

Abstract
Pancreatic serous microcystic adenomas (SCAs) are rare, benign tumors with a striking female preference. Virtually no information is available about chromosomal or genetic anomalies in this disease. We performed extensive molecular characterization of 21 cases of formalin-fixed, paraffin-embedded sporadic SCAs consisting in genome-wide allelic loss analysis with 79 microsatellite markers covering all 22 autosomes, assessment of microsatellite instability, and mutational analysis of the VHL, K-ras, and p53 genes in nine cases for which frozen tissue was available. Although no case showed microsatellite instability of the type seen in mismatch repair-deficient tumors, a relatively low fractional allelic loss of 0.08 was found. Losses on chromosome 10q were the most frequent event in SCAs (50% of cases), followed by allelic losses on chromosome 3p (40% of cases). Moderately frequent losses (>25% of cases) were found on chromosomes 1q, 2q, and 7q. The VHL gene, located on chromosome 3p, had somatic inactivating mutations in two of nine cases (22%), whereas no mutations were found in either K-ras or p53, in agreement with the finding that all 21 cases stained negative for p53 by immunohistochemistry. Our study indicates that the involvement of chromosomal arms 10q and 3p is characteristic of SCAs and that the VHL gene is involved in a subset of sporadic cases.
Pancreatic microcystic serous cystadenomas (SCAs) are rare exocrine tumors composed of small cysts lined by glycogen-rich cells. The disease has a definite female preference, generally has a benign clinical course, and may present itself either sporadically or as part of von Hippel-Lindau (VHL) syndrome. 1,2
There is almost no information available regarding the chromosomal and molecular alterations giving rise to this disease. Mutations in codon 12 of the K-ras gene have not been found in a total of 11 cases of SCAs analyzed. 3-5 Likewise, no tumor was found to stain for p53 protein by immunohistochemistry in seven cases 4 and no mutations in p53 were detected in three cases analyzed. 5 On the assumption that the sporadic and VHL-associated cases of SCA may involve the same molecular targets, another study analyzed 10 sporadic tumors for allelic loss at the VHL locus and for alterations of the VHL gene; six cases (60%) were found to have loss of heterozygosity (LOH) on chromosome 3p and one case had a somatic silent mutation in the VHL gene. 6
The multistep process of carcinogenesis involves the progressive alteration of oncogenes and tumor suppressor genes. As the inactivation of these latter commonly occurs by mutation and/or deletion, the chromosomal regions harboring potential tumor suppressor genes can be identified by LOH analysis using polymerase chain reaction (PCR) amplification of polymorphic microsatellite repeats in tumor and matched-normal DNA. When analysis of LOH is extended to multiple chromosomal arms, a distinct allelotype is generated.
Here we present the allelotype of SCA obtained by genome-wide allelic loss analysis of 21 cases each analyzed with 79 microsatellite markers. This analysis is complemented with by the assessment of microsatellite instability and the mutational analysis of the VHL, K-ras, and p53 genes.
Materials and Methods
Tumor Samples and DNA Extraction
Twenty-one serous cystic adenomas of the pancreas were studied from a total of 72 formalin-fixed paraffin-embedded cases available from the files of the Department of Pathology at the University of Verona. The cases subjected to molecular analysis were selected according to strict criteria consisting in an exclusive microcystic histological subtype, which would permit their microdissection to a high cellularity. Thus, all macrocystic lesions, the microcystic variant with low cellularity in which the epithelial component is only a small proportion of unit volume having a relatively high stromal cell content into the intracystic septae, and those tumors with extensive epithelial denudation of the cysts and/or marked stromal cell component were excluded form the study. All of the 21 SCAs under study were well circumscribed, had a central scar, and had cysts <1 cm in diameter. The median patient age was 61 years (range, 49 to 72 years); 19 tumors were from female patients. The average tumor diameter was 5.6 cm (range, 2.5 to 10 cm). Five tumors originated from the head, nine from the body, five from the body-tail, and two from the tail. As no case was associated with VHL syndrome, all tumors were considered sporadic. Normal and tumor DNA were obtained by microdissection in all 21 cases from paraffin-embedded sections as described. 7 Based on microscopic assessment, neoplastic cellularity of at least 90% was obtained in all cases. The presence of p53 protein nuclear accumulation was examined using anti-p53 monoclonal antibodies as previously described. 8 Nine tumors also had frozen tissue available and in four cases cryostat enrichment to a neoplastic cellularity of >70% was possible.
Allelic Loss Analysis
The primers used were from the ABI Prism Linkage Mapping Set version 1 or 2 (Perkin-Elmer, Emeryville, CA). PCR products were pooled and electrophoresed on an ABI 377 DNA sequencer. Only microsatellites showing two distinct alleles in normal DNA were considered as informative. For these, the intensity of the fluorescent bands of tumor was compared to that of matched-normal DNA. LOH was scored when there was at least a fivefold difference in the ratio of the intensities of the two alleles in tumor and matched-normal DNA. Microsatellites showing differently sized alleles compared with their respective normal sample were scored as instable. All cases were also analyzed for microsatellite instability using the markers BAT-25 and BAT-26. 9 In four cases for which high molecular weight DNA was available, chromosomal arms 3p and 10q were analyzed in detail using markers from the ABI Prism Linkage Mapping Set version 1 (Perkin-Elmer) (arm 10q, 11 markers) or 2 (arm 3p, 11 markers).
Mutational Analysis by PCR-Single-Strand Conformational Polymorphism (SSCP)
General conditions used for PCR amplification and SSCP have been described. 10 The nine cases for which frozen tissue was available were analyzed for mutations in the VHL, K-ras, and p53 genes. For the VHL gene, exon 1 was amplified in two overlapping fragments, whereas exons 2 and 3 were amplified in their entirety. The primer pairs and the size of the amplified fragments are as follows: exon 1A: VHL 1S, CCCGGGTGGTCTGGATCGCG, and VHLN2AS, AGTTCACCGAGCGCAGCACG (228 bp); exon 1B: VHLN3S, CCGAGGAGGAGATGGAGGCC, and VHLN4AS, GCTATCGTCCCTGCTGGGTC (235 bp); exon 2, VHL5S, CTTTAACAACCTTTGCTTGT and VHL6AS, TTACCACAACAACCTTATCT (195 bp); exon 3: VHL7S, GAGGATTTGGTTTTTGCCCT, and VHL8AS, ACAGTGTAAGTTTCAACAGA (229 bp). Primers for amplification of exon 1 of the K-ras gene 11 and exons 5 to 9 of p53 12 were as described. Bands exhibiting aberrant migration were cut from the gel, reamplified, and sequenced on an ABI Prism 377 instrument. Results with SSCP analysis were confirmed in two independent experiments.
Results
Generation of an Allelotype for SCA
Each of the 21 cases was analyzed for allelic loss using 79 microsatellite markers on all 22 autosomes (Table 1) ▶ . At least two markers were used per chromosomal arm with the exception of 5p, 12p, and 12q for which only one marker was used. Sixty-four percent of the 1,659 markers analyzed were informative; 85 losses were observed among the 1,066 informative markers. Representative results are shown in Figure 1 ▶ where examples of allelic loss and retention are shown, which also demonstrates the success of the enrichment procedure used (>90% in all cases as assessed by microscopy). All cases showed at least one allelic loss, although the individual fractional allelic loss varied greatly (range, 0.02 to 0.31); the mean fractional allelic loss was 0.08 calculated as the number of allelic losses divided by the number of informative markers. Detailed results for the individual chromosomal arm in each case is shown in Figure 2 ▶ along with the resulting allelotype of SCAs. Allelic losses on chromosome 10q were the most frequent event in SCAs, observed in ∼50% of cases. Allelic loss involving chromosome 3p was also a relatively frequent event, seen in ∼40% of cases. Moderately frequent losses (>25% of cases) were found on chromosomes 1q, 2q, and 7q. No losses were observed on chromosomes 4p, 8q, 12q, 16p, 18, 20p, or 22, whereas the remaining chromosomes showed varying, low degrees of allelic loss. No case showed microsatellite instability of the type seen in mismatch repair-deficient tumors, as assessed by analysis of the markers BAT-25 and BAT-26.
Table 1.
Microsatellite Markers Used for Study of SCA
| D1S255 | D6S289 | D11S1338 | D18S452 |
| D1S2667 | D6S470 | D11S935 | D18S59 |
| D1S2785 | D6S287 | D11S908 | D18S63 |
| D1S2800 | D6S434 | D11S937 | D18S70 |
| D6S446 | D18S1161 | ||
| D2S2368 | D12S336 | ||
| D2S165 | D7S493 | D12S346 | D19S221 |
| D2S112 | D7S507 | D19S884 | |
| D2S117 | D7S510 | D13S285 | D19S210 |
| D7S669 | D13S158 | D19S414 | |
| D3S1566 | D7S798 | ||
| D3S1289 | D14S275 | D20S112 | |
| D3S1263 | D8S258 | D14S258 | D20S889 |
| D3S1262 | D8S264 | D20S171 | |
| D3S1565 | D8S270 | D15S978 | D20S195 |
| D8S514 | D15S1007 | ||
| D4S2935 | D21S266 | ||
| D4S1592 | D9S171 | D16S423 | D21S263 |
| D4S402 | D9S161 | D16S3075 | |
| D4S1572 | D9S164 | D16S3136 | D22S283 |
| D9S273 | D16S3091 | D22S539 | |
| D5S406 | |||
| D5S433 | D10S189 | D17S921 | |
| D5S424 | D10S249 | D17S831 | |
| D10S212 | D17S949 | ||
| D10S583 | D17S785 | ||
| D10S217 |
Figure 1.

Whole mount H&E-stained section of a SCA and example of microsatellite analysis. In the high magnification picture of the tumor (×100) it is to be noted that the proportion of neoplastic cells approaches 100% as the cell component of the stroma is minimal in this tumor type. Cells were microdissected from appropriate areas to obtain normal and tumor DNA and processed for microsatellite analysis, an example of which is shown. N and T indicate the electropherogram tracings from normal and tumor DNA, respectively. The marker shown is D1S2800 in case SCA21. The success of the enrichment procedure for tumor cells is exemplified by the complete loss of one of the two alleles in the tumor DNA.
Figure 2.
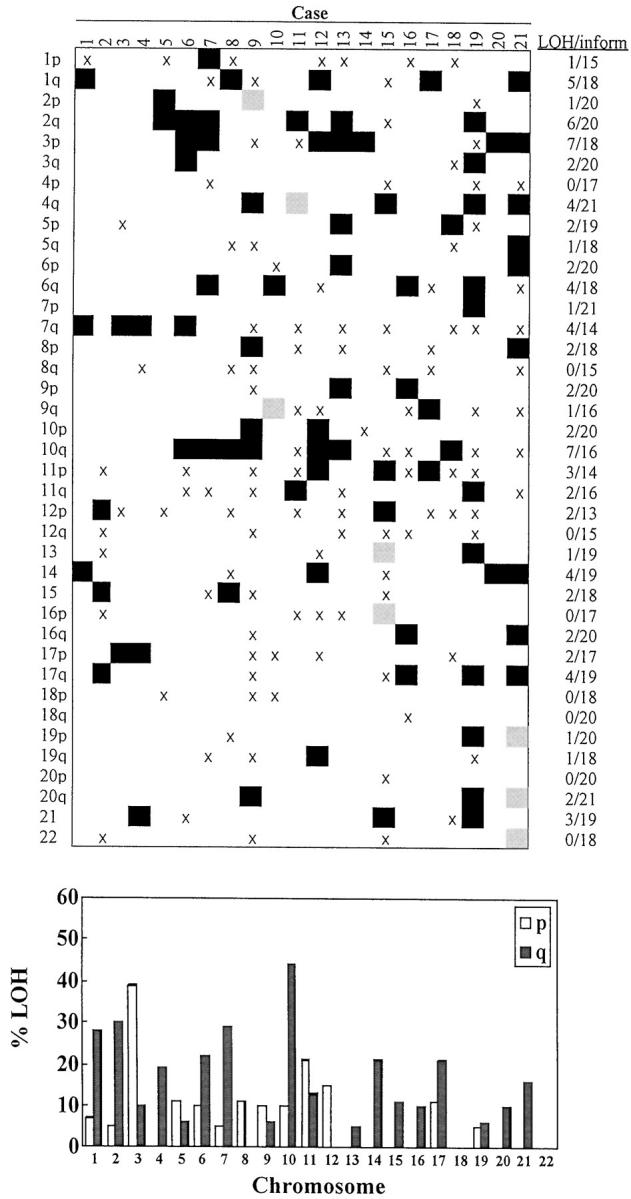
Allelic losses and allelotype of SCA. Detailed allelic losses in SCA for individual chromosomal arms and cases are shown at the top. LOH are indicated as black boxes, instabilities as gray boxes, and noninformative markers by X. Proportion of losses and informative markers is indicated on the right. The allelotype of pancreatic SCA is shown at the bottom. The graph shows the percentage of cases showing allelic loss in at least one informative marker used for each chromosomal arm.
A more detailed microsatellite analysis of the two chromosomal arms, 3p and 10q, found to be most frequently altered in our allelotyping studies was performed on the four cases having frozen tissue available and a neoplastic cellularity >70%. This analysis confirmed the losses on these chromosomal arms seen using paraffin-embedded material, but the small number of cases did not obviously permit definition of a smallest common deleted region (data not shown).
Mutational Analysis of VHL, K-ras, and p53
The nine cases with high molecular weight DNA available were analyzed for alterations in exons 1, 2, and 3 of the VHL gene; exon 1 and 2 of K-ras; and exons 5 to 9 of p53 by PCR-SSCP analysis. Although no case had mutations in either K-ras or p53, two cases (22%) had potentially inactivating mutations in the VHL gene (Figure 3) ▶ . In particular, tumor SCA1 had a T-A substitution at codon 80 (AGT-AGA) resulting in Ser to Arg substitution and case SCA18 had an insertion of T at codon 175 (TAC-TTAC) resulting in a frameshift. Both mutations were somatic in nature. The PCR-SSCP analysis was repeated in two independent experiments with identical results. Our ability to detect mutations in the VHL gene under the conditions used for PCR-SSCP analysis was also demonstrated by the analysis of 15 cases of renal clear-cell carcinoma in which mutations were found in eight cases (53%; data not shown), a frequency expected from literature data. 13 Although case SCA1 was not informative for the marker analyzed at 3p25, tumor SCA18 showed LOH at marker D3S1263.
Figure 3.

Representative examples of PCR-SSCP analysis of VHL. A: Exon 1B. B: Exon 3. Only cases possessing bands with altered mobility are indicated.
Discussion
By genome-wide microsatellite analysis of 21 SCAs we have generated an allelotype for this uncommon pancreatic tumor. Our data show that: 1) SCA displays a moderate number of allelic losses on selected chromosomes with LOH on chromosomes 3p and 10q being the most common events; 2) chromosome arms 1q, 2q, and 7q show moderate frequencies of allelic loss (>25% of cases), whereas most of the remaining chromosomes show much lower degrees of allelic loss varying from 5 to 20%; 3) no allelic losses were detected on chromosomes 4p, 8q, 12q, 16p, 18, 20p, or 22; 4) inactivating mutations in the VHL gene were found in two of nine tumors tested. The fact that no mutations in either K-ras or p53 were found coincides with the data obtained in a smaller number of cases that have been previously analyzed. 3-5 Moreover, all cases also scored negative for nuclear accumulation of p53 as determined by immunohistochemistry (data not shown), further confirming the absence of alterations in p53.
The most common molecular anomaly found in SCA was the loss of genetic material on chromosome 10q, seen in ∼50% of cases. LOH on 10q is also found in approximately one-half of pancreatic ductal cancers, which are characterized by allelic loss on chromosomal arms 9p, 17p, and 18q in nearly all cases. 14 Interestingly, LOH on chromosome 10q has been found in follicular thyroid tumors. 15 Allelic loss on 10q has also been correlated with invasiveness of transitional cell carcinomas of the bladder. 16 One possible candidate gene might be PTEN, located on the long arm of chromosome 10 and which is mutated in several cancers. 17 It is unfortunate that only a small number of cases had high molecular weight DNA available for further localization of the chromosomal regions involved in these losses.
Allelic loss on chromosome 3p was the second most frequent event found in SCA. As microcystic tumors are part of the von Hippel-Lindau syndrome and the VHL gene is located on chromosome 3p25, it is reasonable to believe that this gene may be altered in sporadic SCA. A previous study found LOH on chromosome 3p25 in six of 10 cases analyzed in at least one of four markers tested. 6 Among these 10 cases, one was found to have a somatic silent mutation in the VHL gene. 6 Using three markers for chromosome 3p, we found allelic loss in at least one marker in 39% of cases. Analysis of nine cases by PCR-SSCP found inactivating mutations in the VHL gene in two tumors (22%). Mutations in the VHL gene have been observed in 4 of 23 sporadic hemangioblastomas analyzed 18,19 and in 4 of 48 cases of sporadic pheochromocytomas. 20 Thus the frequency of somatic mutations found in SCA in our analysis is in line with what has been found for other sporadic benign counterparts of familial VHL tumors; ie, mutation of VHL is not an extremely common event. However, alterations in VHL are found in approximately one half of sporadic renal clear-cell carcinomas, 13 a cancer type which is also part of VHL syndrome.
Among the three chromosomes showing relatively frequent allelic losses, arm 7q is of particular interest. Allelic losses on chromosome 7q were observed in ∼30% of SCAs. Genetic losses on the long arm of chromosome 7 have been found at relatively high frequency in other benign tumors, including uterine leiomyoma 21 and, notably, in ovarian serous tumors. 22 A number of different studies, including in vitro experiments and cytogenetic analysis, have suggested that one or more tumor suppressor genes are located on chromosome 7q. 23-25 Interestingly, the gene coding for Cul1 also resides on 7q. 26,27 This gene is part of a complex (Skp1-Cul1-F-Box protein) that has similarities to the VHL elongin complex 28 and its loss results in the dysregulation of cyclin E. 29
One interesting question is whether SCA shares chromosomal or molecular events with pancreatic endocrine tumors or ductal carcinoma. The mean fractional allelic loss of 0.08 in SCA is much lower than that found in ductal cancer (0.36) 30 or pancreatic endocrine tumors (0.15), 31 and the allelotype of SCA does not resemble that of pancreatic ductal or endocrine tumors. 14,32 The chromosomes showing most frequent LOH in our SCAs were 10q (50%) and 3p (40%) followed by 1q, 2q, and 7q (>25%). This pattern is clearly distinct from that observed in pancreatic endocrine tumors, in which the most frequent allelic losses are found on chromosomes 3q, 11p, and 16p with a frequency never exceeding 36%. 31 Pancreatic ductal carcinoma is characterized by highly frequent losses at chromosomes 1p, 9p, 17p, and 18q. 14,32
In conclusion, the anomalies found by genome-wide allelotyping indicate that chromosomal arm 10q may harbor a tumor suppressor gene involved in the pathogenesis of SCA. Our chromosomal deletion map also suggests that SCA tumorigenesis involves molecular pathways different from those occurring in more common pancreatic tumor types, including mutation of the VHL gene in a subset of sporadic cases.
Footnotes
Address reprint requests to Professor Aldo Scarpa, M.D., Dipartimento di Patologia - Sezione di Anatomia Patologica, Università di Verona, Strada Le Grazie, I-37134 Verona, Italy. E-mail: a.scarpa@univr.it.
Supported by grants from the Consorzio Studi Universitari di Verona, Italy; Associazione Italiana Ricerca Cancro, Milan, Italy (to A. S.); co-financed grant from Verona University and Ministero Università e Ricerca Scientifica e Tecnologica (Cofin 9806151968-9906195987-9906218982), Rome, Italy; and European Community grant BIOMED 2 CE Contract No. BMH4-CT98-3805.
References
- 1.Christoferson LA, Guastafson MB, Peterson AG: Von Hippel-Lindau’s disease. J Am Med Assoc 1961, 178:280-282 [DOI] [PubMed] [Google Scholar]
- 2.Horton WA, Wong V, Eldridge R: Von Hippel-Lindau disease: clinical and pathological manifestations in nine families with 50 affected members. Arch Intern Med 1976, 136:769-777 [DOI] [PubMed] [Google Scholar]
- 3.Ebert MP, Hoffmann J, Schneider-Stock R, Kasper HU, Schulz HU, Lippert H, Roessner A, Malfertheiner P: Analysis of K-ras gene mutations in rare pancreatic and ampullary tumours. Eur J Gastroenterol Hepatol 1998, 10:1025-1029 [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa T, Nakao A, Nomoto S, Hosono J, Harada A, Nonami T, Takagi H: Immunohistochemical and molecular biological studies of serous cystadenoma of the pancreas. Pancreas 1998, 16:40-44 [DOI] [PubMed] [Google Scholar]
- 5.Yamaguchi K, Chijiiwa K, Noshiro H, Torata N, Kinoshita M, Tanaka M: Ki-ras codon 12 point mutation and p53 mutation in pancreatic diseases. Hepatogastroenterology 1999, 46:2575-2581 [PubMed] [Google Scholar]
- 6.Vortmeyer AO, Lubensky IA, Fogt F, Linehan WM, Khettry U, Zhuang Z: Allelic deletion and mutation of the von Hippel-Lindau (VHL) tumor suppressor gene in pancreatic microcystic adenomas. Am J Pathol 1997, 151:951-956 [PMC free article] [PubMed] [Google Scholar]
- 7.Sorio C, Baron A, Orlandini S, Zamboni G, Pederzoli P, Huebner K, Scarpa A: The FHIT gene is expressed in pancreatic ductular cells and is altered in pancreatic cancers. Cancer Res 1999, 59:1308-1314 [PubMed] [Google Scholar]
- 8.Chilosi M, Doglioni C, Magalini A, Inghirami G, Krampera M, Nadali G, Rahal D, Pedron S, Benedetti A, Scardoni M, Macri E, Lestani M, Menestrina F, Pizzolo G, Scarpa A: p21/WAF1 cyclin-kinase inhibitor expression in non-Hodgkin’s lymphomas: a potential marker of p53 tumor-suppressor gene function. Blood 1996, 88:4012-4020 [PubMed] [Google Scholar]
- 9.Samowitz WS, Slattery ML, Potter JD, Leppert MF: BAT-26 and BAT-40 instability in colorectal adenomas and carcinomas and germline polymorphisms. Am J Pathol 1999, 154:1637-1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scarpa A, Moore PS, Rigaud G, Inghirami G, Montresor M, Menegazzi M, Todeschini G, Menestrina F: Molecular features of primary mediastinal B-cell lymphoma: involvement of p16INK4A, p53 and c-myc. Br J Haematol 1999, 107:106-113 [DOI] [PubMed] [Google Scholar]
- 11.Scarpa A, Capelli P, Villaneuva A, Zamboni G, Lluis F, Accolla R, Mariuzzi G, Capella G: Pancreatic cancer in Europe: Ki-ras gene mutation pattern shows geographical differences. Int J Cancer 1994, 57:167-171 [DOI] [PubMed] [Google Scholar]
- 12.Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C, Hirohashi S: Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol 1993, 142:1534-1543 [PMC free article] [PubMed] [Google Scholar]
- 13.Brauch H, Weirich G, Brieger J, Glavac D, Rodl H, Eichinger M, Feurer M, Weidt E, Puranakanitstha C, Neuhaus C, Pomer S, Brenner W, Schirmacher P, Storkel S, Rotter M, Masera A, Gugeler N, Decker HJ: VHL alterations in human clear cell renal cell carcinoma: association with advanced tumor stage and a novel hot spot mutation. Cancer Res 2000, 60:1942-1948 [PubMed] [Google Scholar]
- 14.Hahn SA, Seymour AB, Hoque AT, Schutte M, da Costa LT, Redston MS, Caldas C, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE: Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer Res 1995, 55:4670-4675 [PubMed] [Google Scholar]
- 15.Yeh JJ, Marsh DJ, Zedenius J, Dwight T, Delbridge L, Robinson BG, Eng C: Fine-structure deletion mapping of 10q22–24 identifies regions of loss of heterozygosity and suggests that sporadic follicular thyroid adenomas and follicular thyroid carcinomas develop along distinct neoplastic pathways. Genes Chromosom Cancer 1999, 26:322-328 [DOI] [PubMed] [Google Scholar]
- 16.Cappellen D, Gil Diez de Medina S, Chopin D, Thiery JP, Radvanyi F: Frequent loss of heterozygosity on chromosome 10q in muscle-invasive transitional cell carcinomas of the bladder. Oncogene 1997, 14:3059-3066 [DOI] [PubMed] [Google Scholar]
- 17.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R: PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275:1943-1947 [DOI] [PubMed] [Google Scholar]
- 18.Tse JY, Wong JH, Lo KW, Poon WS, Huang DP, Ng HK: Molecular genetic analysis of the von Hippel-Lindau disease tumor suppressor gene in familial and sporadic cerebellar hemangioblastomas. Am J Clin Pathol 1997, 107:459-466 [DOI] [PubMed] [Google Scholar]
- 19.Olschwang S, Richard S, Boisson C, Giraud S, Laurent-Puig P, Resche F, Thomas G: Germline mutation profile of the VHL gene in von Hippel-Lindau disease and in sporadic hemangioblastoma. Hum Mutat 1998, 12:424-430 [DOI] [PubMed] [Google Scholar]
- 20.Eng C, Crossey PA, Mulligan LM, Healey CS, Houghton C, Prowse A, Chew SL, Dahia PL, O’Riordan JL, Toledo SP: Mutations in the RET proto-oncogene and the von Hippel-Lindau disease tumour suppressor gene in sporadic and syndromic phaeochromocytomas. J Med Genet 1995, 32:934-937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng WR, Scherer SW, Koutsilieris M, Huizenga JJ, Filteau F, Tsui LC, Nepveu A: Loss of heterozygosity and reduced expression of the CUTL1 gene in uterine leiomyomas. Oncogene 1997, 14:2355-2365 [DOI] [PubMed] [Google Scholar]
- 22.Haas CJ, Diebold J, Hirschmann A, Rohrbach H, Schmid S, Lohrs U: Microsatellite analysis in serous tumors of the ovary. Int J Gynecol Pathol 1999, 18:158-162 [DOI] [PubMed] [Google Scholar]
- 23.Ogata T, Ayusawa D, Namba M, Takahashi E, Oshimura M, Oishi M: Chromosome 7 suppresses indefinite division of nontumorigenic immortalized human fibroblast cell lines KMST-6 and SUSM-1. Mol Cell Biol 1993, 13:6036-6043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zenklusen JC, Oshimura M, Barret JC, Conti CJ: Inhibition of tumorogenicity of a murine squamous cell carcinoma (SCC) cell line by a putative tumour suppressor gene on human chromosome 7. Oncogene 1994, 9:2817-2825 [PubMed] [Google Scholar]
- 25.Xiao S, Geng JS, Feng XL, Liu XQ, Liu QZ, Li P: Cytogenetic studies of eight primary gastric cancers. Cancer Genet Cytogenet 1992, 58:79-84 [DOI] [PubMed] [Google Scholar]
- 26.Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM: cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell 1996, 85:829-839 [DOI] [PubMed] [Google Scholar]
- 27.Yu ZK, Gervais JL, Zhang H: Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci USA 1998, 95:11324-11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stebbins CE, Kaelin WG, Jr, Pavletich NP: Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science 1999, 284:455-461 [DOI] [PubMed] [Google Scholar]
- 29.Dealy MJ, Nguyen KV, Lo J, Gstaiger M, Krek W, Elson D, Arbeit J, Kipreos ET, Johnson RS: Loss of Cul1 results in early embryonic lethality and dysregulation of cyclin E. Nat Genet 1999, 23:245-248 [DOI] [PubMed] [Google Scholar]
- 30.Seymour A, Hruban R, Redston M, Caldas C, Powell S, Kinzler K, Yeo C, Kern S: Allelotype of pancreatic adenocarcinoma. Cancer Res 1994, 54:2761-2764 [PubMed] [Google Scholar]
- 31.Chung DC, Brown SB, Graeme-Cook F, Tillotson LG, Warshaw AL, Jensen RT, Arnold A: Localization of putative tumor suppressor loci by genome-wide allelotyping in human pancreatic endocrine tumors. Cancer Res 1998, 58:3706-3711 [PubMed] [Google Scholar]
- 32.Chung DC, Smith AP, Louis DN, Graeme-Cook F, Warshaw AL, Arnold A: A novel pancreatic endocrine tumor suppressor gene locus on chromosome 3p with clinical prognostic implications. J Clin Invest 1997, 100:404-410 [DOI] [PMC free article] [PubMed] [Google Scholar]