM-1 blockade prevents leukocyte adhesion and endothelial cell injury in the diabetic retina. A: After treatment with the anti-ICAM-1 antibody, diabetic retinal leukocyte adhesion was markedly decreased in all retinal vessels. B: Analysis with PI revealed a concomitant decrease in retinal endothelial cell injury and death.
The neutralizing anti-CD-18 F(ab′)2 reagent produced a similar result (Figure 6) ▶ . Treatment with CD-18 F(ab′)2 reduced leukocyte adhesion 39.6% in venules (n = 12, P < 0.05) and 42.3% (n = 12, P < 0.005) in capillaries. CD18 blockade also potently reduced endothelial cell PI staining 48.1% (n = 12, P < 0.005) in venules and 42.5% (n = 12, P < 0.005) in capillaries. Unlike the ICAM-1 blockade experiment, arteriolar leukocyte adhesion and endothelial cell PI staining were not affected by CD18 blockade.
Figure 6.

CD18 blockade prevents leukocyte adhesion and endothelial cell injury and death in diabetic retinal venules and capillaries. A: After treatment with anti-CD-18 F(ab′)2, diabetic retinal leukocyte adhesion was markedly decreased in the retinal venules and capillaries. B: Analysis with PI revealed a concomitant decrease in retinal endothelial cell injury and death; however, no change in arteriolar endothelial cell injury and death was observed.
Discussion
Retinal endothelial cell death has previously been shown to colocalize with static leukocytes. 4 The current data extend these findings by showing that adherent leukocytes are causal for diabetic endothelial cell injury and death, and not vice versa. Moreover, molecular targets for inhibiting the process, ICAM-1 and CD18, have been uncovered. Because rodent and human diabetes share many features—including acellular capillaries, pericyte and endothelial cell loss, microaneurysm formation, blood-retinal barrier breakdown, leukocyte buildup, and ICAM-1 up-regulation—we believe the current findings are relevant to human diabetes.
Leukocyte adhesion and endothelial cell death are ongoing in diabetes. 3,4,7 The current data represent a snapshot in time, but the cumulative number of injured and dying endothelial cells in the diabetic retina likely increases with time and becomes sizable. Given the presumably larger total capillary area in the retina, the proportion of injured and dying capillary endothelial cells is smaller than that for the arterioles and venules. This fact does not seem to fit with the preferential development of acellular capillaries in the diabetic retina. We propose that the clustered damage to capillaries, where little endothelial reserve exists, contributes to the formation of acellular capillaries. Moreover, because a single endothelial cell can form a retinal capillary lumen, and only a few endothelial cells are needed to form a retinal capillary, 14 the death of a relatively small number of capillary endothelial cells can take on outsized importance. In contrast, we speculate that discrete and focal damage to larger vessels is less likely to impede flow and cause the death of a vessel, given the relatively smaller scale of the injury and the larger number of surrounding endothelial cells available for repair.
The methods used to assess leukocyte adhesion and endothelial cell death are novel for the retina. Because leukocytes were observed to adhere to the arterioles of the diabetic retina, an unusual result, confirmation was required. The SLO and perfusion methods mutually confirmed the increased leukocyte adhesion in diabetic arterioles, as well as the capillaries 7 and venules. Although the -fold increases were comparable, the differences in absolute numbers were likely due to the optics of the SLO, which precluded visualization of the retinal periphery and mid-periphery.
Although endothelial cell death was previously observed to occur 4 to 6 months after the onset of experimental diabetes 3,4 the current data demonstrate that endothelial cell injury and death can be detected within 1 week of diabetes. Furthermore, the number of injured and dying endothelial cells appears to be much greater than previously recognized. The numerical disparity may be due to the fact that both necrotic and apoptotic cells are quantified with PI labeling. Previous work quantified endothelial cell apoptosis alone. 3 Moreover, because the PI method cannot discriminate between lethal and sublethal injury, it is conceivable that some of the injured PI-stained cells observed in the current study are still viable. Lastly, it is important to note that different time points were examined in the two studies. Nevertheless, it cannot be ruled out that some of the PI staining observed in these studies was artifactual in nature.
Finally, we speculate, as others have before us, that acellular capillaries develop when retinal endothelial cells reach replicative senescence. 3 Acellular capillaries were not observed in our 1-week diabetic rats. Replicative senescence requires time, potentially explaining the delay between the onset of endothelial cell death and the appearance of acellular capillaries. Endothelial cell death and proliferation are known to proceed simultaneously in the diabetic human retina. 15 We speculate that replicative senescence eventually tips the balance in favor of endothelial cell death.
Taken together, the data identify a new pathogenic phenomenon in diabetic retinopathy: leukocyte-mediated endothelial cell injury and death. The results of this study suggest that the inhibition of ICAM-1 and CD18 may avert or suppress acellular capillary formation via the suppression of endothelial cell injury and death. If so, the retinal ischemia and VEGF up-regulation that characterize diabetic retinopathy may be preventable.
Footnotes
Address reprint requests to Anthony P. Adamis, M.D., Massachusetts Eye and Ear Infirmary, 243 Charles Street, Boston, MA 02114. E-mail: tony_adamis@meei.harvard.edu.
Supported by the Roberta W. Siegel Fund (to A. P. A.), Deutsche Forschungsgemeinschaft DFG Jo 324/2–1 (to A. M. J.), the Juvenile Diabetes Foundation International (to A. M. J. and A. P. A.), National Institutes of Health grants EY11627 and EY12611 (to A. P. A.), and the Massachusetts Lions (to A. P. A.).
References
- 1.Adamis AP, Shima DT, Tolentino M, Gragoudas ES, Ferrara N, Folkman J, D’Amore PA, Miller JW: Inhibition of VEGF prevents retinal ischemia-associated iris neovascularization in a primate. Arch Ophthalmol 1996, 114:66-71 [DOI] [PubMed] [Google Scholar]
- 2.Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L, Ferrara N, King G, Smith LEH: Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA 1995, 92:10457-10461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizutani M, Kern TS, Lorenzi M: Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest 1996, 97:2883-2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schröder S, Palinski W, Schmid-Schönbein GW: Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol 1991, 139:81-100 [PMC free article] [PubMed] [Google Scholar]
- 5.Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M: Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci USA 1991, 88:11555-11558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank RN: Aldose reductase inhibition: the chemical key to the control of diabetic retinopathy? Arch Ophthalmol 1990, 108:1229-1231 [DOI] [PubMed] [Google Scholar]
- 7.Miyamoto K, Khosrof S, Bursell S-E, Rohan R, Murata T, Clermont A, Aiello LP, Ogura Y, Adamis AP: Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci USA 1999, 96:10836-10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barouch FC, Miyamoto K, Allport JR, Fujita K, Bursell SE, Aiello LP, Luscinskas FW, Adamis AP: Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest Ophthalmol Vis Sci 2000, 41:1153-1158 [PubMed] [Google Scholar]
- 9.Ofosu-Appiah W, Sfeir G, Smith D, Richard T: Neutrophil-mediated damage to vascular endothelium in the spontaneously hypertensive rat. Clin Immunol Immunopathol 1997, 83:293-301 [DOI] [PubMed] [Google Scholar]
- 10.DeLano FA, Forrest MJ, Schmid-Schönbein GW: Attenuation of oxygen free radical formation and tissue injury during experimental inflammation by P-selectin blockade. Microcirculation 1997, 4:349-357 [DOI] [PubMed] [Google Scholar]
- 11.Diamond DS, Springer TA: A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J Cell Biol 1993, 120:545-556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLeod DS, Lefer DJ, Merges C, Lutty GA: Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol 1995, 147:642-653 [PMC free article] [PubMed] [Google Scholar]
- 13.Haugland RP: Handbook of Fluorescent Probes and Research Chemicals, 6th ed. 1996, :p 149 OR, Molecular Probes, Eugene [Google Scholar]
- 14.Kern TS, Engerman RL: Vascular lesions in diabetes are distributed non-uniformly within the retina. Exp Eye Res 1995, 60:545-549 [DOI] [PubMed] [Google Scholar]
- 15.Sharma NK, Gardiner TA, Archer DB: A morphologic and autoradiographic study of cell death and regeneration in the retinal microvasculature of normal and diabetic rats. Am J Ophthalmol 1985, 100:51-60 [DOI] [PubMed] [Google Scholar]